Korean J. Mineral. Petrol. Vol. 33, No. 3, p. 143~152, 2020 https://doi.org/10.22807/KJMP.2020.33.3.143
Na-, K-버네사이트 층간 구조에 대한 분자동역학 시뮬레이션 연구
박수정·권기덕*
강원대학교 자연과학대학 지질학과
A Molecular Dynamics Simulation Study of Na- and K-birnessite Interlayer Structures
Sujeong Park and Kideok D. Kwon*
Department of Geology of Kangwon National University, Chuncheon 24341, Korea
요 약: 버네사이트(birnessite)는 약 7 Å의 d-spacing을 가지는 대표적인 층상형 산화망간광물로 높은 양이온 교환능력을 가지기 때문에 지하수나 퇴적물 공극 유체의 화학조성을 결정짓는 중요한 역할을 한다. 버네사이 트의 양이온 교환 반응 기작을 규명하기 위해서는 층간 내 양이온의 배위 환경과 결정구조에 대한 원자 수 준의 이해가 매우 중요하다. 이번 연구에서는 원자 수준의 계산광물학 방법인 고전 분자동역학(classical molecular dynamics; MD) 시뮬레이션을 이용하여 기존 실험에서 보고된 화학조성을 가지는 삼사정계 N a-와 K-버네사이트의 결정구조, 층간 양이온의 배위 환경 및 적층 구조를 계산하였다. 계산 결과는 기존 X-선 실 험에서 보고된 격자 상수와 층간 배위 환경을 잘 재현하여 시뮬레이션 방법의 신뢰성을 보여주었으며, X-선 실험만으로는 구분하기 어려운 층간의 양이온과 물 분자 위치를 구별한 원자 수준의 정보를 제공하였다. 망 간 팔면체 층의 적층 순서는 동일하지만 층간 내 N a
+와 K
+의 위치가 서로 상이하고, 층간 양이온의 배위 환 경과 결정구조 간의 상관관계를 보인다. 원자 수준의 분자동역학 시뮬레이션은 버네사이트의 양이온 교환 반 응 기작 규명에 크게 기여할 것으로 기대한다.
핵심어: 삼사정계 버네사이트, 양이온 교환, 적층 구조, MnFn, 분자동역학 시뮬레이션
Abstract:
Birnessite is a layered manganese oxide mineral with ~7Å
of d-spacing. Because of its high cation exchange capacity, birnessite greatly impacts the chemical compositions of ground water and fluids in sediment pores. Understanding the cation exchange mechanisms requires atomistic investigations of the crystal structures and coordination environments of hydrated cations in the interlayer. In this study, we conducted classical molecular dynamics (MD) simulations, an atomistic simulation method of computational mineralogy, for triclinic Na-birnessite and K-birnessite whose chemical formula are from previous experiments. We report our MD simulation results of the crystal structures, coordination environments of Na+ and K+, and the polytypes of birnessite and compare them with available experimental results. The simulation results well reproduced experimental lattice parameters and provided atomic level information for the interlayer cation and water molecule sites that are difficult to distinguish in X-ray experiments. We also report that the polytype of the Mn octahedral sheets is identical between Na- and K-birnessite, but the cation positions differ from each other, demonstrating a correlation between the coordination environment of the interlayer cations and the crystal lattice parameters. This study shows that MD simulations are very promising in elucidating ion exchange reactions of birnessite.Keywords:
Triclinic birnessite, Cation exchanges, Polytype, Molecular dynamics simulations, MnFn*Corresponding author Tel: +82-33-250-8553
E-mail: [email protected]
서 론
층상형 산화망간광물은 심해저 망간단괴나 토양 광 물 또는 암석이나 화석의 표면을 코팅하면서 다양한 지질학적 환경에서 산출된다(Post, 1999; McKeown and Post, 2001; Owocki, 2016). 대표적인 층상형 산화망간광물인 버네사이트(birnessite)는 높은 양이온 교환능력과 산화·환원 능력으로 해양, 지하수나 퇴적 물 공극 내 유체의 화학조성을 결정하는 큰 역할을 한다(Golden et al., 1986; Post and Veblen, 1990;
Trouwborst et al., 2006; Madison et al., 2013).
버네사이트의 화학적 반응성은 그 독특한 결정구조로 부터 이해할 수 있다. 버네사이트 형 광물은 기본적 으로 Mn4+로 이루어진 [MnO6] 팔면체 층이 적층된 구조로, 망간 원자 빈자리(vacancy)나 치환된 Mn3+와 같은 결함으로 인한 높은 구조적 음전하(structural charge) 때문에 [MnO6] 팔면체 층간에 수화된 양이온 을 함유 및 교환할 수 있다(Fig. 1). 다양한 Mn2+, Mn3+와 Mn4+ 산화수가 존재하는 버네사이트는 강한 자연 산화제이기도 하다(Manceau et al., 1997;
Tebo et al., 2004). 이러한 성질 때문에 버네사이트 는 양이온 교환제, 유기물 산화 촉매제, 수계 전해질 기반의 충전 배터리의 양극 물질 등 재료 분야에서도 주목받고 있다(Le Goff et al., 1996; Chitrakar et al., 2011; Iyer et al., 2012; Hou et al., 2014; Jo et al., 2019; Shan et al., 2019). 자연상에서 산출되 는 버네사이트는 결정도가 매우 낮아서 합성 버네사 이트를 이용하여 결정구조와 화학적 반응성 간의 관 계 규명 연구가 진행되어 왔다(Lanson et al., 2000;
Johnson and Post, 2006; Drits et al., 2007; Aldi et al., 2012; Birkner and Navrotsky, 2017).
