Yakhak Hoeji Vol. 64, No. 5 DOI 10.17480/psk.2020.64.5.394
종 설(Review)
의료제품의 장기추적조사 관련 제도현황과 중장기 추진방향
심다영
†,
*·이정은†,
*·최남경**·정선영***·김희진**·김선하**·김묘송***·성희진*·신주영*,#
*성균관대학교 약학대학, **이화여자대학교 융합보건학과,***중앙대학교 약학대학
Legal Basis Review for the Establishment of Long Term Follow-up of Medical Products in Korea and Mid-Term, Long-Term Plan for
Introduction of new Regulation
Da-Young Shim†,*, Jeong-Eun Lee†,*, Nam-Kyong Choi**, Sun-Young Jung***, Hee-Jin Kim**, Seon-Ha Kim**, Myo-Song Kim***, Hee-Jin Seong*, and Ju-Young Shin*,#
*School of Pharmacy, SungKyunKwan University
**Department of Health Convergence, Ewha Woman’s University
***College of Pharmacy, Chung-Ang University
(Received June 5, 2020; Revised September 18, 2020; Accepted September 23, 2020)
Abstract As the safety issues of medical products have been continuously generated, the need for long-term safety management, such as long term follow-up (LTFU), has been emerged. Current laws and regulations in Korea, however, have limitations to implement LTFU and track medical products immediately. This review examines the current status of safety management of medical products in Korea to propose future plan for establishing medical products LTFU system in Korea. We review the laws and regulations for safe use of medical products and compare regulations, definitions and processes in the following laws: Pharmaceutical Affair Act, Medical Device Act, Act on Safety and Support for Advanced Regenerative Medicine and Advanced Biopharmaceuticals. In case of medicinal products except Advanced Biopharmaceuticals, regulations are needed to be developed and enforced even though there is legal basis for LTFU in Pharmaceutical Affair Act. In Medical Devices Act, there is no legal basis for conducting LTFU. Thus, establishing safety management regulation for LTFU in medical devices is essential. On the other hand, LTFU on advanced biopharmaceutical has implemented since this August with the new law. In order to conduct a LTFU and registry management, the regulatory science center should have a systematic process and manpower. Furthermore, to establish a LTFU system for all medical products, running pilot project is essential to evaluate feasibility. Public relations activities and training expertise to induce stakeholder participation is indispensable for successful implementation of LTFU.
Keywords long term follow-up, medical products, safety regulation, monitoring, registry, advanced biopharmaceutical
서 론(Introduction)
의약품, 의료기기 등 의료제품 산업이 지속적으로 성장함에 따라 국내·외 안전성 관리 강화의 필요성이 높아지고 있다. 최 근 2년간 인공유방 보형물, 인보사케이 주 사태 등 의료 제품에 서 발암유발 위험성이 발견되고 의약품의 주요 원료가 바뀌는 등 국민의 건강에 위협을 주는 안전성 문제가 여러 차례 이슈
된 바 있다. 당시 국내 환자 추적조사 체계가 부재하여, 해당제 품을 사용한 환자 소재파악을 위하여는 요양기관의 협조를 얻 어야 하는 등 신속한 대응에 어려움을 겪었다.
1-3)또한 현 체계 에서는 이상사례 발생 시 집단 및 개인 단위에서 명확한 인과 성 평가를 위한 데이터 확보가 어렵다는 문제가 있다.
세포치료제, 유전자치료제, 조직공학치료제 등 살아있는 세포 나 유전물질을 함유한 첨단바이오의약품의 경우에는 투여 후 인 체에 상당기간 잔류하는 특성으로 특성변화, 종양원성 등 의 잠 재적 위험성이 있으나, 아직까지는 구체적으로 밝혀진 바가 없 다.
4-5)또한, 첨단바이오의약품 이외에도 만성질환에 사용되는 의 약품은 장기복용에 따라, 인체이식형 의료기기는 기기 자체 외 에도 시술자의 기술, 시술과정 등 다양한 외부요인들에 의해 안 전성 및 유효성이 영향을 받을 수 있다.
6)따라서 일부 의약품과 의료기기는 임상시험만으로는 장기 안전성 및 유효성의 정보를
†
These authors contributed equally to this work
#
Corresponding author
Ju-Young Shin, School of Pharmacy, Sungkyunkwan University, 2066, Jangan-gu, Suwon-si, Gyeonggi-do, South Korea
Tel: 82-31-290-7702, Fax: 031-290-8800
E-mail: [email protected]
확립하는 데 어려움이 있어 장기적 안전성 모니터링이 필요하다.
