소아 급성골수성백혈병의 진단 및 치료
이재욱ㆍ조 빈
가톨릭대학교 의과대학 소아과학교실
Diagnosis and Treatment of Pediatric Acute Myeloid Leukemia
Jae Wook Lee, M.D., Ph.D. and Bin Cho, M.D., Ph.D.
Department of Pediatrics, College of Medicine, The Catholic University of Korea, Seoul, Korea
Acute myeloid leukemia (AML) is a heterogeneous malignancy that comprises 25-30%
of pediatric leukemias in Korea. Several inherited diseases, such as Down syndrome and Fanconi anemia, predispose towards AML leukemogenesis. Subgrouping of AML is a key diagnostic step, previously done with the French-American-British (FAB) classi- fication and recently complemented by that of the World Health Organization (WHO).
An important feature of AML is the possibility of chloroma at diagnosis, which, if de- tected, requires follow-up evaluation to determine treatment response. Numerous genetic abnormalities with prognostic relevance have recently been found, the most important of which include those of the core-binding factor (CBF) leukemias, and FLT3-ITD mutation. These genetic abnormalities, combined with patient response to initial treat- ment, allow for a scheme of risk stratification, and the current consensus is to treat low risk patients with chemotherapy only, whereas high risk patients may receive allogeneic transplant in first remission, although the benefits of transplant remain inconclusive.
Overall, the outcome of children and adolescents with AML has improved significantly so that many clinical trials now report event-free survival of around 60%. However, much of this improvement stems from better supportive care and transplant methods, and the genetics-based diagnostic advances in AML have yet to result in enhanced treatment.
New therapeutics, including possibly targeted therapy, are necessary to further improve the outcome of pediatric AML.
pISSN 2233-5250 / eISSN 2233-4580 http://dx.doi.org/10.15264/cpho.2015.22.1.8
Clin Pediatr Hematol Oncol 2015;22:8∼14
Received on April 7, 2015 Revised on April 13, 2015 Accepted on April 16, 2015
Corresponding Author: Bin Cho
Department of Pediatrics, College of Medicine, Seoul Saint Mary's Hospital, The Catholic University of Korea, Seocho-gu, Banpo-daero 222, Seoul 137-701, Korea Tel: +82-2-2258-6187 Fax: +82-2-537-4544
E-mail: [email protected]
Key Words: Acute myeloid leukemia, Children, Genetic abnormalities, Chemotherapy, Hematopoietic cell transplantation
Introduction
Acute myeloid leukemia (AML) is the second most com- mon form of leukemia after acute lymphoblastic leukemia in children and adolescents, and accounts for approx- imately 25-30% of pediatric leukemias in Korea. In the past,
patients with AML had a uniformly dismal prognosis.
However, improvements in risk stratification based on ge-
netic and cytogenetic abnormalities harbored by the leuke-
mic blast, as well as response to therapy, and advances in
hematopoietic cell transplantation (HCT) methodology and
supportive care have all contributed to better survival for
childhood AML. Several national and multi-national trials
Fig. 1. (A) Morphology of AML with maturation (M2), showing myeloblasts with abundant cytoplasmic granulation, as well as more
mature myeloid precursors (Wright Giemsa, ×1,000). (B) Morphology of AML, M7 characterized by megakaryoblasts with cytoplasmic bleb formation and sparse cytoplasmic granulation (Wright Giemsa, ×1,000).now report around 60% event-free survival (EFS) and 70%
overall survival (OS).
Predisposition
Most patients with de novo AML do not have significant risk factors for leukemogenesis. However, many inherited diseases may predispose to subsequent diagnosis of AML.
Down syndrome is perhaps the most widely known amongst the inherited diseases with a predilection for acute leuke- mia, including AML, with one large-scale study reporting that Down syndrome patients comprise around 14% of chil- dren with AML [1]. Other genetic diseases that may evolve into AML include Fanconi anemia, Bloom syndrome, Kostmann disease, Shwachman-Diamond syndrome, Diamond-Blackfan anemia, Li-Fraumeni syndrome, and neurofibromatosis type 1. Previous chemotherapy for a different malignancy or im- munosuppressive therapy is also a major risk factor for sec- ondary AML. These cases are currently classified as “thera- py-related myeloid neoplasms” according to the World Health Organization (WHO) classification, and are characterized by cytogenetic abnormalities such as monosomy 5, monosomy 7 and MLL gene rearrangements [2].