층간 내 양이온 교환 반응 기작을 규명하는 연구는 주로 삼사정계(triclinic) 버네사이트를 이용하였다 (Post and Veblen, 1990; Kuma et al., 1994; Le Goff et al., 1996; Lopano et al., 2007; Post et al., 2011; Birkner and Navrotsky, 2017). 삼사정계 버네 사이트는 원자 빈자리보다는 Mn3+ 치환으로 인해 구 조적 음전하를 가지는 특징이 있다. Mn4+로 이루어진 [MnO6] 팔면체 중 약 1/3을 Mn3+이 치환하고 있어 서 팔면체 층이 높은 음전하를 띠고, 층간에 존재하 는 양이온에 의해 전체적인 전하의 균형을 이룬다 (Post et al., 2011). 따라서 층간 내 양이온의 함량은 버네사이트의 평균 산화수와도 밀접한 연관성을 가진 다. 기존의 양이온 교환 실험은 주로 화학분석, 열중 량 분석 및 X-선 회절 분석을 통해 양이온 교환 전 후의 층간 양이온과 물 함량 변화, 그리고 d-spacing 과 같은 결정구조의 변화를 관찰하였다. 나아가 결정 구조 변화와 경험적으로 상관관계를 보이는 수용액 화학, 온도, 양이온의 반경이나 수화에너지 또는 망간 이온의 평균 산화수 등 구조적·화학적 요소를 기반으 로 버네사이트 양이온 교환 반응에 대한 개념적인 반 응모델이 제시되어왔다(Lopano et al., 2007; Lopano et al., 2011). 그러나, 실험으로 이온 교환과정에서 발생하는 반응중간체(reaction intermediates)나 전이 상태(transition states)를 모니터링하는 것은 매우 어 려울 뿐만 아니라, 실험에서 제시된 요소들은 합성과 정에서 독립적인 조절이 어렵고 서로 의존되어 복합 적으로 결정구조에 영향을 주기 때문에, 실험만으로 는 제시된 요소와 결정구조의 독립적인 상관관계를 규명하는 것은 거의 불가능하다. 정확한 반응 기작 규명을 위해서는 각 요소와 결정구조의 관계에 대한 원자 수준의 이해가 매우 중요하다.
계산광물학은 결정구조에 영향을 주는 요소들에 대 한 독립적인 조절이 가능하다. 버네사이트 층간 내 물 함량은 실험으로 조절하기 어렵고, 버네사이트 층 간에 존재하는 양이온과 물 분자의 위치는 X-선 회 절 분석만으로 구분이 어렵다(Aldi et al., 2012). 계 산광물학에서는 물 함량을 쉽게 조절할 수 있으며, 물 분자의 진동수에 해당하는 시간 간격(주로 10-15sec) 으로 양이온과 물 분자 위치에 대한 명확한 구분과 모니터링이 가능하여 반응중간체나 전이 상태를 연구 하기 용이하다. 이번 연구에서는 버네사이트 층간 내
Fig. 1. Crystal structure of triclinic Na-birnessite.
양이온과 물 분자 위치를 규명하기 위해, 시간 간격 에 따른 원자/이온들의 위치와 속도를 계산하는 뉴턴 의 운동방정식을 기반으로 하는 고전 분자동역학 (classical molecular dynamics; MD) 시뮬레이션을 실시하였다. 전자의 상호작용을 고려하는 양자역학 밀 도범함수이론(density functional theory)과는 달리 원 자 또는 이온 간의 상호작용을 계산하는 방법이기 때 문에 신뢰도는 상대적으로 낮지만, 그만큼 계산 비용 이 저렴하여 밀도범함수이론 계산으로는 다룰 수 없 는 큰 공간적·시간적 스케일의 시뮬레이션이 가능하 다. 특히, 버네사이트와 같이 구조적 결함과 무질서가 높은 광물의 연구에는 크기가 큰 광물 모델이 필요하 고 수많은 경우의 수를 보이는 층간 양이온과 물 분 자들의 가능한 위치(configurations)에 대한 통계적인 샘플링 방법이 중요하기 때문에 고전 MD 시뮬레이 션은 매우 효율적인 방법이다(Newton and Kwon, 2020).
고전역학 MD 시뮬레이션의 신뢰도를 결정짓는 가 장 핵심적인 계산요소인 힘 장(force field)은 일련의 수식과 가변변수 모음을 이용하여 원자 또는 이온 간 의 상호작용을 나타낸다. Cygan et al. (2012)은 점토 광물 시뮬레이션에 널리 사용되는 ClayFF(Cygan et al., 2004)를 기반으로 하는 힘 장을 이용하여 Na-와 K-버네사이트 MD 시뮬레이션 연구를 수행한 바 있 다. 그러나 이 연구에 사용된 힘 장에서는 Mn3+와 Mn4+를 구별하지 못하고, 실험에서 제시한 평균 산 화수를 이온의 전하로 모든 망간에 동일하게 적용하 여 사용하였다. 이런 경우, 광물의 평균 산화수에 따 라 시뮬레이션 모델별로 다른 전하를 사용해야 하므 로, 다양한 광물에 적용하기에는 힘 장의 호환성 (transferability)과 신뢰도에 문제가 있다. 그러나, 광물 의 평균 산화수와는 관계없이 독립적인 Mn3+와 Mn4+의 전하를 사용하는 ‘MnFn’ 힘 장(Newton and Kwon, 2018; 2020)의 최근 개발로 층상형뿐만 아니라 다양 한 터널형 산화망간광물에 대한 충분한 신뢰도를 보 장하는 MD 시뮬레이션이 가능하게 되었다. 이번 논 문에서는 MnFn 힘 장을 사용하여 기존 실험에서 제 시된 화학식을 가지는 Na-버네사이트와 K-버네사이 트에 대한 MD 시뮬레이션을 수행하였다. 시뮬레이션 으로 버네사이트 구조의 격자 상수, 원자간 거리 및 적층 구조 결과를 분석하고 기존 실험과의 비교 결과 를 제시한다.
연구방법
버네사이트 구조
MD 시뮬레이션에 사용된 버네사이트의 초기 모델 은 Lopano et al. (2007)의 합성 Na-버네사이트(이하 Na-Bir로 표기) 단위포 구조를 3×6×2로 확장시킨 구 조(supercell)을 사용하였다(Fig. 1). Na-Bir의 확장된 구조는 a = 15.53 Å, b = 17.11 Å, c = 14.67 Å, ∠ = 89.5°, ∠ = 103.2°, ∠ = 89.9°로 삼사정계의 대칭을 가진다. K-버네사이트(이하 K-Bir로 표기)의 경우, Na-Bir와 동일한 구조에 Na+ 대신 K+을 치환하여 사 용하였다. 화학식은 Lopano et al. (2007)과 Post and Veblen (1990)의 Na0.58Mn4+1.42Mn3+0.58O4·1.5H2O 와 K0.46Mn4+1.6Mn3+0.4O4·1.4H2O을 참고하여, 실험조 성과 동일한 양이온 및 물 비율을 가지면서 광물 구조 의 총 전하를 맞추기 위해 각각 Na20(Mn4+52, Mn3+20) O144·52H2O와 K14(Mn4+58, Mn3+14)O144·42H2O로 조정 하여 사용하였다.