7-8)잇따른 안전성 이슈 발생 이후, 국내에서도 의료제품 안전성 관리 강화의 중요성이 대두됨에 따라 여러가지 제도의 강화 움 직임이 나타나고 있다. 2020년 8월 ‘첨단재생의료 및 첨단바이 오의약품 안전 및 지원에 관한 법률 (약칭: 첨단재생바이오법)’
이 시행되었다. 기존의 약사법 하에서 관리되었던 세포치료제, 유전자치료제, 조직공학제제, 첨단바이오융복합제제를 ‘첨단바이 오의약품’으로 지정하고 새로운 법령 체계하에서 제품 특성을 고려한 체계적인 품질, 안전성 및 유효성 관리가 가능하게 되었 다. 이에 따라 장기추적조사 수행을 위한 법적 근거를 마련하였 다. 인체이식형 의료기기의 경우 장기간 인체내 삽입되어 안전 상의 심각한 위해를 초래할 가능성이 있는 의료기기에 대하여 장기간 추적관찰을 위한 시판 후 안전성 규제강화 및 환자등록 연구 추진을 위한 국가 주도의 노력이 국내·외에서 진행 중이 다. 특히 미국 FDA는 의료기기 환자등록연구를 활성화하고 연 구방법을 개발하기 위한 기관인 National Evaluation System for health Technology Coordinating Center (NESTcc) 설립 및 운영을 지원하고 있다.
하지만 체계적인 장기추적조사 수행을 위하여 규제과학센터 의 전문성과 인프라를 갖추고 내부 업무프로세스 확립하는 등 추가적인 준비가 필요하다. 또한 첨단바이오의약품 이외에도 장 기간 사용에서의 안전성 추적관리가 필요한 의약품, 의료기기에 대한 국내 장기추적조사 추진하기 위하여 법적 근거 검토가 필 요하다. 이에 본 연구는 첨단재생바이오법 상의 장기추적조사의 체계적 수행을 위한 방안을 모색하고 나아가 의료제품 전 영역 의 장기추적조사 체계의 중장기 추진방안을 제시하고자 하였다.
연구 방법(Research Methods)
본 연구에서는 의약품을 안전성 관리 규정 적용 범위에 따라 첨단재생바이오 법 상 분류된 첨단바이오의약품(세포치료제, 유 전자치료제, 조직공학제제, 첨단바이오융복합제제)과 그 외 의약 품으로 구분하여 표현하였다. 전 영역 의료제품 장기추적조사를 위한 연구 대상은 첨단바이오의약품, 그 외 의약품, 의료기기이 며 의약외품은 장기적 사용에 대한 안전성 이슈가 극히 드물 것 으로 판단되어 조사대상에서 제외하였다. 의약품과 의료기기를 한 번에 포함하고 있는 융복합제품의 경우 주작용에 따라 의약 품 혹은 의료기기로 분류되어 관리되기 때문에 본 연구에서 따 로 고려하지는 않았다.
조사 내용
첨단바이오의약품을 제외한 의료제품에서 능동감시 등 장기 추적조사 추진 가능성 검토를 위하여 기존의 의료제품 안전관 리제도를 조사하였다. 이후, 장기추적조사의 구체적 내용 및 다 른 의료제품관련 제도하에서의 장기추적조사 수행 가능성 검토 를 위하여 첨단재생바이오법 상 장기추적조사 관련 내용을 조 사하고, 이를 토대로 약사법, 의료기기법 상 장기추적조사 수행
의 법적 근거를 검토하고 내용을 비교·분석하여 제시하였다.
또한 첨단재생바이오법에서 명시하고 있는 규제과학센터의 역 할을 조사하고, 역할수행을 위하여 기관 내 필요한 전문인력과 조직구성(안)을 의료제품 레지스트리 설계 및 운영에 관한 국외 문헌 조사를 통하여 제시하였다. 마지막으로 장기추적조사 추진 을 위한 국내 내·외부 환경을 분석하여 이에 맞는 전략을 제 시하였으며, 국내 의료제품 전 영역의 장기추적조사 체계 추진 을 위한 중장기 로드맵을 제시하였다.
조사 방법
국가법령정보센터 홈페이지를 이용하여 가장 최신의 법령을 조사하였다.
9)장기추적조사 수행 가능성이 있는 기존 의료제품 안전성 관리 제도를 파악하기 위하여 약사법, 의료기기법 및 관 련 하위법령에서 재심사, 재평가, 허가갱신제도, 약사법 상 위해 성관리계획(Risk Management Plan, RMP), 의료기기법 상 의료 기기 추적관리(Medical devices tracking)제도를 조사·분석하였다.
첨단재생바이오법 상 장기추적조사 관련 내용 분석과 그 외 의약품 및 의료기기에 대한 장기추적조사 추진 가능성을 확인 하기 위하여 첨단재생바이오법(장기추적조사), 약사법(위해성관 리계획), 의료기기법(추적관리) 및 관련 하위법령 내용을 조사·
분석하여 법적근거를 검토였다. 그 결과를 각 법령 별 관련조항, 내용, 수행 기간, 적용대상, 조사수행주체, 수집정보 및 주체, 환 자정보 보관주체, 벌칙조항에 관한 사항으로 제시하여 비교하였다.
