저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Master’s Thesis in Neuroscience
Signaling pathways mediated by Toll-like
Receptor 2 involved in the protection of
oligodendrocytes from ischaemic cell
death
Ajou University Graduate School
Major in Neuroscience
Department of Biomedical Sciences
Samma Tasneem Chowdhury
Signaling pathways mediated by
Toll-like Receptor 2 involved in the
protection of oligodendrocytes from
ischaemic cell death
Thesis advisor: Byung Gon Kim, MD, PhD.
I submit this thesis as the Master’s thesis in Neuroscience
August, 2017
Ajou University Graduate School
Major in Neuroscience
Department of Biomedical Sciences
ACKNOWLEDGEMENTS
At first I want to praise the almighty GOD who gave me good health, tolerance and patience to finish my Masters study and thesis work successfully. Foremost, I would like to express my sincere gratitude to my advisor Prof. Dr. Byung Gon Kim for his perpetual support to complete my study in Korea and my thesis research work. I want to appreciate his patience, intelligence, immense knowledge and motivation which enable me to accomplish the thesis work without much difficulty. Despite thesis and the coursework study, the opportunity to work in his Brain Disease Research Center lab - gave me enormous chances to develop my knowledge in the sector of neuroscience research.
Besides my advisor, I would like to thank my rest of the thesis committee, Prof. Dr. Eun Hye Joe and Prof. Dr. Eun Young Kim for their insightful comments and encouragement which incented to fill the gap of my research from various perspectives.
My special gratitude and love for my senior and fellow labmates for their stimulating discussions which helped me to find out the solutions of many difficult problems. In particular, I am grateful to Dr. Yuexian Cui and Dr. Jun Young Choi for their suggestions, ideas and fruitful discussions.
Last but not the least, I would like to thank my family: my parents, my brothers and sister, and my fiancé for supporting me spiritually throughout writing this thesis and my life in general. Apart from their support, encouragements from my closest people are also highly acknowledged.
i
-ABSTRACT-
Signaling pathways mediated by Toll-like Receptor 2 involved in the
protection of oligodendrocytes from ischaemic cell death
Oligodendrocytes (OLs), myelinating glia of central nervous system, extend multiple branches to ensheath axons. Rapid saltatory conduction of nerve impulse depends on myelin sheath. Furthermore, recent studies have shown that axonal integrity cannot be maintained without intact myelin sheath. Therefore, devastating neurological deficits occurs when ischaemic white matter injury causes demyelination and OL loss. Treating OL loss and demyelination can be a therapeutic target for ischaemic white matter stroke. Previously, we found that Toll-like receptor 2 (TLR2) expressed in OLs provide cell-autonomous protective effects on ischaemic cell death. Here, I sought to identify the intracellular signaling pathways downstream of TLR2 that are involved in the protection of OLs from ischaemic cell death. Several protective pathways such as p38 MAP kinase, CREB, NFκB, AKT, and ERK can be activated in response of cellular stresses and participate in cytoprotective effects. Treatment of PAM3CSK4 (PAM3), a well-known TLR2 agonist, induced phosphorylation of these pathways except AKT. The same pathways were activated in response to PAM3 treatment following oxygen glucose deprivation (OGD). To identify a specific pathway causally linked to the PAM3-mediated promotion of OL survival, each pathway was inhibited pharmacologically or genetically using specific inhibitors or siRNA, respectively, and the extent of OL death was measured after using CCK or LDH assay. Among all the tested, pathways p38 MAPK inhibitor and NFκB inhibitor significantly attenuated the pro-survival effect of PAM3. siRNA knockdown experiments confirmed functional implication of the two pathways in the PAM3-mediated increase of OL survival. These results demonstrated that TLR2 provides protective effects on OLs under ischaemic stress through the p38 MAPK and NFκB pathways. Further studies will clarify effector molecules downstream of the two pathways that confer cellular protection on ischaemic OLs.
ii
TABLE OF CONTENTS
ABSTRACT i TABLE OF CONTENTS ii LIST OF FIGURES iv ABBREVIATION v I. INTRODUCTION 1A. Importance of ischaemic white matter stroke research 1 B. Pathophysiology of ischaemic white matter stroke 2
C. Myelination and oligodendrocyte 2
D. Toll-like Receptor 2 and OLs 3
E. Signaling pathways 4
F. Aims of the thesis research 7
II. MATERIALS AND METHODS 8
1. Primary OL culture 8
2. Collection of oligodendrocyte precursor cells (OPCs) 8
3. Oxygen-glucose deprivation (OGD) challenge 9
4. Western blot 9
5. Inhibitor treatment 10
6. siRNA nucleofaction 10
7. Cell viability assay 10
8. Cytotoxicity assay/LDH assay 11
9. Statistical analysis 11
III. RESULTS 12
1. PAM3 activates intracellular signaling pathways
2. Further activation of intracellular signaling pathways after OGD 12 14
iii
4. p38 MAPK and NFκB inhibition reverse the effect of PAM3 on ischaemic cell
18
5. Genetic knockdown of p38 MAPK and NFκB attenuates PAM3 mediated prosurvival effects on OLs under ischaemic stress
23
IV. DISCUSSION 28
V. CONCLUSION 31
iv
LIST OF FIGURES
Figure 1: Expression and activation of signaling pathways in OLs cultures after PAM3 treatment
13
Figure 2: Further activation of signaling pathways in OLs cultures after PAM3 treatment. Following OGD
15
Figure 3: PAM3 mediated cell survival after OGD 17
Figure 4: Dose dependent effect of inhibitors 19
Figure 5: p38 MAPK or NFκB inhibitor block PAM3CSK4-mediated prosurvival effects on OLs under ischaemic stress in CCK assay
21
Figure 6: p38 MAPK or NFκB inhibitor block PAM3CSK4-mediated prosurvival effects on OLs under ischaemic stress in LDH assay
22
Figure 7: Genetic knockdown of signaling molecule 24
Figure 8: Genetic knockdown of p38 MAPK and NFκB attenuates PAM3 mediated prosurvival effects on OLs under ischaemic stress in CCK assay
25
Figure 9: Genetic knockdown of p38 MAPK and NFκB attenuates PAM3 mediated prosurvival effects on OLs under ischaemic stress LDH assay
v
ABBREVIATION
OLs: Oligodendrocytes TLR2: Toll-like Receptor 2 PAM3: PAM3CSK4
OGD: Oxygen glucose deprivation
CREB: cAMP response element binding protein ERK1/2: Extracellular signal-regulated kinase1/2 NFκB: Nuclear factor kappa B
IκB: Inhibitor of kappa B
HBSS: Hank’s Balanced Salt Solution PBS: Phosphate buffered saline
DMEM: Dulbecco's Modified Eagles Medium MGM: Mixed glial media
PDL: poly-D-lysin FBS: Fetal bovine serum LDH: Lactate dehydrogenase CCK: Cell counting kit
1
I. INTRODUCTION
A. Importance of ischaemic white matter stroke research
According to WHO report (2017), ischaemic heart disease and stroke are the most common causes of mortality. Among these two, stroke is the second cause of death where more than 80% are caused by major cerebral arteries blockage which leads to brain infarction involving both white and gray matter (Dewar, Underhill and Goldberg 2003, Cui et al. 2009). The majority of damage caused by stroke is located in subcortical regions and, remarkably, white matter occupies nearly half of the average infarct volume (Wang et al. 2016). Indeed, white matter and OLs are exquisitely vulnerable to ischaemia and is often injured more severely than gray matter (Pantoni, Garcia and Gutierrez 1996, Valeriani, Dewar and McCulloch 2000). Patients with white matter stroke present with a variety of neurological dysfunctions such as cognitive impairment, gait disturbance, and sensori-motor dysfunctions of a variable degree (de Groot et al. 2000, Simpson et al. 2007, Young, Halliday and Kril 2008, Tomonaga et al. 1982, Whitman et al. 2001).
In Alzheimer’s disease (AD), white matter atrophy is a common phenomenon (Villain et al. 2010) evidenced by neuroimaging (Baxter et al. 2006). Pathology of Alzheimer’s at autopsy is associated with cerebral white matter lesions that can also be detected by MRI (Moghekar et al. 2013). This relationship may hint potential association between white matter ischaemia and Alzheimer’s disease. Previously, it was also hypothesized that amyloid pathology may interact with chronic ischaemia in the white matter synergizing in the aggravation of cognitive deficits (Moody et al. 1997, Black, Gao and Bilbao 2009). Recent studies reported that early deterioration in AD patients may be partly accounted for extensive ischaemic changes in the white matter (Burton et al. 2004, Defrancesco et al. 2014). Therefore, it is conceivable that preventing or ameliorating the ischaemic white matter changes may be able to delay clinical deterioration in AD patients. To study the Parkinson’s disease onset, assessing the white matter changes are clinically useful (Kamagata et al. 2013). Brain white matter hyperintensities (WMH) commonly observed
2
on older adults associated with balance and gait impairment on brain imaging and have also been linked to cognitive deficits imaging (Herman et al. 2013). Increased WMH burden in PD is associated with worse cognitive status, regardless of age or vascular risk factors (Caligiuri et al. 2015, Kandiah et al. 2013). Thus, chronic ischaemia to the white matter may be a common factor that can contribute to aggravation of various neurodegenerative disorders (Lou et al. 2012, Debette and Markus 2010, Kinsella et al. 2009). Those clinical and preclinical condition suggest that ischaemic stroke in white matter is a serious medical condition that draws medical attention.