치환된 Mn3+ 팔면체는 b축을 따라 일렬로 배열되 며, 두 개의 Mn4+ 팔면체 열에 의해 분리된다고 보 고된 바 있다(Drits et al., 1997) (Fig. 2). 화학조성 에 따라 치환된 Mn3+의 함량은 전체 Mn의 1/3보다 적기 때문에, Mn3+ 팔면체 열에는 치환되지 않고 남 아있는 Mn4+가 존재한다. Mn3+ 팔면체 열에 존재하는 Mn4+의 위치는 Newton and Kwon (2018)을 참고하 여 임의로 선택하였다. 총 전하의 균형을 맞춰주기 위해 층간에는 Mn3+와 동일한 함량의 양이온이 존재 하며, 층간의 양이온과 물 분자의 배열은 Mn3+와 Mn4+팔면체의 배열을 반영하여 b축을 따라 층간 중 간에 임의로 배열하였다.
원자간 포텐셜(Interatomic Potentials)
이번 연구에서 사용한 MnFn 힘 장은 Newton and Kwon (2018, 2020)이 개발한 산화망간광물에 대한 힘 장이다. MnFn 힘 장으로 묘사되는 시스템의 총 에너지는 광물을 구성하고 있는 원자간 비결합 에너
지(Enon-bonded)와 결합에너지(Ebonded)의 합으로 다음과
같이 표현된다(식 1).
(1)
Enon-bonded는 쿨롱에너지(ECoul)와 Lennard-Jones(LJ)
에너지(ELJ)로 식 2로 묘사된다.
E
total
=Enon bonded –
+Ebonded
(2)
ECoul은 원자 간의 정전기적 인력을 나타내며, e는 기본 전하, o 는 진공상태의 유전율(8.85419×10-12F/
m2), qi와 qj는 원자 i와 j의 부분 전하, rij는 두 원자 (i, j) 간의 거리를 의미한다. ELJ는 반데르발스 에너지 를 의미하는 함수로, Do,ij는 퍼텐셜 우물의 깊이, Ro,ij
는 두 원자가 평형을 이루었을 때의 거리를 나타낸다.
식 1의 Ebonded는 Ebondstretch 항과 Eanglebend 항으로 조 화(harmonic)함수로 표현되고, 물 분자(H2O)에만 적 용하며, 각각 O-H 결합의 신축 진동과 H-O-H 결합 의 굽힘 진동에너지를 나타낸다(식 3).
(3)
rij는 두 원자(i-j)의 결합길이, ijk는 세 원자(i-j-k)의 결합각을 의미하고, 평형상태의 원자간 거리는 ro, 결 합각은 o로 표현하며, k는 용수철 상수에 해당한다.
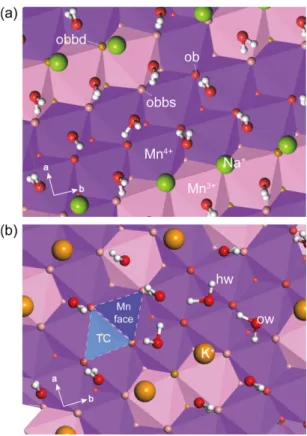
Mn 팔면체 층에서 가교 산소(ob) 및 Mn3+와 결합 하는 가교 산소(obbs, obbd)의 부분 전하는 Newton and Kwon (2018, 2020)에 따라 구분하여 사용하였 다(Fig. 2a). MnFn 힘 장에서 ob는 Mn4+와 Mn4+를 결합하는 가교 산소를 의미하며, 결합하는 망간 원자 중 Mn3+가 하나일 경우 obbs, 두 개일 경우 obbd로 구분한다. Mn4+가 Mn3+로 치환되면 –1e의 순 전하 (net charge)가 발생하는데, MnFn 힘 장의 Mn4+, Mn3+의 부분 전하는 각각 +2.1e과 +1.575e로 –0.475e 의 부족한 전하를 Mn3+와 결합하는 가교 산소에 할 당해주어 전체 이온 전하와 구조적 전하의 균형을 이 루었다. 이번 연구에서는 MnFn 힘 장으로부터 치환 된 Mn3+의 함량에 따라 Mn3+ 및 Mn3+와 결합하고 있는 가교 산소(obbs, obbd)의 부분 전하를 0.03%
이내로 수정하여 구조 전하를 맞춰주었으며, 수정된 부분 전하는 각각 Mn3+= +1.5754e, ob = -1.05e, obbs
= -1.1295e, obbd = -1.208e이다.
시뮬레이션 및 분석 방법
버네사이트의 MD 시뮬레이션은 BIOVIA Materials
Studio 2020의 Forcite pl us 모듈을 사용하였다. 광물 의 벌크 구조를 묘사하기 위해 3차원 주기적 경계조 건(periodic boundary condition)을 적용하였다. 원자 간의 상호작용 제한 반경(cutoff radius)을 격자 상수 중 가장 짧은 축 길이의 1/2로 설정하여 원자간 상호 작용 에너지가 중복되어 계산되는 것을 방지하였다.
원자 간 비결합 상호작용 에너지를 계산하기 위해 Ewald summation (Ewald, 1921) 방법을 사용하였으 며, 정확도는 1.0×10-5 kcal/mol로 설정하였다. MD 시 뮬레이션의 초기 구조로는 MnFn 힘 장으로 얻어진 정적(static) 최적화 구조(geometry optimized structure) 를 사용하였다. 구조최적화 계산을 위한 에너지의 수 렴 허용 오차(tolerance)는 2.0×10-5 kcal/mol을 사용 하였다. 이후 MD 시뮬레이션 계산을 위해 Verlet- velocity 알고리즘을 사용하였다(Verlet, 1967). 시뮬레 이션 계산 절차는 먼저 설정된 온도 압력에서의 구조 E
non bonded –
=ECoul
+ELJ
e
2
4o
--- qi
qj
r
ij
--- D
o ij
Ro ij
rij
---
12
2Ro ij
rij
---
6
–i j
+
i j
=
E
bonded
=Ebondstretch
+E bend
k1
rij
–ro
2
k2
ijk
–o
2
i j k
+
i j
=
Fig. 2. Atom types (ob, obbs, obbd, hw, ow) assigned to (a) Na-birnessite and (b) K-birnessite MD simulations. Polyhedra represent Mn
3+or Mn
4+octahedra. TC = tridentate cavity; Mn face = Mn
octahedron face.