또한, 장기추적조사 수행을 위한 첨단재생바이오법 상 명시되 어 있는 규제과학센터의 역할을 검토하고, 해당 업무를 체계적 으로 수행하기 위하여 규제과학센터 내 필요한 전문인력과 조 직구성(안)을 제시하였다. 이를 위하여, 미국 보건의료연구원 (AHRQ, Agency for Healthcare Research and Quality)에서 발간 한 레지스트리 구축, 유지, 데이터 평가 등을 위한 참고서
「Registries for Evaluating Patient Outcomes: A User’s Guide」
에서 제시한 ‘Planning Registries’, ‘Use of Registries in Product Safety Assessment’ 관련 내용을 참고하였으며, 해당 내용을 국 내 규제과학센터의 역할에 적합하게 변경하여 제시하였다.
보다 전문적인 현황 파악과 국내실정에 맞는 방안마련을 위
하여 현업 종사자 및 관련 전문가 의견 수렴을 위한 전문가 자
문회의를 진행하였다. 약물역학연구자, 장기추적관찰 경험이 있
는 임상의, 규제전문가와 세포치료제 제조판매회사, 의료기기회
사, 국내·외 제약사, 의료제품 별 협회 관계자 등 총 18명이
참석하였다. 국내 장기추적조사 체계 수립, 환자등록 시 개인정
보 보호 관련, 규제과학센터 설립 및 운영에 관한 고려사항을
주요안건으로 자문회의는 총 2회에 걸쳐 진행되며 연구내용 전
반에 관한 검토 및 자문을 받았다. 또한 전문가 논의를 거쳐 국
내 의료제품 전 영역 장기추적조사 체계도입 및 운영을 위한 현
재 국내환경의 강점(Strength), 약점(Weakness), 기회(Opportunity),
위협(Threat) 요인을 논의하고 SWOT분석을 수행하였으며, 이를
토대로 강점의 극대화 전략 및 약점 극복전략을 제시하였다.
결 과(Results)
국내 의료제품 장기추적조사 관련 법제도 비교·분석
기존의 의약품, 의료기기 안전성 관리를 위한 제도를 조사한 결과 약사법과 의료기기법에서 재심사, 재평가, 허가갱신 제도 가 공통으로 포함되어 있었으며, 능동적으로 관리대상 제품에 대한 정보를 수집하기 위한 제도로는 약사법 상 위해성관리계 획(RMP), 의료기기법 상 의료기기 추적관리(Medical devices tracking) 제도가 있었다. 구체적인 내용은 Table 1에 제시되어 있 다. 기존의 제도 중 위해가능성이 높은 제품을 관리대상으로 지 정하여 제품 및 사용 환자정보를 능동적으로 수집하여 관리한 다는 점에서 위해성관리계획, 의료기기 추적관리제도에서 첨단 바이오의약품 이외의 의료제품 대상 장기추적조사 추진 가능성 을 검토하고, 첨단재생바이오법의 추적관리조사 내용과 비교하였다.
첨단재생바이오법에 따르면 첨단바이이오의약품의 종류, 비임 상시험 및 임상시험 결과, 투여대상 및 방법, 기 축적된 안전성 정보를 고려하여 장기간 안전성을 관찰할 필요가 있다고 판단 되는 제품에 대하여 장기추적조사 대상으로 지정할 수 있다. 장 기추적조사 대상으로 지정된 첨단바이오의약품의 품목허가 및 수입자, 임상시험계획 승인을 받고자 하는 자는 허가 및 승인 신청 시 조사계획을 수립하여 제출하고, 장기추적조사를 수행을 담당하여 매년 장기추적조사의 내용 및 결과 등을 식품의약품 안전처장(이하, 식약처장)에 보고하여야 한다. 품목허가권자 및 수입자, 임상시험승인받은자는 판매·공급내역을, 해당 제품을 투여하는 의사·치과의사 등은 매 사용시 마다 환자동의를 받 아 환자정보 및 투여정보를 규제과학센터 전산망에 등록하여야
한다. 줄기세포치료제는 5년 이내, 유전자치료제는 15년 이내, 동물 조직·세포 포함 첨단바이오의약품는 30년 이내로 장기추 적조사기간을 부여한다.
약사법 상 위해성관리계획이란 신약 등의 품목허가를 받고자 하는 자는 품목허가 신청 시 또는 시판 후 안전성 중점검토를 위해 해당 품목에서 안전성 및 유효성 중점 검토항목, 의약품 감시 계획, 위해성 완화 조치방법 등의 내용을 포함하여 계획을 수립하는 것을 말한다. 이후 대상 의약품의 품목허가권자는 계 획서에 따라 위해성 관리계획을 이행하고 평가결과와 그에 따 른 안전성·유효성 등에 대한 종합적인 의견을 정기적으로 식 약처장에게 보고해야 한다. 첨단바이오의약품 또한 위해성관리 계획 제출 및 이행 대상에 포함된다.