B. Pathophysiology of ischaemic white matter stroke
If we dissect white matter pathology, myelinated and unmyelinated axons, glial cells and blood vessels are the components of white matter. The structural integrity of axons and myelin sheath are weakened by several kinds of proteases activated by white matter ischaemia (Arai and Lo 2009, Lo, Dalkara and Moskowitz 2003). During developmental period, OPCs from the ventricular zone migrate to their final destination and then to from myelin sheaths once they start differentiation. It remains to be fully elucidated how oligodendrogenesis occurs. It would be reasonable to assume that multiple cell–cell trophic interactions may be critical for OLs to start enwrapping axons. Recent findings propose that there might lie an oligovascular niche in white matter wherein cell–cell trophic coupling between oligodendrocyte and cerebral endothelium and helps sustain ongoing oligodendrogenesis (Arai and Lo 2009). Mechanism that can provide protective effect to OLs that remain in the white matter during and after ischaemia can facilitate remyelination of axon thus can maintain structural intregity. In this regard cell based treatment would be promising for restorative therapy in white matter stroke.
C. Myelination and oligodendrocyte
In humans, myelination is an ongoing process that starts in utero, peaks during the first postnatal year, and continues into adulthood (Benes 1989). Myelination of adult brain
3
contribute to maintain axonal integrity, plasticity and circuitry function (Fields 2008, Fancy et al. 2011, Young et al. 2013, Zatorre, Fields and Johansen-Berg 2012). Experience in early life can also affect the number of OLs and levels of myelination. Region-specific decreases in white matter is associated with childhood neglect (Teicher et al. 2004), whereas enhanced white matter tract development in adulthood is exhibited by the extensive training in childhood (Bengtsson et al. 2005). Furthermore, higher numbers of OLs, larger white matter volume, increased myelination and increased cognitive abilities can be seen in young rodents (Sturrock, Smart and Tricklebank 1983, Szeligo and Leblond 1977, Markham and Greenough 2004, Sirevaag and Greenough 1987) and rhesus monkeys (Sanchez et al. 1998) raised in enriched environments. In fact, myelination levels also influenced by genetic factors, and diseases such as depression, schizophrenia, bipolar disorder, multiple sclerosis (MS), and leukodystrophies (Aston, Jiang and Sokolov 2005, Davis et al. 2003, Uranova et al. 2001, Tkachev et al. 2003). These novel findings challenge the dogma that myelination occurs only during development and is fixed once the development is ended.
In the CNS, OLs synthesize huge quantities of myelin, a lipid-rich, multilayered spiral-wrapped membrane. It insulates nerve axons and help to maintain nerve conduction. Myelin damage results conduction failure. It can cause cognitive and motor defects eventually leading paralysis. Mechanism that can reduces OLs death can promote myelination and reduce disease outcome.
D. Toll-like Receptor 2 and OLs
Toll-like receptors (TLRs) are transmembrane pattern-recognition receptors (PRRs) that commence signals in response to different pathogen-associated molecular patterns (PAMPs) (Kawai and Akira 2007b). TLRs are leading PRRs that have a major role in the induction of innate immunity against infesting microbial pathogens. TLRs have generally been considered receptors expressed on antigen presenting cells of the immune system. B cells, dendritic cells, monocytes and macrophages are such kind of immune cells where they mediate innate immunity (Okun et al. 2009). Studies are very limited regarding the expression and function of TLRs in oligodendrocytes, compared to other cell types.
4
Expression of TLR2 on OLs was first reported by Bsibsi and colleagues (Bsibsi et al. 2002) showing the predominant expression of TLR2 and 3. Lehnardt and colleagues (Lehnardt et al. 2006) confirmed TLR2 expression on OLs. It also published that TLR2 is expressed by OLs and is up-regulated in MS lesions (Sloane et al. 2010). Cerebral ischaemia results in the upregulation of TLR2, TLR4 and TLR9 mRNA in mouse CNS (Ziegler et al. 2007). Above mentioned evidences provide a possible neuroprotective role of TLR2 mediated signaling for oligodendrocytes by increasing myelination of neurons in the CNS. Another report indicated the involvement of TLR signaling in oligodendrocytes during damage repair after spinal cord injury (Kigerl et al. 2007). Our lab reported that TLR2 provides an endogenous protective effects on ischemic demyelination and OL death (Choi et al. 2014) and this beneficial function of TLR2 may not be directly related to its canonical role in mounting innate immune responses (Akira and Takeda 2004), but may be because of stimulation of cell survival pathways (Li, Jiang and Tapping 2010, Okun, Griffioen and Mattson 2011).
E. Signaling pathways
Previous study from our lab showed that TLR2 provide endogenous protective effects on ischaemic demyelination and OL degeneration (Choi et al. 2014). The aim of my thesis research is to find out the downstream signaling molecules that are involved in TLR2-mediated cell survival in OLs. I performed literature data mining and selected a set of potential intracellular signaling pathways involved in the process. Mitogen-activated protein kinases (MAPKs) are serine-threonine protein kinases which play like intracellular pathways to control numerous cellular functions including cellular stress response, survival, proliferation, migration, and differentiation (Cuenda and Rousseau 2007). The MAPK pathway is a group of proteins in the cell that relates a signal from a receptor on the surface of the cell to the DNA in the nucleus of the cell. All MAPKs consisting of Thr-Xaa-Tyr motif in the primary amino acid sequence of their activation loop, where Xaa differs between the various subfamily members. It can be divided into four sub-families: extracellular-regulated kinases (ERKs), p38, c-Jun N-terminal kinases (JNKs), and big MAPKs (BMKs). ERK1 and ERK2 are activated in response to various mitogenic factors,
5
differentiation stimuli and cytokines. ERK1/2 signaling plays a critical role in cell proliferation by regulating cell growth and cell cycle progression. p38 MAPK a unique Thr-Gly-Tyr (TGY) dual phosphorylation motif (Thornton and Rincon 2009). p38 is activated in response to many inflammatory cytokines including interleukin (IL)-1, -6, -8, and tumor necrosis factor alpha (TNFα) (Dambach 2005, Schieven 2009). In addition, p38 is responsible for cytokine gene activation and their post-transcriptional regulation. Cytokine-induced activation of the p38 signaling cascade, the pathway can be activated by other stimuli including osmotic shock, stress, UV radiation, reactive oxygen species, DNA damage, cytokines, chemokines, growth factors and GPCR ligands (Thornton and Rincon 2009).
CREB (cAMP response element-binding protein) is a transcription factor. It binds to certain cAMP response elements (CRE) DNA sequences, thereby can upregulate or downregulate the transcription of the downstream genes. CREB1 is critical for a variety of cellular processes, including proliferation, adaptive response and differentiation. CREB family members are believed to be important for learning and memory and contribute to neuronal adaptation to drugs of abuse and hormonal control of metabolic processes, including regulation of gluconeogenesis by hormones glucagon and insulin.
NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells) is a protein complex that controls transcription of DNA, cytokine production and cell survival. It consists of two subunits: p65/RelA and p50/p105, and is inactive in the cytoplasm by remaining a complex with its inhibitory unit, IκB. NFκB is found in all animal and almost all cell types and is involved in cellular responses to stimuli such as cytokines, stress, free radicals, UV radiation, heavy metals, oxidized LDL, and bacterial or viral antigens. NFκB plays a crucial role in balancing the immune response to infection (κ light chains are critical components of immunoglobulins). Phosphorylation of IκB results in its ubiquitination and rapid degradation by proteasomes, freeing NFκB p65 to translocate to the nucleus and bind to DNA, leading to expression of target genes (Wang et al. 2006). NFκB is important in conducting cellular responses because it belongs to "rapid-acting" transcription factors, i.e., transcription factors that are present in cells in an inactive state and do not require new protein synthesis in order to become activated. Reactive oxygen
6
species (ROS), tumor necrosis factor alpha (TNFα), interleukin 1-beta (IL-1β), bacterial lipopolysaccharides (LPS) are the known inducer of NFκB, and however they are variable in action.
The AKT pathway or PI3K-Akt Pathway is a signal transduction pathway that promotes survival and growth in response to extracellular signals. Key proteins involved are PI3K (phosphatidylinositol 3-kinase) and AKT (Protein Kinase B). In inactive stage AKT resides in the cytosol, when the cell is stimulated then it translocates to the plasma membrane. It has a high affinity for second messenger PI(3,4,5)P3, binding to it
preferentially over other phosphoinositides. The AKT-PI3K pathway is critical for cell survival because activated AKT influences many factors that are involved in apoptosis, either by transcription regulation or direct phosphorylation. AKT inhibits transcription factors in the nucleus, which promote the expression of cell death genes, and enhances transcription of anti-apoptotic genes.