적 평형을 이루기 위해 0.5 fs (10-15 sec)의 시간 간 격으로 100 ps (10-12 sec) 동안 수행되었고, 구조적 평형이 이루어진 이후부터는 추가적으로 200 ps를 더 계산하여 0.05 ps 간격으로 얻은 4000개의 구조 결과 를 분석에 사용되었다. Na-Bir와 K-Bir의 계산에 적 용된 앙상블(ensemble)은 정온-정압 앙상블(NPT; N=
원자의 개수, P=압력, T=온도)로, 온도와 압력을 298.15 K, 1.0 atm로 일정하게 유지시키기 위해 각 각 Nosé-Hoover 온도조절(Nosé, 1991)과 Parrinello- Rahman 압력조절(Parrinello and Rahman, 1981)을 사용하였다. Nosé-Hoover 온도조절에 필요한 Q ratio 는 0.01로 설정해주었다.
원자 간의 거리와 배위수를 분석하기 위하여 사용 된 동경 분포 함수(RDF, radial distribution function) 는 선택된 하나의 원자(i)를 기준으로 일정 반경(r)에 위치하는 두께가 dr인 껍질 내에 다른 원자(j)가 존재 하는 확률을 나타내는 확률 밀도 함수이다(식 4).
(4)
rij는 원자 i와 j의 거리를 나타내며 ρ는 시스템의 전체 개수밀도(total number density)이다. 동경분포 함수[g(r)]는 반경 r과 원자 i를 제외한 N개의 원자 j 에 대한 거리 rij가 같을 때(즉, r = rij)의 가중치를 1 로 두고 개수를 더하는 함수이다. < >는 N개의 원자 i에 대하여 평균을 낸 것이다. 식 (4)로부터 식 (5)을 유도하여 배위수(coordination number, C.N.)를 계산 한다.
(5)
이번 연구에서는 g(r)의 첫 번째 껍질의 최대값을 보이는 거리를 배위하는 원자 간의 평균 거리로 보았 으며, 최대값 이후 바로 나타나는 최소값을 첫 번째 껍질의 끝으로 두어 그 지점에서의 배위수를 계산하 였다. 결정축(a, b, c)의 길이는 평형을 이룬 이후에 얻은 4000개의 자료에 대한 평균값이며 결정각(∠,
∠, ∠)은 확장된 구조의 각도로 마찬가지로 평균 을 내어 사용하였다.
결과 및 토의
격자 상수 및 (001) d-spacing
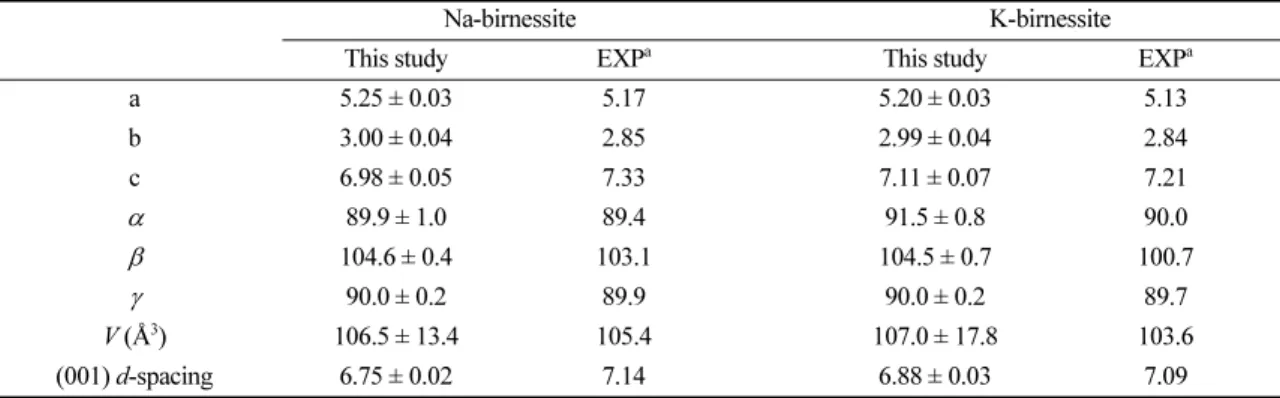
MD 시뮬레이션으로 얻은 Na-Bir와 K-Bir의 격자 상수를 Na+에 대한 K+ 교환 반응 실험에서 얻어진 기존 실험결과와 비교하였다(Table 1). Na-Bir와 K- Bir는 모두 삼사정계를 유지하였으며, c축을 제외한 다른 격자 크기는 모두 기존 실험결과보다는 큰 값을 가졌다. Na-Bir의 경우 합성 Na-Bir에 대한 X-선 회 절 분석결과(Lopano et al., 2007)와 비교했을 때, a, b, c축의 길이가 각각 1.5%, 5.3%, -4.8%, ∠, ∠
, ∠는 각각 0.6%, 1.4%, 0.1%의 오차율을 보였 다. Post et al. (2002), Lanson et al. (2002) 그리 고 Lopano et al. (2007)가 보고한 Na-Bir의 (001) d-spacing은 약 7.14 Å이지만, 계산으로 얻어진 값은 6.75 Å으로 -5.5%의 상대적으로 큰 오차를 보였다.
이번 연구에서 얻은 d-spacing 값은 기존 Newton g r 1
4r
2
dr--- r r –
rj
j 1 =
N
=
C.N. 4 r
2
g r rdr
0r
1
=
Table 1. Calculated lattice parameters of Na-birnessite and K-birnessite. All distances are in Å; the angles are in degrees
Na-birnessite K-birnessite
This study EXPa This study EXPa
a 5.25 ± 0.03 5.17 5.20 ± 0.03 5.13
b 3.00 ± 0.04 2.85 2.99 ± 0.04 2.84
c 6.98 ± 0.05 7.33 7.11 ± 0.07 7.21
89.9 ± 1.0 89.4 91.5 ± 0.8 90.0
104.6 ± 0.4 103.1 104.5 ± 0.7 100.7
90.0 ± 0.2 89.9 90.0 ± 0.2 89.7
V (Å3) 106.5 ± 13.4 105.4 107.0 ± 17.8 103.6
(001) d-spacing 6.75 ± 0.02 7.14 6.88 ± 0.03 7.09
a
Experimental results (Lopano et al., 2007).
and Kwon (2018)의 MD 시뮬레이션 연구로 얻어진 값(6.82 Å)과 매우 흡사하다.