의료기기법상 추적관리제도는 사용 중 발생하는 부작용 또는 결함이 인체에 치명적인 위해를 줄 수 있는 의료기기를 관리대 상으로 지정하고 그 소재파악을 목적으로 한다. 국내 추적관리 대상 품목은 52종류로 지정되어 있으며, 해당 품목으로 허가를 받는 의료기기는 추적관리대상이 된다. 추적관리대상 의료기기 의 제조업자·수입업자·판매업자·임대업자·수리업자(이하, 취급자)는 제품의 제조·판매(구입) ·임대 또는 수리 등에 대 한 내용을 기록하고 매월 식약처장에게 제출하여야 한다. 해당 제품을 취급하는 의사·한의사·치과의사 등(이하, 사용자)은 의 료기기 이용 환자에 대한 추적 가능한 사항을 기록하여 보관하 여야 한다. 취급자와 달리 사용자는 정기적 보고의 의무는 없고 식약처장이 제출을 요구하는 경우에 한하여 제출하면 된다. 장 기추적조사, 위해성관리계획, 의료기기 추적관리제도의 상세한 내용은 Table 2에 제시하였다.
Table 1. Current safety management regulation of medical products
Regulation Medicinal products Medical devices
Reexamination
신약 또는 이미 허가된 의약품과 유효성분의 종류 또는 배합비율 이 다른 전문의약품, 이미 허가된 의약품과 유효성분은 동일하나 투여경로가 다른 전문의약품, 이미 허가된 의약품과 유효성분 및 투여경로는 동일하나 명백하게 다른 효능·효과를 추가한 전문의 약품, 그 밖에 식품의약품안전처장이 재심사를 받을 필요가 있다 고 인정한 의약품을 대상으로 품목 허가 받은 날부터 품목에 따라 4~6 년 이 지난 날부터 3개월 이내 식약처장의 재심사를 받아야 함.
재심사 신청 시 정기보고 결과자료 및 이를 종합적으로 분석, 평가 한 자료를 제출
신개발의료기기 (국내 대상 질환 환자 수가 적 고 용도상 특별한 효용가치를 갖는 경우) 또는 희소의료기기 (작용원리, 성능, 사용목적 등이 기 허가품목 중 없는 경우)에 해당하는 의료기 기인 경우 식약처장은 해당 품목이 시판된 후 4~7 년 범위 이내 일정기간 내에 그 안전성과 유 효성에 대하여 재심사 받을 것을 명 할 수 있음.
Renewal of Marketing
Approval
의약품의 품목허가 및 품목신고의 유효기간은 5년으로 하며, 품 목허가 및 신고 받은 의약품에 대해 5년마다 품목허가·신고의 갱신이 필요. 갱신 신청 시 유효기간 내 수집된 안전관리에 관한 자료 및 조치계획, 국외 사용현황 및 안전성 관련 조치에 관한 자 료, 품질관리·표시기재·제조 및 수입 실적에 관한 자료를 제출 해야함.
제조허가ㆍ제조인증ㆍ제조신고 및 수입허가 ㆍ수입인증ㆍ수입신고(이하 “제조허가 등”)의 유효기간은 5년으로 하며, 유효기간이 끝난 후 에 계속하여 해당 의료기기를 제조 또는 수입 하려면 식약처장에게 제조허가 등을 갱신 받아 야 함[시행일: 2020. 10. 08.].
Active monitoring
Risk management plan
신약, 희귀의약품, 시판 후 중대한 부작용 발생으로 인해 RMP제출 이 필요하다고 식약처장이 인정하는 의약품, 신청인이 RMP제출 이 필요하다고 인정하는 의약품, 재심사대상 의약품으로 지정되 는 전문의약품, 줄기세포 치료제 등의 제조판매·수입 품목허가를 받으려는 자는 안전성 및 유효성 중점 검토항목, 의약품 감시 계획, 위해성 완화 조치방법 등을 포함하는 종합적인 의약품 안전관리 계 획(이하 “위해성 관리계획”)을 식약처장에게 제출하여야 함.
Medical devices tracking
인체에 1년 이상 삽입되는 의료기기, 의료기관
외의 장소에서 사용가능한 생명유지용 의료기
기 중에서 사용 중 부작용 또는 결함이 발생하
여 인체에 치명적 위해를 줄 수 있어 그 소재를
파악해 둘 필요가 있는 의료기기를 별도로 정
하여 관리할 수 있음.