TLR2 has relation with all 5 pathways explained above. ERK1/2 and PI3K/AKT provided protective effect through TLR2 has been shown previously (Li et al. 2010). p38 MAPK depends on upregulation of TLR2, TLR4 in skeletal muscle cell and also organism like Trichomonus-vaginalis (Chang, Park and Kim 2006, Zbinden-Foncea et al. 2012). As a part of innate immunity there are many evidence that provide NFκB activation via TLR2 (Kawai and Akira 2007a). In cancer therapy, to produce proinflammatory cytokines TLR2 mediated activation of NFκB and IL-β are essential in immune cell to stimulate CD4(+) T cells to differentiate to T helper (Wang, Miyahara and Wang 2008). Previous studies showing that mammalian TLR2 signaling activates p38 MAPK, CREB, NFκB and several transcription factor like AP1, IFN-γ, IRAK and IRF (O'Neill, Golenbock and Bowie 2013). p38 MAPK is necessary for OLs development and differentiation (Chew et al. 2010, Bhat, Zhang and Mohanty 2007) myelination (Fragoso et al. 2007, Haines et al. 2008) and also prevent apoptosis in mature cultured OLs (Hamanoue, Sato and Takamatsu 2007). CREB is the well-known transcription factor helps in OLs differentiation (Bhat et al. 2007) and white matter renewal (Miyamoto et al. 2013) provide protection against white matter damage (Watanabe et al. 2006). For OLs differentiation ERK1/2 is an important molecule (Younes-Rapozo et al. 2009, Xiao et al. 2012). NFκB also a transcription factor important
7
for OLs maturation and myelination (Raasch et al. 2011). Phosphatidylinositol 3-kinase (PI3K) and its downstream target, AKT, affect cell survival, proliferation, and differentiation (Zhang, Chopp and Zhang 2013, Vojtek et al. 2003). The PI3K/Akt, p38MARK and ERK1/2 signals are involved in OPC differentiation (Chew et al. 2010, Fyffe-Maricich et al. 2013, Santra et al. 2012, Rafalski et al. 2013).
F. Aims of the thesis research
Considering the importance of OLs and TLR2 in white matter stroke, in this thesis research I tried to find out the pathway that is involved in the TLR2 mediated cell survival from ischaemia by determining protein level. To investigate the mechanism of TLR2,
kinase inhibitors was treated to block different signaling pathways then I have
conducted cell viability and cytotoxicity assays to determine the specific pathway
that helps TLR2 to provide cell protective outcome.
8
II. Materials and methods
1. Primary OL culture
Primary OL cultures were prepared from Sprague-Dawley rats. The brains of post-natal day (P) 0–P1 pups (14–15 per litter) of mixed gender were removed from the skull and placed in the Hank’s Balanced Salt Solution (HBSS) in on ice. Overlying meninges were removed with forceps and both cortices were isolated from the telencephalon. The tissue was collected (3 brain) in a 15ml conical tube containing 200ul HBSS. The tissue was dissociated with a 1 ml micropipettor and a digestion solution was prepared containing 1 ml 0.25% trypsin (Hyclone), 1 ml mixed glial media (MGM, Dulbecco's Modified Eagles Medium (DMEM,Hyclone) containing 10% FBS and 1% GlutaMAX and 1% Penicillin/Streptomycin) and 60 ug/ul DNAseI. Then warmed to 37°C for 20 min, the cells were triturated with a 1 ml micropipettor and the reaction was stopped by filling the conical tube with MGM. The single cell suspension was centrifuged at 1,000 r.p.m. for 5 min. The pellet was resuspended with MGM after decanting the media and plated on poly-D-lysine (PDL)-coated T-75 cell culture flasks (Sigma, SIAL06) per animal. The cells were then placed in a 37°C tissue culture incubator for 9-10 days, changing whole media 1 day after culture and half of the media every 48 hours later until 9 days.
2. Collection of oligodendrocyte precursor cells (OPCs)
After 9 to 10 days in vitro, mixed glial cultures were shaken at 100 rpm at 37°C for 30min to remove microglia. Second shaking was then performed at 250 rpm at 37°C for 16 to 18 hours to remove OLs from astrocyte monolayer. The cell suspension was plated on bacterial grade Petri dish for 1 hour to separate OLs from remaining microglia and astrocytes. Now enriched for OPCs, centrifuged at 1,000 r.p.m. for 5 min in MGM was done. After suction the media, the pellet was resuspended with serum-free OL differentiation media consisting of 1mg/ml bovine insulin, GlutaMAX, 33mg/ml Holo-transferrin, B27 supplementation,100X OL supplementation and the total cell number was determined using Luna dual fluorescence cell counter. The OPCs were then seeded onto plate under a variety of experimental conditions, as described below.
9
3. Oxygen-glucose deprivation (OGD) challenge
Cultured OLs were transferred to an anaerobic chamber kept at 37°C and culture medium was replaced with glucose-free differentiation media that had been saturated with N2 gas for 1 hour. Glucose-free DMEM collected from Hyclone and differentiation media was deprived from GlutaMAX. The cells were exposed to OGD for 6 hours and then transferred to a normoxic chamber and the media were replaced by fresh ones containing glucose. Then kept in the normoxic chamber for 18 hour for maintain total 24 hour.
4. Western blot
OLs were plated in six-well plates at a density of 10,00,000cells/well and allowed to attach overnight before OGD and pharmacological treatments. Cells were then lysed in 1X RIPA buffer (CellNest) containing protease and phosphatase inhibitor cocktail (Thermo scientific) 10ul per ml and 5 pulse ultrasonification. The harvested cells were centrifuged at 12000 r.p.m. for 20 min at 4°C. Protein containing supernatant was collected and protein concentration of the supernatant was measured using Bradford assay. Equal amount of proteins were heat denatured with Laemmli’s sampling buffer at 95°C for 5 min and electrophoresed on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels. Next, the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA) and blocked for 1 hour with Tris-buffered saline containing 5% skim milk then incubated overnight with primary antibodies in 5% skim milk at 4°C and subsequently incubated for 1 hour at RT with HRP-linked secondary antibody. The following antibodies were used: rabbit-phospho p38 MAPK (Cell signaling, 1:1000), rabbit p38 MAPK (Cell signaling, 1:1000), rabbit phospho-CREB (Cell signaling, 1:1000), rabbit CREB (Cell signaling, 1:1000), rabbit phosphor-p44/42 MAPK(Erk1/2) (Cell signaling, 1:1000), rabbit p44/42 MAPK (Erk1/2) (Cell signaling, 1:1000), rabbit phospho-Akt (Cell signaling, 1:1000), rabbit Akt (Cell signaling, 1:1000), rabbit IκB-α (Santa cruz, 1:1000), rabbit NFκB p65 (Santa cruz, 1:1000), secondary antibody: goat anti-rabbit (Cell signaling, 1:2000). As loading control, mouse β-actin (Sigma, 1:50000) was used.
10
5. Inhibitor treatment
Inhibitor compounds were dissolved in DMSO to obtain 100 mM stock solutions. Cells were plated in triplicate at 10,000 cells/well in a 96-well white plate and incubated overnight. Cells were treated with different concentration of p38 MAP kinase (SB202190), ERK inhibitor (U0126), NFκB inhibitor (BAY11-7085), JNK (SP600125) for 24 hours with or without OGD and PAM3.
6. siRNA nucleofaction
Rat specific p38 MAPK, NFκB p65, ERK1, ERK2, CREB-1 siRNA and control siRNA were obtained from Santa Cruz. Amaxa Nucleofactor® electroporation system (Lonza) was used to transfect siRNA into OLs. After collecting OLs, cells were resuspended with Nucleofactor® solution and siRNAs then transferred to an electroporation cuvette. Finally, resuspended OLs in a cuvette were electroporated using the Nucleofactor II® machine (program number: A-033). OL media were replaced immediately after electroporation. OLs were plated onto culture dishes for immunoblotting, CCK or LDH assays.
7. Cell viability assay
Cell viability was determined using the WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt], commercially called Cell Counting Kit-8, was purchased from Dojindo Laboratory according to the manufacturer’s protocol. Briefly, cells were plated in triplicate at 10,000 cells/well in precoated 96-well plate (Biocoat Poly-D-Lysine cellware, Corning) and incubated overnight. Cells were then treated with OGD for 6 hours with or without PAM3CSK4 and inhibitors for 18 hours or 24 hours. Then 10 μl CCK-8 solutions was added to each well containing 100ul of medium and incubated in same incubator condition for 2 to 4 hours. The absorbance at 450 nm was measured using a microplate reader using Bradford assay. In addition to the assay, cell morphology changes were visualized under light microscope.
11
8. Cytotoxicity assay/LDH assay
Cell death was measured by lactate dehydrogenase (LDH) assay kit (Takara Bio, Inc, Madison, WI, USA). 20000 Cells were seeded in 96 well plate, after overnight incubation, added OL-differentiation media without glucose for OGD insult and then media was changed again with OL-differentiation media and after 18 hour 100 ul of media was collected to a clear 96 well plate. Then 100 ul of solution C added to each wells and incubated in the dark for 30 min. The absorbance at 490 was measured using a microplate reader using Bradford assay. The LDH level corresponding to complete cell death was determined in sister cultures exposed 1.5% Triton X-100 for 24 hours (complete death, CD). Baseline LDH levels were determined in a condition of only media without any cells (baseline, BL). Percentage of cell death in each experimental conditions was calculated using the following formula: % of OLs death = (experimental value- BL) x 100/(CD -BL).