K-Bir 시뮬레이션 결과와 합성 K-Bir 실험결과 (Lopano et al. 2007)를 비교했을 때 a, b축은 각각 1.3%, 5.3%의 오차율을 보였으며, ∠, ∠, ∠는 1.7%, 3.8%, 0.3%의 오차율을 보이며 구조를 재현했 다(Table 1). K-Bir의 c축의 길이는 7.11 Å, (001) d-spacing은 6.88 Å으로, 각각 기존 X-선 회절 분석 결과보다 1.4%, 3.0% 작게 계산되었다(실험값 = 7.21 Å, 7.09 Å). K-Bir의 d-spacing 값은 기존에 여 러 실험을 통하여 7.03–7.26 Å 범위로 보고되었으며 (Kuma et al., 1994; Feng et al., 1997; Lopano et al., 2007; Post et al., 2011), 계산 결과 Na-Bir 와 마찬가지로 실험결과보다 약 2–5% 작게 계산되는 경향을 보였다. 층간에 수화된 양이온을 함유하는 버 네사이트의 결정구조는 층간수의 함량에 d-spacing이 크게 영향을 받을 수 있다. Post and Veblen (1990) 에서는 Na-Bir에 대한 K+ 교환 시 양이온과 물 함량 이 동시에 변하는 것을 관찰하였으나, 실험에서 보고 된 물 함량은 열 중량분석을 통하여 층간수 뿐만 아 니라 버네사이트 입자 간에 존재하는 흡착수까지 포 함할 수 있으므로 정확한 층간수의 함량은 알 수 없 다. 실험에서 제시된 물 함량을 그대로 사용한 이번 MD 시뮬레이션에서 계산된 Na-Bir와 K-Bir의 d- spacing의 상대적으로 큰 오차는 기존 실험에서 제시 된 물 함량의 불확실성에 기인한다고 판단된다.
양이온 교환 실험에서 보고된 (001) d-spacing을 비교해보면, 교환 반응 전 Na-Bir의 d-spacing은 7.14 Å인데 상대적으로 이온반경이 큰 K+으로 교환 이후 d-spacing은 7.09 Å로 더 작다(Post and Veblen, 1990; Lopano et al., 2007). 이 실험결과는 양이온 의 반경과 d-spacing 크기 사이의 양의 상관관계를 보여준 이번 MD 계산 결과와는 정반대되는 경향으 로, 이에 관해서는 추가적인 연구가 필요하다. 층간 내 분포하는 이온의 크기뿐만 아니라 양이온이나 층 간수 함량, 양이온의 배위 환경 및 물 분자들의 구조 가 버네사이트의 결정구조에 큰 영향을 끼칠 수 있다.
따라서, Na+와 K+의 교환실험 결과를 명확하게 설명 하기 위해서는 이온반경 이외에 수화된 층간 내 양이 온과 물 분자의 구조 영향에 관한 연구가 필요하다.
층간 양이온(Na+, K+) 배위 환경
우선, 망간 팔면체 층(이하 Mn 층으로 표기)을 이
루는 원자(Mn4+, Mn3+, Olayer)들의 거리를 기존 실험 결과와 비교하였다. 계산된 Na-Bir의 Mn 층에서의 Mn과 Olayer의 평균 거리는 1.95 Å으로 실험값인 1.93 Å (Post et al., 2002) 및 1.96 Å (Lanson et al., 2002)과 매우 유사한 값을 가졌다. Mn4+-Olayer와
Mn3+-Olayer의 평균 길이는 각각 1.95 Å와 2.03 Å였다.
K-Bir의 경우, 전체 Mn-Olayer 평균 거리는 1.95 Å이 며, Mn4+-Olayer와 Mn3+-Olayer의 평균 거리는 각각 1.95 Å와 2.03 Å을 가져 Na-Bir의 시뮬레이션 결과와 일치하였다. Post et al. (2011)이 보고한 K-Bir의 전 체 Mn-Olayer 거리는 1.91–2.02 Å으로, 계산 결과가 기존 실험결과를 잘 재현하였다.
층간 양이온이 Mn 층의 산소(Olayer) 또는 물 분자
산소(Owater)와 결합하는 거리 및 배위수를 계산하기
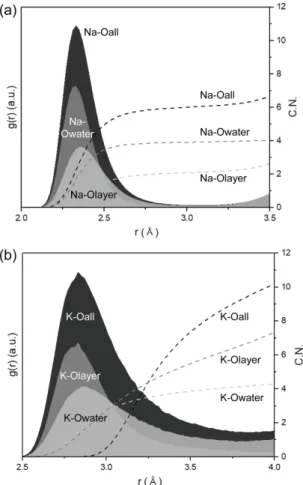
Fig. 3. Radial distribution functions (solid areas)
between the interlayer cation and O and the
cumulative coordination numbers (C.N.) (dashed
lines) of the cation in (a) Na-birnessite and (b)
K-birnessite.
위해 RDF 분석을 수행하였다(Fig. 3). 일반적으로 버 네사이트 내 Na+과 K+는 모두 층간 가운데 위치하면 서 주변의 산소들과 배위하는 모습을 볼 수 있다(Fig.
4). Na-Bir 층간 내 Na+ 이온과 전체 산소(Oall) 평균 거리[d(Na+-Oall)]는 2.33 Å이며, 물 분자의 산소 (Owater)와 가교 산소(Olayer)에 해당하는 d(Na+-Owater)는 2.31 Å, 그리고 d(Na+-Olayer)는 2.35 Å으로 계산되었다 (Fig. 3a). 기존의 Na-Bir 시뮬레이션 결과인 d(Na+- Oall) = 2.33 Å, d(Na+-Owater) = 2.32 Å, d(Na+-Olayer) = 2.45 Å와 근접한 값을 나타낸다(Newton and Kwon, 2018). 물 분자와 양이온의 X-선 산란 포텐셜이 비슷 하여, X-선 회절 분석을 통한 층간 Na+과 Owater 간 의 거리 및 Na+과 Olayer의 거리를 구분하기 어렵다.
이번 시뮬레이션에서는 d(Na+-Na+), d(Na+-Owater), d(Na+-Olayer), 그리고 d(Owater-Owater)와 d(Owater-Olayer)를
명확하게 구분할 수 있다. Lopano et al. (2007)에서 제시한 2.59 Å와 2.85–2.95 Å의 층간 내 양이온 또 는 산소 간의 거리는 이번 계산 결과에서의 각각 d(Owater-Olayer) = 2.63 Å와 d(Owater-Owater) = 2.97 Å에 해 당한다. 기존 합성 Na-Bir에 대한 실험에서 층간 Na+ 이온은 주변 6개의 산소와 배위한다고 보고되었 다(Lopano et al., 2007). 이번 연구에서도 Na+ 이온 은 주위 산소와 평균 5.7 배위수를 가졌으며, 주로 Mn 층에 존재하는 2개의 Olayer와 주변 3–4개의
Owater와 배위를 하였다(Fig. 3a). 평균적으로 Na+와
배위하는 2개의 Olayer는 ob, obbs, obbd 중 obbd와 가장 빈번하게 배위를 이루었고, ob와는 배위를 이루 지 않았다. 이는 Na+이 Mn4+보다 Mn3+ 근처에 우세 하게 위치한다는 것을 의미한다(Fig. 2a).