장기추적조사 체계 운영지원을 위한 규제과학센터 설립 시 고 려사항
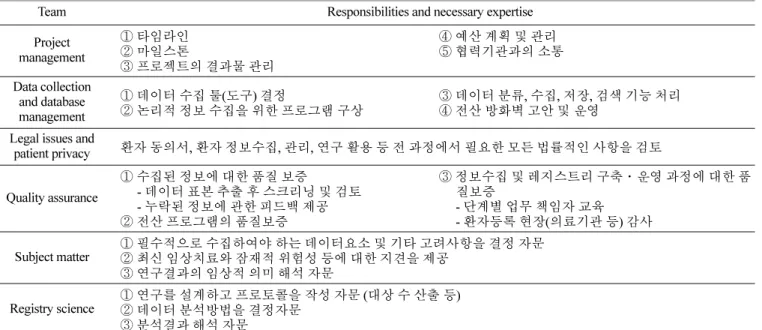
첨단재생바이오법 제 32조(첨단바이오의약품 규제과학센터 설 립·지정)와 관련 하위법령에 따르면 규제과학센터는 첨단바이 오의약품 투여 및 판매·공급내역의 등록·관리에 필요한 통합 전산망을 구축하여 운영하며, 장기추적조사 지원에 관한 업무를 수행한다. 통합 전산망 구축·운영 및 장기추적조사를 수행을 체계적으로 지원하기 위하여 규제과학센터 내부적으로 체계적 인 업무 프로세스와 전문인력구성이 필요하며, 규제과학센터 설 립 초기에 관리대상으로 지정되는 제품의 예상 수와 특성을 고 려하여 조직과 구성인력, 규모 등을 적절히 갖추어야한다.
10)미 국 AHRQ의 「Registries for Evaluating Patient Outcomes: A User’s Guide」에 따르면, 의료정보 수집 및 보관을 위한 레지스 트리 운영을 위해서는 총 6개 영역의 전문인력이 필요하다. 통 합 전산망을 구축하여 장기추적조사 체계를 운영하면서 환자정 보를 수집·관리하기 위하여 총괄운영팀, 전산팀, 법률팀, 품질 관리팀이 필요하다. 또한, 안전성 정보를 수집하고 수집된 정보 를 분석하기 위하여 임상팀과 통계분석팀이 필요하다.
11)AHRQ 의 가이드북 내용을 바탕으로 국내 장기추적조사 지원에 관한 업무를 수행하기 위한 규제과학센터의 조직구성(안)을 Table 3
에 제시하였다. 현행 법률상 안전성 정보 수집 및 분석을 수행 하는 주체는 품목허가권자 및 수입자, 임상시험승인받은자로 조 사되어, 이 내용을 고려하여 국내 규제과학센터의 임상팀과 통 계분석팀은 해당 내용에 대한 사전검토 및 자문역할을 수행하 는 것으로 제안하였다.
의료제품 장기추적조사 추진을 위한 국내 환경 SWOT분석 및 전략방안
SWOT 분석은 장기추적조사 제도 도입 및 운영을 주관하는 규 제기관과 정부의 관점에서 진행하였다. 내부환경을 의료제품 안 전성 관리에 관한 현행 법·제도, 규제과학센터의 내부체계, 통 합전산망 등 기존의 인프라로 규정하였다. 외부환경은 그 외의 장기추적 조사 추진에 직간접적으로 영향을 미칠 수 있거나 원 활한 장기추적조사를 위하여 고려해 볼 수 있는 사항을 외부환 경으로 규정하여 분석을 수행하였다. 장기추적조사 추진에서 강 점(Strength), 기회(Opportunity)로 작용하는 내·외부적 요소에는 전국민건강보험제도와 국가 전산망, 바코드, Radio-Frequency Identification (RFID), 의료기기 Unique Device Identification (UDI) 의무화 도입 등의 인프라와 기존의 시판 후 안전성관리 프로세스가 있다. 또한, 급성장하는 첨단 바이오의약품 시장, 세 Table 2. Legal basis for long term follow-up of medical products
Law
Act on Safety and Support for Advanced Regenerative Medicine and Advanced
Biopharmaceuticals
Pharmaceutical Affair Act Medical Device Act
Atricle 제30조 (첨단바이오의약품 장기추적 조사 및 투여내역 등록)
제37조의 3 (의약품의 시판 후 안전관리) 시행규칙 제 4조 제1항 제11호
( 위해성관리계획)
제29조 (추적관리대상 의료기기) 제30조 (기록의 작성 및 보족)
Subjects
1. 사람 줄기세포 포함 세포치료제 2. 동물의 조직·세포 포함 세포치료
제
3. 유전자치료제 등
1. 신약, 재심사대상 의약품 2. 희귀의약품
3. 시판 후 부작용 발생 혹은 신청인이 필요하다고 인정하는 경우
4. 첨단바이오의약품
1. 인체에 1년 이상 삽입되는 의료기 기 (48종)
2. 의료기관 외 장소에서 사용가능한 생명유지용 의료기기 (4종)
Director 품목허가를 받은 자 및 수입자
임상시험계획 승인을 받은자 품목허가를 받은 자 및 수입자 취급자, 사용자
Reporting period 매년 1회 보고 품목허가 후 첫 2년은 매 6개월마다 보고,
이후 매년 보고 취급자: 매달 보고
( 사용자 정기 보고 의무 없음)
Duty
▶ 의사·치과의사·약사