9. Statistical analysis
All numerical values and error bars in the quantification graphs are expressed as mean ± SEM. Statistical comparison of mean values was performed using one-way ANOVA followed by Tukey’s posthoc test for three and more independent groups. All quantification graphs were generated using GraphPad Prism software version 5.0 (GraphPad Software)
12
III. RESULTS
1. PAM3 activates intracellular signaling pathways
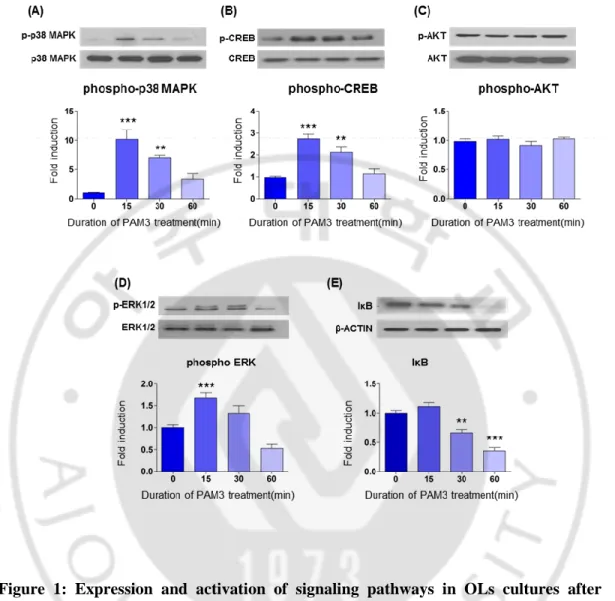
To find out relationship between those signaling pathways expressed on OLs and TLR2, I treated OLs with PAM3. PAM3 was treated for 15, 30 and 60 minutes. Relative level of expression was checked by western blotting. PAM3 induced activation of p38 MAPK, ERK, CREB, NFκB pathways.
A rapid and transient increase in p38 MAPK and CREB activation observed at 15 min following PAM3 treatment (Fig.1 A, B) which also significant in 30 min and continue up to 60 min treatment. Degradation of the inhibitory subunit IκB during PAM3 treatment, meaning activation of NFκB. Degradation was significant from 30 min to 60 min of treatment (Fig.1 E). ERK pathway also significantly activated after 15 min treatment and consequent activation occur at 30 min treatment, but after 60 min expression level tends to decrease than normal (Fig.1 D). Level of AKT remained same after treatment in all condition (Fig.1 C). So AKT is not included in TLR2 mediated activation. Almost all the pathways except AKT activated at 30 min time point, so for future experiment 30 min was used for PAM3 treatment.
13
Figure 1: Expression and activation of signaling pathways in OLs cultures after PAM3 treatment.
(A, B, D, E) western blot results depict expression and activation of p38 MAPK, CREB, ERK, IκB after PAM3 treatment in cultured OLs lysates. (C) AKT level remained unchanged after adding PAM3. Corresponding quantification graphs are showing the level of phosphorylation. Western data was analyzed by imageJ. Graphs expressed as mean ± SEM. *, ** and *** indicate p < 0.05, p < 0.01 and p <0.001 by one-way ANOVA followed by Tukey’s posthoc analysis compared with control group. N = 5 culture for each group.
14
2. Further activation of intracellular signaling pathways after OGD
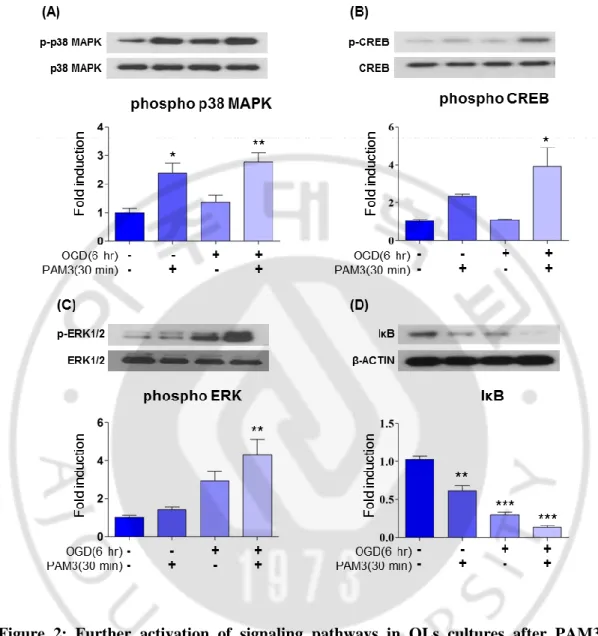
To investigate potential involvement of those pathways (except AKT) with OGD and TLR2, I checked the expression of signaling molecule by western blotting after OGD insult for 6 hour and treating cell with PAM3 for 30 minute after OGD. Because in 30 min time point all molecule showed significant expression. CCK assay illustrated that there was no significant difference in cell number between 2 hour and 6 hour OGD (3A). Both 2 and 6 hours OGD can create around 50% cell death. So protein was extracted after 30 min PAM3 treatment following 6 hour OGD.
OGD can activate p38 MAPK phosphorylation compares to control. Significant phosphorylation of p38 MAPK was found after OGD and PAM3 treatment (Fig.2 A). Both PAM3 and OGD treatment was increased CREB phosphorylation more significantly than only OGD and PAM3 itself (Fig.2 B). Only OGD cannot activate CREB. Expression was significant after PAM3 together with OGD treatment in ERK1/2 protein level (Fig.2 C). In case of NFκB, degradation of IκB was significant after OGD (Fig.2 D), more significant degradation occurs after adding PAM3 only and PAM3 after OGD. Activation of these four pathways after simultaneous treatment with OGD and PAM3 suggested the relationship between those pathways with TLR2 and ischaemia. Ischaemia induced activation of TLR2 phosphorylates p38 MAPK, CREB, ERK1/2 and NFκB.
15
Figure 2: Further activation of signaling pathways in OLs cultures after PAM3 treatment. Following OGD
(A, B, C, D) western blot results showing expression and activation of p38 MAPK, CREB, ERK, IκB after OGD and PAM3 treatment in cultured OLs lysates. Corresponding quantification graphs are demonstrating the level of phosphorylation. Western data was analyzed by imageJ. Graphs expressed as mean ± SEM. p < 0.05, p < 0.01 and p <0.001 indicated *, ** and *** by one-way ANOVA followed by Tukey’s posthoc analysis compared with control group. N = 5 culture for each group.
16
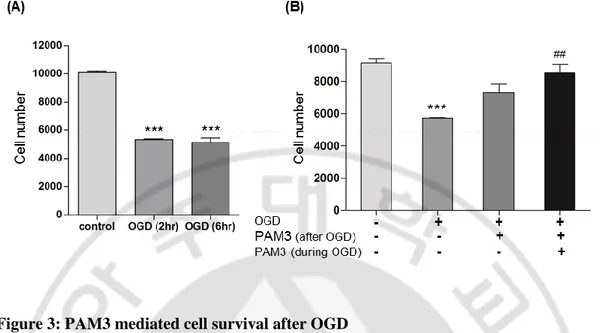
3. in vitro PAM3-mediated cell survival
Previous data suggested that OLs from TLR2 deficient mice release high amount of LDH compare to WT OLs (Choi et al., 2014). So in WT type mouse TLR2 provide endogenous effect. Same effect exerted by PAM3 in in vitro cultured OLs. Cells treated with OGD 2hour or 6 hour showed 50% cell death. Addition of PAM3 can show agonism effect of TLR2 by saving around 30% OGD treated cells. Interestingly PAM3 treatment in both during and after OGD showed highly protective effect and large number of cell compare with OGD group (Fig.3 B). Experiment with 2 hour OGD was done in previous study. But to be consistent with western blot and also to see the more intense effect of OGD, 6 hour OGD insult was carried on throughout the experimental procedure.
17
Figure 3: PAM3 mediated cell survival after OGD
A. Quantification graph of CCK assays to examine OL cell number after 2 hour and 6 hour OGD (N=5 from 3 culture).
B. Quantification graph of CCK assays to examine PAM3 mediated OLs survival after OGD (N=5 from 3 culture). * for compare with control and # for compare with OGD. Graphs expressed as mean ± SEM. p <0.001 indicated ***.