Classification of AML
Historically, AML has been classified according to the French-American-British (FAB) system which categorizes AML according to morphology, from minimally differentiated (M0) to megakaryoblastic leukemia (M7). Each subtype of AML is morphologically distinct. For example, AML with maturation (M2) is noted for the presence of myeloblasts with abundant cytoplasmic granulation and Auer rods, co- existing with promyelocytes and myelocytes (Fig. 1A). In contrast, AML, M7 is characterized by megakaryoblasts which often have cytoplasmic bleb or pseudopod formation, and little granulation (Fig. 1B). Bone marrow fibrosis, resulting in a difficult diagnostic aspiration, is a pathologic hallmark of AML, M7 at diagnosis.
In contrast to the morphology-based FAB classification of AML, the WHO classification is notable for grouping AML according to recurrent genetic abnormalities [2]. Included within this group are the genetic abnormalities that make up the core-binding factor (CBF) leukemias, t(8;21)(q22;q22) (RUNX1-RUNX1T1), and inv(16)(p13.1q22) or t(16;16)(p- 13.1;q22)(CBFB-MYH11), which are frequently observed in children with AML, as well as abnormalities that are rela- tively scarce, such as t(6;9)(p23;q34)(DEK-NUP214).
Both the FAB classification and the WHO classification
Fig. 2. (A) 11 year old male patient
with AML1-ETO (+) AML with periorbital EMI (arrow) resulting in proptosis at diagnosis. (B) 13 year old female patient with AML1-ETO (+) AML with sacral EMI (arrow).have distinct advantages. The FAB classification is important in that it allows the specific diagnosis of AML subtypes that are morphologically unique, such as acute erythroid leuke- mia (M6) and acute megakaryoblastic leukemia (M7), while the WHO classification perhaps aids in better clarifying overall prognosis, based on the genetic features of the leu- kemic blast. Hence, an attempt at subgrouping AML ac- cording to both classification schemes may have comple- mentary benefits in the final diagnosis of the child with AML.
Extramedullary Involvement
A distinct feature of AML is extramedullary involvement (EMI) of leukemia, manifested as chloromas, or granulocytic sarcomas. Large-scale studies indicate that the incidence of EMI in children with de novo AML is 7-23% [3,4]. EMI may occur anywhere in the body, and may be overt at diagnosis resulting in symptoms such as proptosis or paraplegia due to spinal cord compression (Fig. 2A, B), or may include asymptomatic organ involvement. As EMI may be found re- gardless of patient symptoms, imaging evaluations such as whole body MRI at diagnosis may aid in detecting baseline EMI prior to therapy. Due possibly to its rarity in childhood AML, the prognostic importance of EMI remains unclear, when compared to the clear association between certain ge- netic abnormalities and overall prognosis. A Japanese study of 240 children with AML found that the presence of EMI when combined with an initial white blood cell (WBC)
count of greater than 100×10
9/L predicted lower EFS [4].
Certainly for patients with EMI, important post-treatment evaluations, such as after remission induction and prior to and after HCT, should include evaluation of EMI as well as the bone marrow.
Initial Treatment
Improvements in HCT methods and enhanced supportive care, especially the treatment of infectious complications, have led to increased survival for pediatric AML, so that many large scale studies report 50-60% EFS and 60-70% OS [5-10].
The key elements of remission induction therapy have remained constant and usually consist of an anthracycline such idarubicin given for 3 days and cytarabine given for 7 days. Either etoposide or thioguanine may be added to this regimen, and the Medical Research Council (MRC) AML 10 trial has shown that both have equivalent therapeutic ef- ficacy [11]. Central nervous system (CNS) directed intra- thecal treatment (for example, cytarabine) should also be given with induction treatment.
An important consideration for patients who start in-
duction chemotherapy is hyperleukocytosis, that is a WBC
count of greater than 100×10
9/L at diagnosis. These pa-
tients comprise almost 20% of children with AML, and are
associated with FAB M1, M4 or M5 morphology, inv(16)
(p13.1q22) and FLT3-ITD genetic abnormalities [12]. Hyper-
leukocytosis is clinically significant because it contributes to
early mortality through pulmonary and intracranial hemor- rhage resulting from leukostasis of the large myeloblasts.
Options in this setting include leukapheresis to rapidly re- duce the WBC count, but data on its efficacy is conflicting and at least one study has shown that such procedures do not result in lower early mortality [12]. Optimal manage- ment of such patients may require a combination of leuko- depletion and aggressive treatment of coagulopathy through transfusions of platelets and fresh frozen plasma, and judi- cious supportive care has shown to result in improved sur- vival [13]. At best, children with AML and initial hyper- leukocytosis still remain an oncologic emergency without an ideal treatment standard, and which requires intensive support to prevent early mortality.