: 투여대상자의 인적사항 및 의학적 과거력, 투여내역 등을 투여일로 부터 10일 이내에 규제과학센터에
▶ 품목허가 받은 자 및 수입 자, 임상 등록 시험계획 승인받은 자
: 판매·공급내역을 규제과학센터 에 등록
▶ 품목허가권자
: 안전관리책임자를 지정하여 위해성 관리계획에 따라 안전정보 수집 및 기록작성. 수집된 안전성 정보의 평 가 결과를 식약처장에게 정기보고
▶ 취급자
: 제조·판매(구입) ·임대·수리 내용 등에 대한 기록. 매월 식약처 장에게 제출
▶ 사용자
: 환자에 대한 추적이 가능하도록 하는 기록. 식약처장 요구 시 10일 안에 제출
Patients’ Data
ownership 규제과학센터에서 보유 품목허가권자가 보유 의료기관에서 보유
Fines
제63조(과태료)
첨단바이오의약품 투여 또는 판매·
공급내역을 등록하지 아니한 자에게 는 1천만원 이하의 과태료를 부과함.
제 95조(벌칙)
안전관리업무를 실시하지 아니한 자는 1 년 이하의 징역 또는 1천만원 이하의 벌금에 처함.
제 54조(벌칙)
추적관리대상 의료기기 기록의 작성
및 보존·제출 의무를 위반한 자는
500 만원 이하의 벌금에 처함.
계적으로 환자중심 안전인식 확산 추세로 인하여 안전성 관리 규제가 강화되고 이를 위하여 Real World Data/Real World Evi- dence (RWD/RWE)활용의 요구도 증가는 장기추적조사 추진에 타당성을 부여한다. 이와 동시에 데이터 기반 신 산업 육성을 위한 데이터 3법, 즉 ‘개인정보 보호법’, ‘신용정보의 이용 및 보 호에 관한 법률’, ‘정보통신망 이용촉진 및 정보보호 등에 관한 법률’의 개정안 통과 및 시행으로 자료원 별 의료데이터 공유 및 활용체계가 활성화될 전망이다. 또한 정보 보안 기술력 강화
로 장기추적조사를 위한 의료정보 수집·관리 및 활용이 용이 해졌다. 의료기기의 경우 한국이 국제 협력 포럼인 International Medical Device Regulators Forum (IMDRF)의 2021년 의장국으 로 선임됨에 따라 국제 조화된 제도를 위한 활동에 박차가 가 해질 전망이다.
그러나 약점(Weakness), 위협(Threat) 요인 또한 존재한다. 우 선적으로 장기추적조사 체계를 운영할 주관기관인 규제과학센 터의 전문인력 및 조직체계, 내부 업무 프로세스가 미완성 단계 Table 3. Registry team for long term follow-up with different kinds of expertise and experience
Team Responsibilities and necessary expertise Project
management
① 타임라인
② 마일스톤
③ 프로젝트의 결과물 관리
④ 예산 계획 및 관리
⑤ 협력기관과의 소통
Data collection and database management
① 데이터 수집 툴(도구) 결정
② 논리적 정보 수집을 위한 프로그램 구상 ③ 데이터 분류, 수집, 저장, 검색 기능 처리
④ 전산 방화벽 고안 및 운영 Legal issues and
patient privacy 환자 동의서, 환자 정보수집, 관리, 연구 활용 등 전 과정에서 필요한 모든 법률적인 사항을 검토
Quality assurance
① 수집된 정보에 대한 품질 보증
- 데이터 표본 추출 후 스크리닝 및 검토 - 누락된 정보에 관한 피드백 제공
② 전산 프로그램의 품질보증
③ 정보수집 및 레지스트리 구축·운영 과정에 대한 품 질보증
- 단계별 업무 책임자 교육 - 환자등록 현장(의료기관 등) 감사 Subject matter ① 필수적으로 수집하여야 하는 데이터요소 및 기타 고려사항을 결정 자문
② 최신 임상치료와 잠재적 위험성 등에 대한 지견을 제공
③ 연구결과의 임상적 의미 해석 자문
Registry science ① 연구를 설계하고 프로토콜을 작성 자문 (대상 수 산출 등)
② 데이터 분석방법을 결정자문
③ 분석결과 해석 자문
The contents of this table referred to AHRQ ’ s ‘ Registries for Evaluating Patient Outcome: A user ’ s guide, 3
rdFig. 1. SWOT analysis for implementing long term follow-up system in Korea.