K-Bir의 층간 K+ 이온과 주변 산소에 대한 RDF
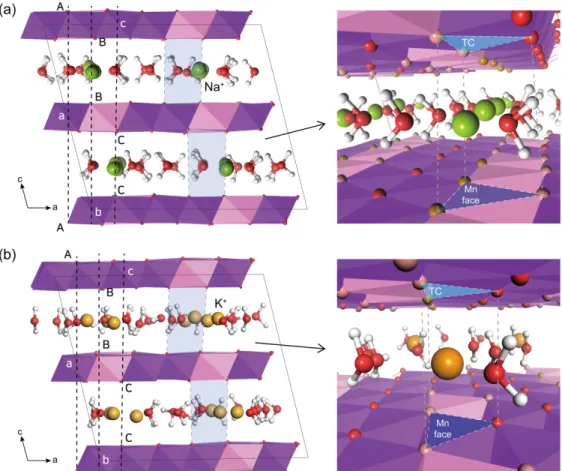
Fig. 4. Polytype sequences (AbC=CaB=BcA=AbC...) of Mn octahedral sheets and coordination environment of inter-
layer cations in MD-simulated (a) Na-birnessite and (b) K-birnessite (green : Na
+; orange : K
+; TC = triden-
tate cavity; Mn face = Mn octahedron face).
분석결과, 전체 산소(Oall)의 거리[d(K+-Oall)]는 2.83 Å 로 물 분자 산소(Owater)와의 거리[d(K+-Owater)]와 가교
산소(Olayer)와의 거리[d(K+-Olayer)]는 각각 2.83 Å와
2.87 Å으로 계산되었다(Fig. 3b). 계산을 통해 얻은 K+과 주변 산소들의 거리는 모두 Lopano et al.
(2007)에서 보고된 K-Bir 층간 내 구분되지 않은 K+ 또는 산소와의 거리인 2.85–2.95 Å에 포함되는 거리 이다. d(K+-Olayer) = 2.87 Å는 X-선 실험을 통해 관찰 한 가교 산소(Olayer)와 층간의 중앙에 위치하는 K+의 거리인 2.85 Å와도 거의 일치한다(Lopano et al., 2007). 층간에 존재하는 K+은 전체 산소에 대하여 약 10–11 배위 산소를 가지며 물 분자와 4 배위수를 이 루고, 6–7개의 가교 산소와 배위한다. 또한, obbd하 고 가장 우세하게 배위하는 Na+와는 다르게, K+은 obbs와 obbd가 비슷한 배위수를 가지며, ob와도 일부 배위를 이루는 것을 알 수 있다(Fig. 2b). 이러한 계 산 결과는 기존 실험연구에서 층간 내 K+는 주변의 6개의 물 분자와 Mn 층에 존재하는 6개의 가교 산 소(윗층과 아랫층 3개씩)를 배위한다는 결과와 일치 한다(Post et al., 2011).
층간 양이온의 종류에 따른 주변 물 분자의 배위 구조의 차이도 결정구조에 큰 영향을 미칠 수 있다.
Johnson and Post (2006)는 양이온 교환과정 동안 발생하는 층간 내 물 분자 구조(O-H 신축 모드)의 변화를 적외선 분광법(IR spectroscopy)을 이용하여 관 찰하였으며, 층간수가 버네사이트의 양이온 교환 및 구조적 안정성에 중요한 역할을 한다고 보고하였다.
이번 연구에서 Na-Bir 층간 내 Na+과 물 분자는 a- b 평면을 따라 규칙적인 배열을 보이고, 물 분자 수 소의 방향은 a-b 평면에 대해 수직을 이루며 Mn 층 을 향하는 것을 볼 수 있다(Fig. 4a). 반면에, K-Bir 의 K+과 물 분자의 배열은 Na-Bir와 비교했을 때 상 대적으로 불규칙하고, 일부 물 분자 수소의 방향은 a- b 평면에 대해 수평을 이루며 층간 영역을 향하는 것 을 볼 수 있었다(Fig. 4b). 이러한 층간 양이온과 물 분자 배열은 층간에 존재하는 양이온의 종류와 함량, 물의 함량에 크게 영향을 받을 것으로 예상된다. 또 한, 이처럼 양이온의 종류에 따른 배위 환경의 차이 는 결과적으로 층간 내 양이온과 물 분자의 배열을 결정짓고, 이러한 층간 구조의 변화는 결정구조에도 큰 영향을 미칠 수 있다. 양이온의 종류와 물 함량에 따 른 물 분자의 쌍극자 모멘트(dipole moment) 분석은
물 함량, 양이온의 배위수, 그리고 d-spacing 간의 상 호관계를 규명하는 데 중요한 정보를 제공할 수 있을 것으로 판단된다.
적층 형태(Polytype)
Drits et al. (2007)는 합성 버네사이트의 경우 층 간 원자 빈자리의 유무, 적층된 층의 개수, 가교 산소 의 위치에 따라 적층 구조를 나누었다. 층간 양이온이 Na+인 버네사이트는 Post and Veblen (1990)에 의해 one-layer monoclinic polytype (1M1) 이라고 밝혀졌으 나, 이후 Lanson et al. (2002)에 의해 monoclinic이 아닌 one-layer triclinic polytype으로 밝혀진 바 있 다. Fig. 4a는 이번 시뮬레이션으로 얻은 Na-Bir의 적층 구조로, Drits et al. (2007)의 표현법을 사용하 여 나타낸 것이다(AbC=CaB=BcA=AbC). 층간을 기 준으로 하는 대칭면은 없었으며, 망간 팔면체의 면(Mn face)과 세 개의 가교 산소로 이루어진 tridentate cavity (TC)가 마주보며 양이온에 대한 삼각기둥 (prismatic)의 배위 구조를 보인다. 망간 팔면체 면과 TC가 마주보는 구조는 Lanson et al. (2002)과 Jul ien et al. (2003), Lopano et al. (2007)에서도 보고된 바 있다. Na+는 c축을 따라서는 층간 가운데 위치하 며, a-b 평면에서는 삼각기둥을 이루는 코너에 해당 하는 가교 산소 위에 위치한다(Figs. 2a and 4a).