18
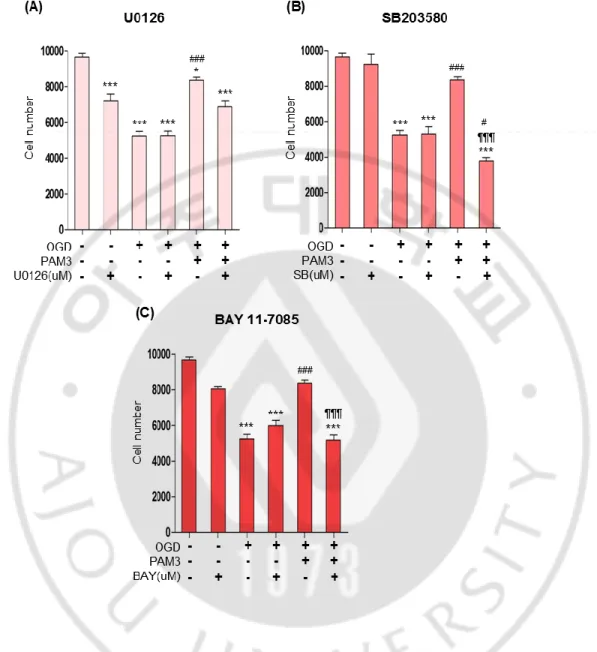
4. p38 MAPK and NFκB inhibition reverse the effect of PAM3 on ischaemic cell
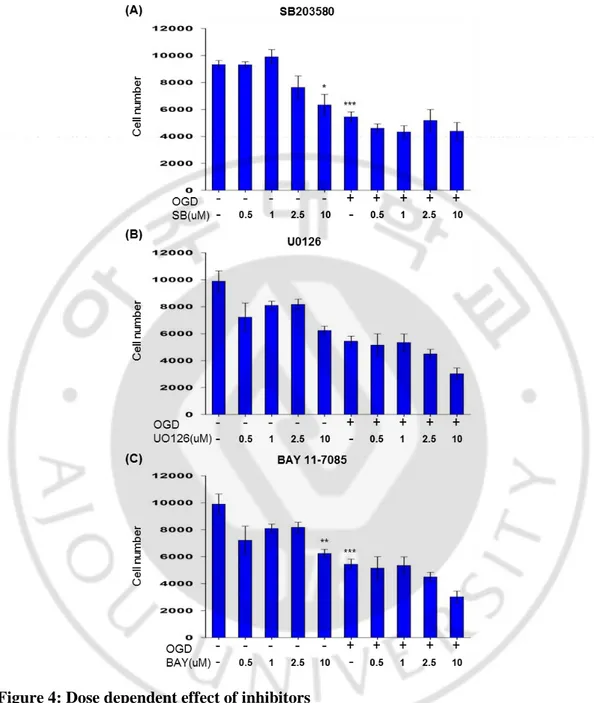
We explore the mechanism of TLR2 mediated cell survival by inhibiting intracellular signaling pathways. We hypothesized that if inhibition of a signaling pathway eliminated the effect of PAM3, PAM3 may be altering that pathway in some way to provide prosurvival effect against ischaemia. Cells were plated in a 96 well plate for overnight. To fix concentration for next experiment they were than treated with a wide range of concentrations (0.5, 1, 2.5, 5, 10 uM) of inhibitors for kinases and receptors with or without OGD.
We selected inhibitors, which are widely used previously. For p38 MAPK data demonstrated that SB203580 is the most extensively studied compound and specific for all isoform of p38 specially α and β (Lee et al. 1994). Regulation of genes greatly elucidate by SB203580, inhibitor of p38 MAPK (Ono and Han 2000). p38MAPK inhibition by SB203580 can attenuates OPC differentiation (Chew et al. 2010). U0126 reduced phospho-active ERK levels and effect was unaltered in normal tissues (Marampon et al. 2011). U0126 can revert the slow Wallerian degeneration by inhibiting ERK1/2 MAPK (Evans et al. 2013). BAY 11-7085 can blocks NFκB pathway and known to inhibit IKK, and thereby inhibits IκB phosphorylation and NFκB activation (Manna, Manna and Sarkar 2007, Dai et al. 2005, Hammar et al. 2005).
Cells were seeded in 96-well plate, after overnight incubation cells were treated with or without OGD and inhibitor was added during and after OGD. SB203580 showed dose dependent effect on naïve cell and almost dose independent manner on OGD treated cell (Fig.4 A). 1 uM concentration was less toxic then others concentration. However, U0126 effect was dose dependent on naïve and OGD treated cells but was causing significant cell death on naïve cells even in 0.5 uM concentration (Fig.4 B). So U0126 considered as toxic for cell or as ERK1/2 is the upstream of all MAP pathways, so inhibiting this pathway might be harmful for cell. In case of NFκB inhibitor BAY11-7085 was not dose dependent but 1uM could be consider as less toxic for cell (Fig.4 C). So for future experiment I have
19
Figure 4: Dose dependent effect of inhibitors
Cells were treated with increasing concentrations of potent and selective inhibitors. Graph showing effects of different doses inhibitor of P38 MAPK, ERK1/2 and NFκB on OLs. Cell was count by CCK assay. Graphs expressed as mean ± SEM. (N=5 from 3 to 4 culture). Graphs expressed as mean ± SEM. p < 0.05, p < 0.01 and p <0.001 indicated *, ** and ***.
20
selected 1uM concentration for all inhibitors and treated cell simultaneously with PAM3, and measure cell viability and cytotoxicity by CCK and LDH assays.
PAM3 was then added to the cell during and after OGD with 1uM concentration of all inhibitors. CCK assay represent that SB203580 and BAY 11-7085 can reverse the effect of PAM3 significantly compare with the PAM3 only effect on OGD (Fig.5 B, C). All 3 inhibitor presenting significant cell death after OGD compare with control group both in CCK assay. Though U0126 decreased cell number in control condition significantly, I want to confirm relation between ERK1/2 and PAM3. But in OGD after addition of ERK1/2 inhibitor demonstrated significant cell death which can be moderate by PAM3 addition. It proves that ERK is not mediated by TLR2 (Fig. 5 A).
Significant cell death was caused by OGD with SB203580. After adding PAM3 result showing that cell number was significantly decreased compare with PAM3 even compare with OGD only group (Fig. 5 B). Same effect exerted by BAY 11-7085, NFκB inhibitor. Addition of PAM3 could not provide protective effect and cell number was significantly decreased compare with PAM3 only group (Fig. 5 C). So these two inhibitor block PAM3 mediated survival which proved the relationship of these two pathways with TLR2 mediated cell survival.
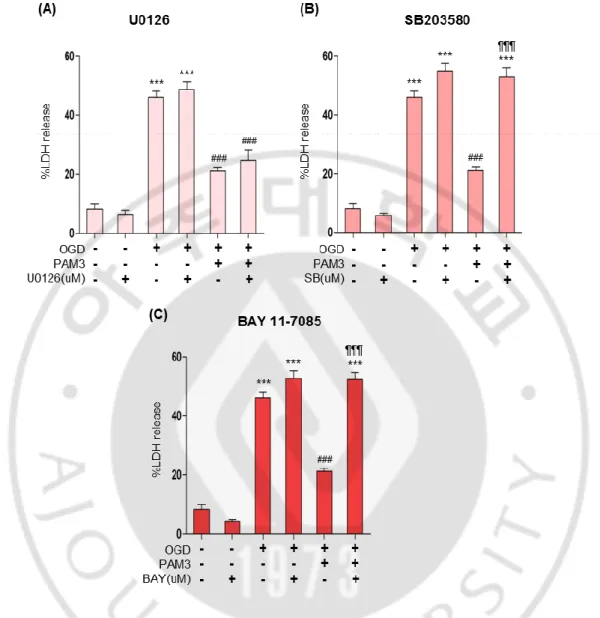
Same effect like CCK assay was elucidated by LDH assay (Fig.6). After OGD, LDH level was significantly increased meaning significant cell death. PAM3 decreased the LDH level by providing protective effect. LDH level of PAM3 treatment in U0126, ERK1/2 inhibitor group was same like only PAM3 treated group (Fig. 6 A). Both group showing significant decreased of LDH. That indicate PAM3 can improve cell survival and ERK1/2 was not provide PAM3 mediated cell survival.
However, significantly high level of LDH even after adding PAM3 exerted by SB203580, p38 MAPK inhibitor and BAY 11 7085,NFκB inhibitor group (Fig. 6 B,C). proved that in absence of p38 MAPK and NFκB, PAM3 not able to provide any protective effect. These results can confirm the involvement of these 2 pathways with PAM3.
21
Figure 5: p38 MAPK or NFκB inhibitor block PAM3CSK4-mediated prosurvival effects on OLs under ischemic stress in CCK assay.
Quantification graphs demonstrated the PAM3 mediated effects of p38 qnd NFκB inhibitors on Ols. Cells were count by CCK assay. Here, * for compare to control, # for compare to OGD, ¶ for compare to OGD and PAM3. *,#; **,## and ***, ###, ¶¶¶ indicate
p < 0.05, p < 0.01 and p < 0.001 by one-way ANOVA followed by Tukey’s posthoc
22
Figure 6: p38 MAPK or NFκB inhibitor block PAM3-mediated prosurvival effects on OLs under ischaemic stress in LDH assay.
Quantification graphs demonstrated the PAM3 mediated effects of p38 and NFκB inhibitors on OLs. Cells were count by CCK assay. Here, * for compare to control, # for compare to OGD, ¶ for compare to OGD and PAM3. p < 0.05 and p < 0.001 indicate *,# and ***, ###, ¶¶¶ by one-way ANOVA followed by Tukey’s posthoc analysis. (N=5 from 3 to 4 culture for each group).