이며, 국가 주도 환자 등록 레지스트리 운영 및 Database (DB) 관리와 분석 경험이 부족하다. 또한 식약처 외의 부처에서도 식 약처와 무관하게 장기추적조사를 위한 다수의 연구를 진행하고 있어 다 부처 간 소통과 협력이 필요한 실정이다. 그리고 의료 기기의 경우 현행 의료기기법 상 제조회사의 환자정보 접근 및 활용에 대한 법적 근거가 부족하여 장기추적조사 수행의 법적 근거가 사실상 부재하다. 외부적 위협요인으로는 새로운 제도 추가로 인한 제약업계 및 의료기관의 거부감이 발생하며, 전문 가 및 대중의 인식부족 및 개인정보에 민감한 사회 분위기는 환 자의 참여율을 저조하게 만들 수 있는 가능성이 있다. 또한 주 치의 개념이 확립되지 않은 국내 상황에서 10년 이상 환자를 추 적관찰하는 것에 어려움이 있을 수 있으며, 자문회의에 참석한 의료제품 회사 관계자들에 따르면 장기추적조사 수행 주체인 제 조회사의 경험과 전문성이 부족한 실정이다. 마지막으로 최근 의료제품 시장에는 신기술이 빠르게 도입되고 있는 것 또한 고 려대상이다. 앞서 논의되었던 SWOT분석 내용은 모두 Fig. 1에 제시되어 있다.
SWOT 분석 결과내용을 토대로 현행제도와의 통합 및 기존의 인프라 활용 범위 확대 등 현재 국내환경의 강점을 극대화하여 효율적으로 장기추적조사를 추진할 수 있는 방안을 논의하였다.
또한 원활한 장기추적조사 추진에서 방해요인으로 작용할 수 있 는 현재 상황이나 고려되어야 하는 사항을 검토하여 극복 방안 을 모색하여 장기추적조사체계 추진에서의 국내 환경의 SO (역 량확대), WO (기회포착), ST (강점활용), WT (위기대응) 전략방 안을 도출한 결과를 Fig. 2에 제시하였다.
고 찰(Discussion)
전세계적으로 의료제품 안전성 관리가 강화되고 있으며, 특히 장기적 위해가능성이 높은 의료제품을 이용하는 환자에게서 실제 임상에서의 안전성 정보를 능동적으로 수집하여 모니터링하는 장 기추적조사가 진행되고 점차 범위를 확대해 나가고 있다.
12-14)국 내에서도 시판 후 안전성 관리 강화의 필요성을 인식하고, 이를 위한 각종 연구와 제도개선을 위하여 노력하고 있다.
15-17)또한 첨단바이오의약품 시장의 확대에 따른 장기적 안전성에 대한 우 려가 커짐에 따라 첨단재생바이오법이 제정되어 첨단바이오의 약품 장기추적조사의 발판을 마련하였다. 그러나 체계적인 장기 추적조사를 수행하기 위해서는 구체적인 계획과 시범사업 수행 을 통한 보완작업 과정이 필요하며, 첨단바이오의약품 이외의 의료제품에 대한 장기추적조사는 여전히 구체화되어 있지 않다.
이에 본 연구는 첨단재생바이오법에 근거한 장기추적조사 체계 구축 및 운영을 위한 구체적 방안을 제시하고 첨단바이오의약 품 이외의 의료제품 영역에서의 장기추적조사 추진 가능성을 검 토하고 그 방안을 모색하고자 하였다.
먼저 장기추적조사 전반을 주관하는 규제과학센터 내 인력과
체계적인 프로세스를 갖추는 것이 필요하다. 장기추적 대상으로
지정될 품목에 대해 소요비용, 우선적용 순위, 필요 인력 추계
및 계획을 통하여 센터 내 조직을 구성하고 전문인력 등 내부
체계를 갖추는 것이 우선되어야 한다. 이후, 장기추적조사를 위
해 필요한 인프라로써 전자증례기록지(Electronic Clinical Report
Form, eCRF) 개발, 통합 전산망 구축, 데이터 표준화 작업등이
Fig. 2. Strategies for implementing long term follow-up system in Korea.
필요하다. 또한, 내부적으로 관련 SOP, 업무 지침서 등을 개발 하고, 장기추적조사 관련 자문역할 수행이 가능하도록 규제기관 내 전문성을 갖어야 한다. 장기추적조사 수행 주체인 의료제품 제조회사를 위한 가이드라인을 마련하여 체계적인 조사가 진행 될 수 있도록 하고 관계자 대상 홍보 및 교육을 진행하여 장기 추적조사가 원활히 수행될 수 있게끔 할 수 있다.
장기추적조사 시범사업은 우선적으로 법적근거가 마련된 첨 단바이오의약품을 대상으로 수행하여 그 결과를 바탕으로 체계 를 보완하고 중 장기적인 목표를 도출해 나가는 것이 필요하다.