K-Bir는 Na-Bir와 동일한 적층 순서를 보였으며, 망간 팔면체 면과 TC가 마주보는 삼각기둥 형태의 층간 배위 환경을 보여준다(Fig. 4b). K+으로 교환된 버네사이트의 적층 순서는 Na-Bir와 같으나, 기존 실 험에 의하면 층간 K+의 경우 삼각기둥의 코너 산소 위가 아닌 Mn face 위 또는 TC 위에 위치한다고 보고한 바 있다(Post and Veblen, 1990; Drits et al., 2007). 이번 계산 결과에서도 층간 내 대부분의 K+의 위치가 Na+ 위치와는 달리 삼각기둥의 가운데 위치하는 것을 확인할 수 있었다(Figs. 2b and 4b).
Na+와 K+의 이온반경의 차이가 약 0.5 Å임에도 불구 하고 비슷한 물 함량을 가지는 Na-Bir와 K-Bir의 d- spacing는 약 0.1 Å 밖에 차이가 나지 않는데, 층간 내 물 분자의 배위 위치도 중요하겠지만 양이온 종류 에 따른 위치의 차이에서 그 이유를 찾을 수 있을 것이다. 층간 내 화학조성 차이에 따른 버네사이트의 결정구조의 다양성은 층간의 양이온의 배위 환경으로 부터 해석할 수 있다.
결 론
산화망간에 특화된 MnFn 힘 장을 사용한 Na-Bir, K-Bir MD 시뮬레이션으로부터 얻은 격자구조와 층 간 내 양이온의 배위 환경은 기존 실험결과를 잘 재 현하였으며, 이는 사용된 계산방법이 삼사정계 버네 사이트 구조를 원자 수준으로 이해하는 데에 있어 적 합함을 보여준다. 기존 실험에서 제시된 화학식을 가 지는 Na-Bir와 K-Bir 모델에 대한 MD 시뮬레이션 결과, d-spacing의 경향은 기존 실험결과와 반대되는 경향을 보여주었다. 이는 실험에서 보고된 물 함량에 대한 불확실성에 기인한 것으로 판단되며, 층간 물 함량에 따른 d-spacing 변화에 대한 추가 시뮬레이션 연구가 필요하다. 층간 내 존재하는 Na+는 주변 6개 의 산소와 팔면체 배위를 이루었고, K+은 주변 산소 와 총 10–11 배위를 이루고 각각 6개, 4–5개의 가교 산소와 물 산소를 배위하면서 기존 실험결과를 잘 재 현하였다. Na-Bir와 K-Bir의 망간 팔면체 층의 적층 형태는 Mn 팔면체 face와 TC가 마주보면서 삼각기 둥의 배위 환경을 이루었고, 층간 Na+는 Mn3+ 팔면 체의 가교 산소의 위, K+는 TC 위에 위치하여 X-선 회절 기반의 기존 실험결과와 잘 일치함을 확인하였 다. 이번 MD 시뮬레이션 결과는 버네사이트의 결정 구조의 다양성은 양이온의 종류(화학조성)에 따른 층 간 내 위치와 배위 환경의 변화에 기인함을 의미하며, 복잡한 버네사이트의 구조를 규명하는 데 유용하게 사용될 수 있음을 보여준다. 특히, 이번 MD 시뮬레 이션에서는 양이온 종류와 층간 물 함량 변화에 따른 원자 수준의 물 분자 배열의 변화를 발견할 수 있었 다. 이는 물 분자가 버네사이트 구조, 나아가 그 안정 성을 결정짓는 중요한 역할을 할 수 있음을 의미한다.
추후 물 분자의 수소 결합 및 쌍극자 모멘트 분포와 같은 층간 물 분자의 배열과 방향성에 대한 MD 시 뮬레이션 연구는 버네사이트 양이온 교환과정 규명에 크게 기여할 것이다.
사 사
이 연구는 한국연구재단의 지원(2019R1A2C2084299) 을 받았으며 KISTI 슈퍼컴퓨터 자원(KSC-2020-CRE- 0050)을 일부를 사용하여 수행되었다. 원고 향상에 도 움을 주신 익명의 심사자와 최헌수 박사께 감사드린 다.
REFERENCES
Aldi, K.A., Cabana, J., Sideris, P.J. and Grey, C.P., 2012, Investigation of cation ordering in triclinic sodium birnes- site via 23Na MAS NMR spectroscopy. American Miner- alogist, 97(5-6), 883-889.
BIOVIA, Dassault systèmes, 2020, BIOVIA Materials Stu- dio, BIOVIA Materials Studio 2020, San Diego: Dassault Systèmes.
Birkner, N. and Navrotsky, A., 2017, Thermodynamics of manganese oxides: Sodium, potassium, and calcium bir- nessite and cryptomelane. Proceedings of the National Academy of Sciences, 114(7), E1046-E1053.
Chitrakar, R., Makita, Y., and Sonoda, A., 2011, Cesium ion exchange on synthetic birnessite (Na
0.35MnO
2·0.6H
2O).
Chemistry Letters, 40(10), 1118-1120.
Cygan, R.T., Liang, J.J., and Kalinichev, A.G., 2004, Molec- ular models of hydroxide, oxyhydroxide, and clay phases and the development of a general force field. The Journal of Physical Chemistry B, 108(4), 1255-1266.
Cygan, R.T., Post, J.E., Heaney, P.J., and Kubicki, J.D., 2012, Molecular models of birnessite and related hydrated layered minerals. American Mineralogist, 97(8-9), 1505- 1514.
Drits, V.A., Lanson, B., and Gaillot, A.C., 2007, Birnessite polytype systematics and identification by powder X-ray diffraction. American mineralogist, 92, 771-788.
Drits, V.A., Silvester, E., Gorshkov, A.I., and Manceau, A., 1997, Structure of synthetic monoclinic Na-rich birnessite and hexagonal birnessite: I. Results from X-ray diffrac- tion and selected-area electron diffraction. American Min- eralogist, 82, 946-961.
Ewald, P.P., 1921, The computation of optical and electro- static lattice potentials. Annalen der Physik, 64, 253-287.
Feng, Q., Yanagisawa, K., and Yamasaki, N., 1997, Synthe- sis of birnessite-type potassium manganese oxide. Journal of materials science letters, 16(2), 110-112.
Golden, D.C., Dixon, J.B., and Chen, C.C., 1986, Ion exchange, thermal transformations, and oxidizing proper- ties of birnessite. Clays and Clay Minerals, 34, 511-520.
Hou, J., Li, Y., Mao, M., Ren, L., and Zhao, X,. 2014, Tre- mendous effect of the morphology of birnessite-type manganese oxide nanostructures on catalytic activity.