23
5. Genetic knockdown of p38 MAPK and NFκB attenuates PAM3 mediated prosurvival effects on OLs under ischaemic stress
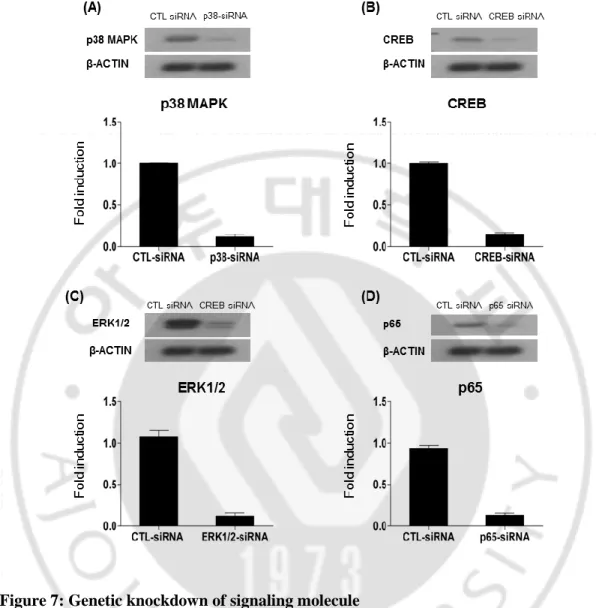
Cells were transfected with specific siRNA for those pathways from Santa Cruz. All siRNA are pool of 3 target-specific 19-25 nt siRNAs designed to knockdown gene expression. ERK1-siRNA and ERK2-siRNA separately purchased and for getting effect of ERK1/2 I treated both siRNA in same time. To knockdown NFκB, I used p65-siRNA, because p65 known as one of the prominent isoform of NFκB. Cells were transfected by nucleofaction. For effective knockdown, I checked the level of each protein after nucleofaction with different concentration of siRNA and then checked expression by western lot. ERK1/2 antibody was used to check ERK1-siRNA and ERK2-siRNA. p65 antibody was used for checking p65 knockdown.
After nucleofaction cells were seeded in 6-well plate and incubated for 48 hour. Then protein was collected and checked by immunoblotting. All molecules were effectively knockdown by transfecting 300nM concentration of each siRNA. siRNA for control was used to compare knockdown level of other siRNA (Fig. 7).
The siRNA transfected cell were seeded in 96-well plate. Day after seeding, cells treated with or without OGD and PAM3. Then cell number measure by CCK assay or cell death measure by LDH assay.
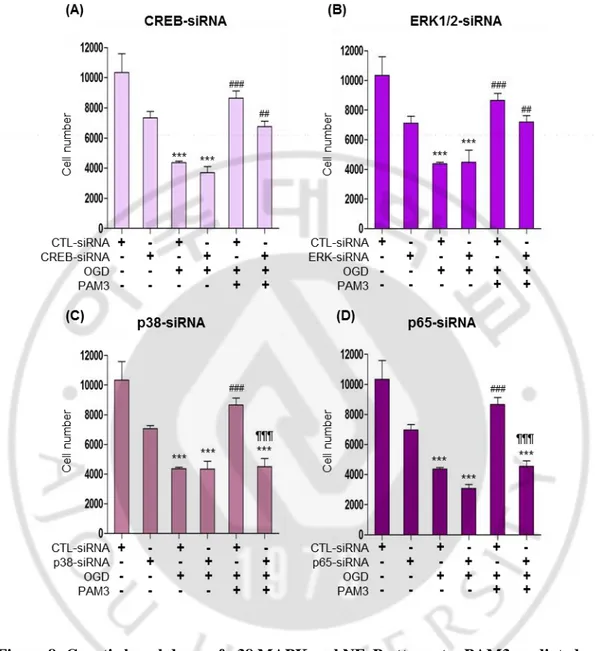
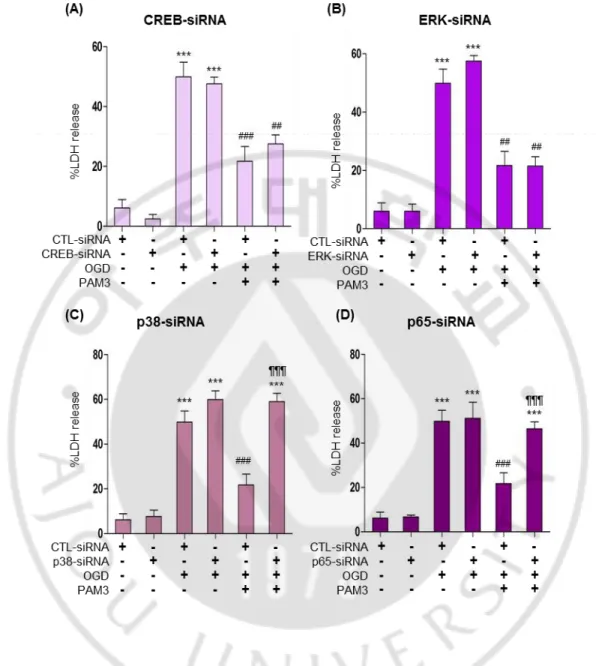
Fig.8 indicated that 50% cell survived after OGD insult and cells can be revived after PAM3 in CTL-siRNA group. Same results exhibited by CREB-siRNA and ERK1/2-siRNA. Graph showing cell number significantly increase after PAM3 addition in CREB and ERK1/2 knockdown group (Fig. 8 A, B). There is no significant difference between control and CREB or control and ERK1/2 regarding PAM3 treatment. Genetic knockdown of CREB and ERK1/2 was not able to block PAM3 mediated cell survival. Knockdown of p38 and p65 by siRNA can block PAM3 effect by decreasing cell survival. Significant cell death occurs in knockdown group of p38 and p65 compare with the PAM3 in control group (Fig. 8 C, D). Interestingly p38 knockdown group with OGD and PAM3 showing significant decrease of cell compare with OGD only.
24
Figure 7: Genetic knockdown of signaling molecule
(A, B, C, D) showing knockdown of CREB, ERK, p38 MAPK, p65 subunit of NFκB. Corresponding quantification graphs demonstrating the levels of phosphorylation of each pathway analyzed by imageJ.
25
Figure 8: Genetic knockdown of p38 MAPK and NFκB attenuates PAM3 mediated prosurvival effects on OLs under ischaemic stress CCK assay.
Quantification graphs demonstrated the PAM3 mediated effects of p38 and NFκB knockdown on Ols. Cells were count by CCK assay. Here, * for compare to control, # for compare to OGD, ¶ for compare to OGD and PAM3. *,# and ***, ###, ¶¶¶ indicate p < 0.05 and p < 0.001 by one-way ANOVA followed by Tukey’s posthoc analysis. (N=5 from 3 to 4 culture for each group).
26
Again in LDH assay, CREB and ERK1/2 knockdown group presented with PAM3 mediated cell survival by expressing low LDH after PAM3 addition like control siRNA group (Fig. 9 A, B). No significant difference between PAM3 addition in control siRNA and CREB-siRNA or control siRNA and ERK1/2-siRNA. LDH level of p38-siRNA and p65-siRNA remained same after PAM3 treatment compare with the OGD group of p38-siRNA and p65-p38-siRNA (Fig. 9, C, D). It was compelling compare with control and only PAM3 group. So both CCK and LDH assay suggested that p38 MAPK and NFκB contribute to the TLR2-mediated cell protection.
27
Figure 9: Genetic knockdown of p38 MAPK and NFκB attenuates PAM3 mediated prosurvival effects on OLs under ischaemic stress LDH assay.
Quantification graphs demonstrated the PAM3 mediated effects of p38 qnd NFκB knockdown on OLs. Cells were count by CCK assay. Here, * for compare to control, # for compare to OGD, ¶ for compare to OGD and PAM3. *,#; **,## and ***, ###, ¶¶¶ indicate
p < 0.05, p < 0.01 and p < 0.001 by one-way ANOVA followed by Tukey’s posthoc
28
IV. DISCUSSION
OLs play important role in CNS like forming myelin and conduction by Na channel and node of Ranvier, remyelinate injured axon and reduced outcome of neurodegenerative diseases like white matter stroke, Alzheimer’s disease, and Parkinson’s disease. So finding the specific molecule that can provide protection during and after ischaemia is therapeutically important. OL survival in an ischaemic condition is promoted by TLR, through potentially multiple downstream signaling molecules. Based on the cell viability and cytotoxicity assays, I found that pharmacological and genetic inhibition of p38 MAPK and NFκB led to attenuation of pro-survival effects of PAM3 treatment that was already established in our previous publication (Choi et al. 2014). Therefore, I conclude that the p38 MAPK and NFκB are two key signaling pathways downstream to TLR2 that provide protective effects in OLs response with ischaemia. In contrast, the cells treated with CREB or ERK1/2 inhibitor or knockdown of these 2 pathway failed to block the effects of TLR2 atomism on OL. Therefore, it would be seasonable to argue that CREB and ERK1/2 are not key downstream pathways mediating TLR2 activation to enhanced OL survival.