Genetic Abnormalities and Prognosis
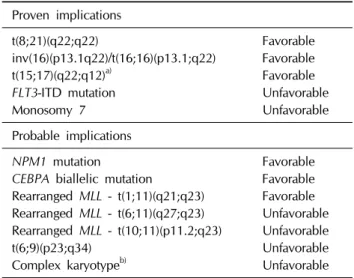
Amongst the cytogenetic abnormalities with a distinctly good prognosis are those of the CBF leukemias, that is, t(8;21)(q22;q22), and inv(16)(p13.1q22) or t(16;16)(p13.1;q22).
Studies from the MRC and Berlin-Frankfurt-Münster (BFM) group both report OS rates above 90% for patients with these genetic abnormalities [14,15]. One factor, however, which may influence this good prognosis is concurrent mu- tation of the KIT proto-oncogene. A large Children’s Oncology Group (COG) study found that KIT mutation status did not adversely affect outcome of children with CBF leukemia [16], in contrast to several other studies which found that children with mutated KIT had lower survival than those patients with wild type KIT [17,18].
Gene mutations are also prognostic in childhood AML.
For adult AML patients, mutations of the nucleophosmin gene (NPM1) confer a good prognosis when found with a normal karyotype and wild type FLT3 [19]. Results from the Pediatric Oncology Group (POG)-9421 study indicate that the incidence of childhood AML patients with mutated NPM1 is around 8%, and that patients with these mutations have a relatively favorable EFS of 69% when found in the setting of wild type FLT3 [20]. Biallelic mutations of the CEBPA gene have also been linked to favorable outcome, with 5-year EFS of 70% [21].
In contrast, several genetic abnormalities are associated
with a poor prognosis. Internal tandem duplication of the juxtamembrane domain of FLT3 (FLT3-ITD) has clearly been linked to lower survival by several large scale pediatric studies [22,23]. Monosomy 7 has also been proven to be a poor prognostic factor in childhood AML. An international study of 258 AML children with either monosomy 7 or dele- tion 7q found that patients with monosomy 7 had an OS rate of 30% [24]. In particular, patients with monosomy 7 and inv(3)(q21q26.2) or t(3;3)(q21;q26.2), genetic abnor- malities that are considered recurrent according to the WHO classification, had a dismal 5% survival rate.
AML patients with a complex karyotype, that is 3 or more structural or numerical abnormalities in the absence of a WHO designated recurrent genetic abnormality, may also have lower survival, as shown by studies from the MRC and BFM groups [14,15]. The genetic abnormality t(6;9)(p22;q34) is defined as recurrent according to the WHO classification, but is rarely found in childhood, with an incidence of less than 1%. This abnormality is more of- ten found in older children and in male patients. Patients with this abnormality often also have the FLT3-ITD muta- tion, but recent studies indicate that patients with t(6;9) (p22;q34) have a high relapse rate and low survival, irre- spective of the FLT3 mutation status [25,26].
The cryptic translocation t(5;11)(q35;p15.5)(NUP98-NSD1) is often found in conjunction with the FLT3-ITD mutation [27], and a recent study based in part on pediatric patients from 4 consecutive trials found that patients with both NUP98-NSD1 and FLT3-ITD mutation had significantly lower CR rate and OS compared with patients with FLT3-ITD mutation alone [28].
Genetic abnormalities involving the MLL gene (11q23)
are found in 15-20% of pediatric AML, with the prognosis
dictated by the MLL rearrangement depending on its trans-
location partner. An international study on 756 AML chil-
dren with MLL rearrangements found that patients with
t(1;11)(q21;q23) had an extremely favorable outcome with
92% EFS, while patients with t(6;11)(q27;q23) or t(10;11)
(p11.2;q23) had less than 20% EFS [29]. A recent study has
shown a relatively high incidence of t(10;11) and t(6;11)
amongst pediatric patients with MLL-rearranged AML, indicat-
ing that the MLL rearrangement is more likely to predict a
poor prognosis in the pediatric patient population [30].
Table 1. Genetic abnormalities that influence prognosis in
childhood AMLProven implications
t(8;21)(q22;q22) Favorable
inv(16)(p13.1q22)/t(16;16)(p13.1;q22) Favorable
t(15;17)(q22;q12)a) Favorable
FLT3-ITD mutation
UnfavorableMonosomy 7 Unfavorable
Probable implications
NPM1 mutation
FavorableCEBPA biallelic mutation
Favorable Rearranged MLL - t(1;11)(q21;q23) Favorable Rearranged MLL - t(6;11)(q27;q23) Unfavorable Rearranged MLL - t(10;11)(p11.2;q23) Unfavorablet(6;9)(p23;q34) Unfavorable
Complex karyotypeb) Unfavorable
a)Found in AML M3, b)3 or more structural or numerical abnor- malities in the absence of a WHO designated recurrent genetic abnormality.