인체이식형 및 생명유지용 의료기기의 경우 추적관리제도가 있 지만 그 대상이 심혈관계 및 근골격계 관련 제품 등에만 한정 되어 있으며, 오직 소재파악을 목적으로 제품정보와 환자의 연 락처, 주소지만을 기록한다. 게다가 환자정보는 의료기관에서만 기록하여 보관하므로 장기추적조사 수행의 주체인 제조회사에 서는 환자정보를 수집하여 활용할 법적 근거가 부족한 것으로 조사되어 추진을 위한 법적 근거 마련이 우선적으로 필요해 보 인다. 하지만 최근 인공유방 발암성 위험 사태 등에 비추어 보 았을 때 장기추적조사를 시급히 도입해야 할 필요성이 있으므 로, 일부 제품에 대하여 거점 병원위주로 시범사업을 진행하여 의료기기 특성을 고려한 장기추적조사 수행 방안을 마련하는 것 이 필요하다. 의료기기의 경우 기존의 추적관리제도를 위한 UDI, 통합시스템 등 인프라를 갖추고 있기에 이를 활용한 방안도 고
려해 봄 직 하다. 장기추적조사체계가 안정화될 때쯤 첨단바이 오의약품 이외의 의약품 중에서도 우선순위를 고려하여 특정 품 목에 대하여 기존의 RMP제도 하에서 장기추적조사 실시 의무 를 부과하는 것도 고려해 볼 수 있을 것이다. 또한, 향후 통합 장기추적조사 도입 시 제품 분류 기준과 조사대상 지정 기준을 명확히 하고, 제품 특성에 따른 조사항목, 조사빈도 등을 구체 적으로 제시하는 것이 효율적인 체계 운영을 위하여 필요하다.
이후, 본 연구에서는 고려하지는 않았으나 의료기기와 의약품을 한꺼번에 포함하는 융복합제품에 대하여도 그 특성을 고려한 장 기추적조사에 관한 추가적 논의가 필요해 보인다. 따라서, 본 연 구진은 연구결과를 바탕으로 국내 의료제품 장기추적조사를 통 한 통합안전관리망 구축을 목표로 하는 장기추적조사 체계 추 진을 위한 중장기 로드맵을 Fig. 3에 제안하였다.
본 연구에서는 첨단바이오의약품, 의약품, 의료기기의 장기추
적조사 추진 방향을 도출하고자 의료제품 각 영역별 법적 근거
를 검토하였다. 관련분야 전문가와 이해관계자의 자문과 의견수
렴을 통해 중장기 추진방향을 제시함으로써 국내 의료제품 장
기추적조사 시행 및 체계 안정화의 발판이 되기를 기대한다. 또
한 본 연구 내용을 바탕으로 체계 도입과정에 있어서 우선순위
를 파악할 수 있다는 점에 그 의의가 있었다. 또한 기존의 안전
성 관리제도와 장기추적조사의 연결성을 강화하기 위하여 활용
가능한 제도와 인프라를 검토하였으므로 이를 활용한다면 새로
Fig. 3. Mid-term and long-term roadmap for implementing long term follow-up system in Korea.
운 제도 도입으로 인해 추가되는 부담을 최소화할 수 있는 효 율적인 장기추적조사를 진행할 수 있을 것이라고 사료된다.
결 론(Conclusion)
현재, 국내 의료제품의 안전성 이슈가 지속적으로 발생하고 있어 시판 후 안전성 관리 강화와 일부 제품의 장기적 사용에 서의 안전성 모니터링에 대한 요구가 커지고 있으며, 의료제품 에 관한 안전성 관리는 지속적으로 강화될 것으로 전망된다. 첨 단재생바이오법 시행으로 첨단바이오의약품의 장기추적조사가 시행될 예정이지만 그 외의 의약품, 의료기기에 관한 장기추적 조사 진행을 위해서는 제도적 개선이 필요한 상황이다. 새롭게 추진되는 첨단바이오의약품 장기추적조사를 수행하기 위하여 규 제과학센터 내 업무프로세스를 확립하고 전문성을 갖추는 것이 필요하다. 시범사업 수행으로 세부사항을 보완해가며 점차 적용 대상 범위를 확대해 나가는 한편, 전문가 교육 및 홍보 등을 통 하여 장기추적조사의 필요성에 대한 사회적 공감을 이끌어 내 고, 이해관계자들의 적극적인 참여를 유도하여 원활한 장기추적 조사가 이루어 질 수 있는 방안을 마련하는 것이 필요하다.
감사의 말씀(Acknowledgment)
본 연구는 2019년도 식품의약품안전처의 용역과제예산으로 수 행되었으며 이에 감사의 말씀을 드립니다(과제고유번호: 19172 미래의343).
Conflict of Interest
모든 저자는 이해 상충을 가지고 있지 않음을 선언한다.
References