Previously, it was thought that TLR2 expression is limited in immune cells as a part of innate immunity. Recent studies improve the knowledge of functional spectrum of TLR2 beyond the boundaries of immune cells. Bsibsi and colleagues first reported that TLR2 can express in OLs. However, role of this receptor was quite confusing because of its relation of immunity. Both merits and demerits of this receptor had been reported. Many studies were done on retinal myelination and spinal cord repair by transplanting TLR2 activated OLs, and the results of these studies indicated neuroprotective effect of TLR2. The protective outcomes of TLR2 activation is still under debate because injection of zymosan,-TLR2 agonist, resulted in cytotoxicity of neurons in the brain (Schonberg, Popovich and McTigue 2007). Few researches have been performed on white matter stroke where OLs are the most vulnerable cell type. Interestingly, our lab found that TLR2 and its agonist PAM3 provide intrinsic prosurvival effects on OLs that were, exposed to ischaemia.
Both p38 MAPK and NFκB pathways are well known to be involved in pro-inflammatory processes. The NFκB pathway has been implicated in other pathological
29
conditions such as autoimmune or toxic demyelination and spinal cord injury (Brambilla et al. 2005, Khoshnan and Patterson 2011). White matter astrogliosis was accompanied by activation of the pro-inflammatory transcription factor nuclear factor NFκB in reactive astrocytes that could provide neuroprotective effect (Saggu et al. 2016). p38 MAPK has been implicated in OLs differentiation and proliferation (Chew et al. 2010, Bhat et al. 2007). The result in my thesis is intriguing in that these two pathways are functionally linked to the pro-survival effects conferred by TLR2 activation in OLs while they are traditionally regarded as proinflammatory pathways. It is conceivable that traditional proinflammatory pathways may be linked to processes supporting cell survival in non-immune cells such as OLs. Thus, intracellular signal pathways may be highly specific to each cell type involved. Cell-type specific functional dissection of TLR2 pathways would be available in near future.
Endogenous ligand for TLR2, which becomes available in response to cellular stress conditions, can initiate TLR2 activation followed by downstream signaling pathways such kind of activity. Our recent studies showed that HMGB1 expressed in cultured OLs and also in vivo white matter model indicated that HMGB1 is the potent ligand for TLR2 receptor (Choi et al. 2017). HMGB1 also effectively induces IL-8 release only from TLR2 overexpressing cells (Yu et al. 2006). HMGB1 is a endogenous ligand for Toll-like receptor 2 (TLR2) signaling on bone marrow-derived Glioblastoma multiforme-infiltrating dendritic cells as well (Curtin et al. 2009). The intracellular signaling pathways activated downstream of HMGB1 correspond to some of the canonical TLR2 pathways. HMGB1 treatment of cultured OLs induced rapid degradation of IκB-α, indicating activation of the NFκB pathway, a TLR2-mediated intracellular signal pathway linked to proinflammatory gene activation (Choi et al. 2017)
.
There are some possible limitations to the experiments with the inhibitors. One is that the inhibitors may have certain nonspecific actions that target pathways other than what is intended. I tried to choose inhibitors that are selective for each kinase or receptor, but there are very few inhibitors that are entirely selective. For example, SB202190 is an inhibitor of p38 MAPK which only can inhibit α subunit where SB203580 can inhibit both α andβ. Inhibiting relA or p65 subunit of NFκB is more convenient because effects of p65 are
30
already proven and the antibodies that detect it are also available. Another limitation is that the concentrations chosen may not adequately inhibit the intended target. We used a wide range of concentrations from 0.5 uM to 10 uM, but it is still possible that a higher concentration may be needed to adequately inhibit the target in the cells. A major limitation to this experiment, as well as the other experiments, is that our model, a mixed glial cell line, may not perfectly represent human OLs. In in vitro culture, I separated OLs by mechanical shaking from other cell type. Although this method can highly enrich OL or OPC progenitors, there are still other cell types remaining in a very small quantity. However, I assume that there is very little , if any, cell-to-cell interactions that may have influenced experimental outcomes in our study because the purity of our culture is higher than 95% (Choi et al. 2014). Finally, the fact that I started with the five signaling pathways that were chosen based on literature data mining suggests that there might be other signaling pathways than the five ones that may be causally linked to TLR2 signaling support of OL survival. In this regard, a more systematic approach to screen potential pathways that may be influenced by TLR2 activation would be more informative. Indeed, I started an expression profiling experiment using cultured OL under OGD stress with or without PAM3 using RNA-seq approach. The data from this experiment would complement those from the current thesis research to provide more definitive answers on the downstream intracellular signaling pathways activated by TLR2 agonism. I speculate that these answers would contribute to the development of a novel therapeutic approach to prevent OL loss and demyelination following ischaemic white matter stroke.
To summarize, my thesis research demonstrate that p38 MAPK and NFκB pathways are causally linked to the pro-survival effects of TLR2 activation on cultured OLs under OGD stress. Further studies will be utilizing an in vivo animal model of white matter stroke to verify that the two signaling pathways play pivotal roles in supporting the extent of ischaemic demyelination in vivo. I will also try to analyze gene expression profiling data for systematic analysis of potential pathways involved in this process. Furthermore, I will identify any effector molecule specifically associated with promotion of OLs. These efforts will bring me closer to the ultimate goal to find an effective therapy for white matter stroke.
31
V. CONCLUSION
I demonstrated that PAM3 can activate several protective signaling pathways like p38 MAPK, CREB, ERK1/2 and NFκB but not AKT. All other selected pathways except AKT, significantly phosphorylated by TLR2 and ischaemia. OGD causes apoptotic cell death, which can reduce by TLR2. So TLR2 provide protection against apoptotic OLs death against ischaemia. From those pathways that activated by ischaemia and TLR2, inhibition or genetic knockdown of p38 MAPK and NFκB can block TLR2 mediated cell survival from OGD that mimic ischaemia. So in vivo application of inhibitor or siRNA can be effective to study this effect precisely. Also studying downstream molecule of those pathways, which can affect size of infarction caused by ET1 or L-NIO can be effective to find out more potential drugs for ischaemic white matter stroke.
32
VI. REFERENCES
1. Akira, S. & K. Takeda (2004) Toll-like receptor signalling. Nat Rev Immunol, 4, 499-511.
2. Arai, K. & E. H. Lo (2009) Oligovascular Signaling in White Matter Stroke.
Biological & pharmaceutical bulletin, 32, 1639-1644.
3. Aston, C., L. Jiang & B. P. Sokolov (2005) Transcriptional profiling reveals evidence for signaling and oligodendroglial abnormalities in the temporal cortex from patients with major depressive disorder. Mol Psychiatry, 10, 309-22.
4. Baxter, L. C., D. L. Sparks, S. C. Johnson, B. Lenoski, J. E. Lopez, D. J. Connor & M. N. Sabbagh (2006) Relationship of cognitive measures and gray and white matter in Alzheimer's disease. J Alzheimers Dis, 9, 253-60.
5. Benes, F. M. (1989) Myelination of cortical-hippocampal relays during late adolescence. Schizophr Bull, 15, 585-93.
6. Bengtsson, S. L., Z. Nagy, S. Skare, L. Forsman, H. Forssberg & F. Ullen (2005) Extensive piano practicing has regionally specific effects on white matter development. Nat Neurosci, 8, 1148-1150.
7. Bhat, N. R., P. Zhang & S. B. Mohanty (2007) p38 MAP kinase regulation of oligodendrocyte differentiation with CREB as a potential target. Neurochem Res, 32, 293-302.
8. Black, S., F. Gao & J. Bilbao (2009) Understanding white matter disease: imaging-pathological correlations in vascular cognitive impairment. Stroke, 40, 8. 9. Brambilla, R., V. Bracchi-Ricard, W.-H. Hu, B. Frydel, A. Bramwell, S.
Karmally, E. J. Green & J. R. Bethea (2005) Inhibition of astroglial nuclear factor κB reduces inflammation and improves functional recovery after spinal cord injury. Journal of Experimental Medicine, 202, 145-156.
10. Bsibsi, M., R. Ravid, D. Gveric & J. M. van Noort (2002) Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp
33
11. Burton, E. J., I. G. McKeith, D. J. Burn, E. D. Williams & J. T. O'Brien (2004) Cerebral atrophy in Parkinson's disease with and without dementia: a comparison with Alzheimer's disease, dementia with Lewy bodies and controls. Brain, 127, 791-800.
12. Caligiuri, M. E., P. Perrotta, A. Augimeri, F. Rocca, A. Quattrone & A. Cherubini (2015) Automatic Detection of White Matter Hyperintensities in Healthy Aging and Pathology Using Magnetic Resonance Imaging: A Review. Neuroinformatics, 13, 261-76.
13. Chang, J. H., J. Y. Park & S. K. Kim (2006) Dependence on p38 MAPK signalling in the up-regulation of TLR2, TLR4 and TLR9 gene expression in Trichomonas vaginalis-treated HeLa cells. Immunology, 118, 164-70.
14. Chew, L. J., W. Coley, Y. Cheng & V. Gallo (2010) Mechanisms of regulation of oligodendrocyte development by p38 mitogen-activated protein kinase. J
Neurosci, 30, 11011-27.