ACS applied materials & interfaces, 6(17), 14981-14987.
Iyer, A., Del-Pilar, J., King’ondu, C.K., Kissel, E., Garces, H.F., Huang, H., El-Sawy, A.M., Dutta, P.K. and Suib, S.L., 2012, Water oxidation catalysis using amorphous manganese oxides, octahedral molecular sieves (OMS-2), and octahedral layered (OL-1) manganese oxide struc- tures. The Journal of Physical Chemistry C, 116(10), 6474-6483.
Jo, M.R., Kim, Y., Yang, J., Jeong, M., Song, K., Kim, Y.I.,
Lim, J.M., Cho, M., Shim, J.H., Kim, Y.M., Yoon, W.S.,
and Kang, Y.M., 2019, Triggered reversible phase trans- formation between layered and spinel structure in manga- nese-based layered compounds. Nature communications, 10(1), 1-9.
Johnson, E.A., and Post, J.E., 2006, Water in the interlayer region of birnessite: Importance in cation exchange and structural stability. American Mineralogist, 91, 609-618.
Julien, C., Massot, M., Baddour-Hadjean, R., Franger, S., Bach, S. and Pereira-Ramos, J.P., 2003, Raman spectra of birnessite manganese dioxides. Solid State Ionics, 159(3- 4), 345-356.
Kuma, K., Usui, A., Paplawsky, W., Gedulin, B., and Arrhenius, G., 1994, Crystal structures of synthetic 7 Å and 10 Å manganates substituted by mono-and divalent cations. Mineralogical Magazine, 58, 425-447.
Lanson, B., Drits, V.A., Feng, Q., and Manceau, A., 2002, Structure of synthetic Na-birnessite: Evidence for a tri- clinic one-layer unit cell. American Mineralogist, 87, 1662-1671.
Lanson, B., Drits, V.A., Silvester, E. and Manceau A., 2000, Structure of H-exchanged hexagonal birnessite and its mechanism of formation from Na-rich monoclinic buse- rite at low pH. American Mineralogist, 85, 826-838.
Le Goff, P., Baffier, N ., Bach, S. and Pereira-Ramos, J.P., 1996, Synthesis, ion exchange and electrochemical prop- erties of lamellar phyllomanganates of the birnessite group. Materials Research Bulletin, 31(1), 63-75.
Lopano, C.L., Heaney, P.J., Post, J.E., Hanson, J., and Komarneni, S., 2007, Time-resolved structural analysis of K-and Ba-exchange reactions with synthetic Na-birnessite using synchrotron X-ray diffraction. American Mineralo- gist, 92, 380-387.
Lopano, C.L., Heaney, P.J., Bandstra, J.Z., Post, J.E., and Brantley, S.L., 2011, Kinetic analysis of cation exchange in birnessite using time-resolved synchrotron X-ray dif- fraction. Geochimica et Cosmochimica Acta, 75(14), 3973-3981.
Newton, A.G., and Kwon, K.D., 2018, Molecular simula- tions of hydrated phyllomanganates. Geochimica et Cos- mochimica Acta, 235, 208-223.
Newton, A.G., and Kwon, K.D., 2020, Classical mechanical simulations of layer-and tunnel-structured manganese oxide minerals. Geochimica et Cosmochimica Acta.
Nosé, S., 1984, A molecular dynamics method for simula- tions in the canonical ensemble. Molecular Physics, 52, 255-268.
Madison, A.S., Tebo, B.M., Mucci, A., Sundby, B. and Luther, G.W., 2013, Abundant porewater Mn (III) is a major component of the sedimentary redox system. sci- ence, 341(6148), 875-878.
Manceau, A., Silvester, E., Bartoli, C., Lanson, B., and Drits, V.A, 1997, Structural mechanism of Co2+ oxida- tion by the phyllomanganate buserite. American mineral-
ogist, 82(11-12), 1150-1175.
McKeown, D.A. and Post, J.E., 2001, Characterization of manganese oxide mineralogy in rock varnish and den- drites using X-ray absorption spectroscopy. American Mineralogist, 86(5-6), 701-713.
Owocki K., Kremer B., Wrzosek B., Krolikowska A., and Kaźmierczak J., 2016, Fungal ferromanganese mineralisa- tion in Cretaceous dinosaur bones from the Gobi desert, Mongolia. PLOS ONE 11, e0146293.
Parrinello, M., and Rahman, A., 1981, Polymorphic transi- tions in single crystals: A new molecular dynamics method. Journal of Applied physics, 52, 7182-7190.
Post, J.E., 1999, Manganese oxide minerals: Crystal struc- tures and economic and environmental significance. Pro- ceedings of the National Academy of Sciences of the USA, 96, 3447-3454.
Post, J.E., and Veblen, D.R., 1990, Crystal structure deter- minations of synthetic sodium, magnesium, and potas- sium birnessite using TEM and the Rietveld method.
American Mineralogist, 75, 477-489.
Post, J.E., Heaney, P.J., and Cho, Y., 2011, Neutron diffrac- tion study of hydrogen in birnessite structures. American Mineralogist, 96, 534-540.
Post, J.E., Heaney, P.J., and Hanson, J., 2002, Rietveld refinement of a triclinic structure for synthetic Na-birnes- site using synchrotron powder diffraction data. Powder Diffraction, 17, 218-221.
Shan, X., Guo, F., Charles, D.S., Lebens-Higgins, Z., Razek, S.A., Wu, J., Xu, W., Yang, W. Page, K.L., Neuefeind, J.C., Feygenson, M., Piper L.F.J. and Teng, X., 2019, Structural water and disordered structure promote aqueous sodium-ion energy storage in sodium-birnessite. Nature communications, 10(1), 1-11.
Tebo, B.M., Bargar, J.R., Clement, B.G., Dick, G.J., Murray, K.J., Parker, D., Verity, R. and Webb, S.M., 2004, Bio- genic manganese oxides: properties and mechanisms of formation. Annu. Rev. Earth Planet. Sci., 32, 287-328.
Trouwborst, R.E., Clement, B.G., Tebo, B.M., Glazer, B.T., and Luther, G.W., 2006, Soluble Mn (III) in suboxic zones. science, 313(5795), 1955-1957.
Verlet, L., 1967, Computer “Experiments” on classical flu- ids. I. Thermodynamical properties of Lannard-Jones mol- ecules. Physical Review, 159, 98-103.
Received September 10, 2020 Review started September 11, 2020 Accepted September 23, 2020
[ 저 자 정 보 ]