15. Choi, J. Y., Y. Cui, S. T. Chowdhury & B. G. Kim (2017) High-mobility group box-1 as an autocrine trophic factor in white matter stroke. Proceedings of the
National Academy of Sciences.
16. Choi, J. Y., Y. Cui, Y. M. Kang, J. H. Kim, S. J. Lee & B. G. Kim (2014) Role of toll-like receptor 2 in ischemic demyelination and oligodendrocyte death.
Neurobiol Aging, 35, 1643-53.
17. Cuenda, A. & S. Rousseau (2007) p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta, 8, 24.
18. Cui, X., M. Chopp, A. Zacharek, C. Roberts, M. Lu, S. Savant-Bhonsale & J. Chen (2009) Chemokine, vascular and therapeutic effects of combination Simvastatin and BMSC treatment of stroke. Neurobiol Dis, 36, 35-41.
19. Curtin, J. F., N. Liu, M. Candolfi, W. Xiong, H. Assi, K. Yagiz, M. R. Edwards, K. S. Michelsen, K. M. Kroeger, C. Liu, A. K. Muhammad, M. C. Clark, M. Arditi, B. Comin-Anduix, A. Ribas, P. R. Lowenstein & M. G. Castro (2009) HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS
34
20. Dai, Y., M. Rahmani, P. Dent & S. Grant (2005) Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-κB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Molecular and
cellular biology, 25, 5429-5444.
21. Dambach, D. M. (2005) Potential adverse effects associated with inhibition of p38alpha/beta MAP kinases. Curr Top Med Chem, 5, 929-39.
22. Davis, K. L., D. G. Stewart, J. I. Friedman, M. Buchsbaum, P. D. Harvey, P. R. Hof, J. Buxbaum & V. Haroutunian (2003) White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch Gen Psychiatry, 60, 443-56.
23. de Groot, J. C., F. E. de Leeuw, M. Oudkerk, J. van Gijn, A. Hofman, J. Jolles & M. M. Breteler (2000) Cerebral white matter lesions and cognitive function: the Rotterdam Scan Study. Ann Neurol, 47, 145-51.
24. Debette, S. & H. S. Markus (2010) The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. Bmj, 26.
25. Defrancesco, M., K. Egger, J. Marksteiner, R. Esterhammer, H. Hinterhuber, E. A. Deisenhammer & M. Schocke (2014) Changes in white matter integrity before conversion from mild cognitive impairment to Alzheimer's disease. PLoS One, 9. 26. Dewar, D., S. M. Underhill & M. P. Goldberg (2003) Oligodendrocytes and
ischemic brain injury. J Cereb Blood Flow Metab, 23, 263-74.
27. Evans, C., S. J. Cook, M. P. Coleman & J. Gilley (2013) MEK Inhibitor U0126 Reverses Protection of Axons from Wallerian Degeneration Independently of MEK–ERK Signaling. PloS one, 8, e76505.
28. Fancy, S. P., J. R. Chan, S. E. Baranzini, R. J. Franklin & D. H. Rowitch (2011) Myelin regeneration: a recapitulation of development? Annu Rev Neurosci, 34, 21-43.
29. Fields, R. D. (2008) White matter in learning, cognition and psychiatric disorders.
35
30. Fragoso, G., J. D. Haines, J. Roberston, L. Pedraza, W. E. Mushynski & G. Almazan (2007) p38 mitogen-activated protein kinase is required for central nervous system myelination. Glia, 55, 1531-41.
31. Fyffe-Maricich, S. L., A. Schott, M. Karl, J. Krasno & R. H. Miller (2013) Signaling through ERK1/2 Controls Myelin Thickness during Myelin Repair in the Adult Central Nervous System. The Journal of Neuroscience, 33, 18402-18408.
32. Haines, J. D., G. Fragoso, S. Hossain, W. E. Mushynski & G. Almazan (2008) p38 Mitogen-activated protein kinase regulates myelination. J Mol Neurosci, 35, 23-33.
33. Hamanoue, M., K. Sato & K. Takamatsu (2007) Inhibition of p38 mitogen-activated protein kinase-induced apoptosis in cultured mature oligodendrocytes using SB202190 and SB203580. Neurochem Int, 51, 16-24.
34. Hammar, E. B., J.-C. Irminger, K. Rickenbach, G. Parnaud, P. Ribaux, D. Bosco, D. G. Rouiller & P. A. Halban (2005) Activation of NF-κB by extracellular matrix is involved in spreading and glucose-stimulated insulin secretion of pancreatic beta cells. Journal of Biological Chemistry, 280, 30630-30637.
35. Herman, T., K. Rosenberg-Katz, Y. Jacob, E. Auriel, T. Gurevich, N. Giladi & J. M. Hausdorff (2013) White matter hyperintensities in Parkinson's disease: do they explain the disparity between the postural instability gait difficulty and tremor dominant subtypes? PLoS One, 8, 31.
36. Kamagata, K., Y. Motoi, H. Tomiyama, O. Abe, K. Ito, K. Shimoji, M. Suzuki, M. Hori, A. Nakanishi, T. Sano, R. Kuwatsuru, K. Sasai, S. Aoki & N. Hattori (2013) Relationship between cognitive impairment and white-matter alteration in Parkinson's disease with dementia: tract-based spatial statistics and tract-specific analysis. Eur Radiol, 23, 1946-55.
37. Kandiah, N., E. Mak, A. Ng, S. Huang, W. L. Au, Y. Y. Sitoh & L. C. Tan (2013) Cerebral white matter hyperintensity in Parkinson's disease: a major risk factor for mild cognitive impairment. Parkinsonism Relat Disord, 19, 680-3.
36
38. Kawai, T. & S. Akira (2007a) Signaling to NF-kappaB by Toll-like receptors.
Trends Mol Med, 13, 460-9.
39. --- (2007b) TLR signaling. Semin Immunol, 19, 24-32.
40. Khoshnan, A. & P. H. Patterson (2011) The role of IκB kinase complex in the neurobiology of Huntington's disease. Neurobiology of disease, 43, 305-311. 41. Kigerl, K. A., W. Lai, S. Rivest, R. P. Hart, A. R. Satoskar & P. G. Popovich
(2007) Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J Neurochem, 102, 37-50.
42. Kinsella, G. J., E. Mullaly, E. Rand, B. Ong, C. Burton, S. Price, M. Phillips & E. Storey (2009) Early intervention for mild cognitive impairment: a randomised controlled trial. J Neurol Neurosurg Psychiatry, 80, 730-6.
43. Lee, J. C., J. T. Laydon, P. C. McDonnell, T. F. Gallagher, S. Kumar, D. Green, D. McNulty, M. J. Blumenthal, J. R. Heys, S. W. Landvatter & et al. (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis.
Nature, 372, 739-46.
44. Lehnardt, S., P. Henneke, E. Lien, D. L. Kasper, J. J. Volpe, I. Bechmann, R. Nitsch, J. R. Weber, D. T. Golenbock & T. Vartanian (2006) A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol, 177, 583-92.
45. Li, X., S. Jiang & R. I. Tapping (2010) Toll-like receptor signaling in cell proliferation and survival. Cytokine, 49, 1-9.
46. Lo, E. H., T. Dalkara & M. A. Moskowitz (2003) Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci, 4, 399-415.
47. Lou, G., Q. Zhang, F. Xiao, Q. Xiang, Z. Su, L. Zhang, P. Yang, Y. Yang, Q. Zheng & Y. Huang (2012) Intranasal administration of TAT-haFGF((1)(4)(-)(1)(5)(4)) attenuates disease progression in a mouse model of Alzheimer's disease. Neuroscience, 223, 225-37.
48. Manna, S., P. Manna & A. Sarkar (2007) Inhibition of RelA phosphorylation sensitizes apoptosis in constitutive NF-kappaB-expressing and chemoresistant cells. Cell Death & Differentiation, 14, 158-170.
37
49. Marampon, F., G. L. Gravina, A. Di Rocco, P. Bonfili, M. Di Staso, C. Fardella, L. Polidoro, C. Ciccarelli, C. Festuccia, V. M. Popov, R. G. Pestell, V. Tombolini & B. M. Zani (2011) MEK/ERK inhibitor U0126 increases the radiosensitivity of rhabdomyosarcoma cells in vitro and in vivo by downregulating growth and DNA repair signals. Mol Cancer Ther, 10, 159-68.
50. Markham, J. A. & W. T. Greenough (2004) Experience-driven brain plasticity: beyond the synapse. Neuron glia biology, 1, 351-363.
51. Miyamoto, N., T. Maki, L.-D. D. Pham, K. Hayakawa, J. H. Seo, E. T. Mandeville, J. B. Mandeville, K.-W. Kim, E. H. Lo & K. Arai (2013) Oxidative Stress Interferes With White Matter Renewal After Prolonged Cerebral Hypoperfusion in Mice. Stroke; a journal of cerebral circulation, 44, 3516-3521. 52. Moghekar, A., S. Li, Y. Lu, M. Li, M. C. Wang, M. Albert & R. O'Brien (2013)
CSF biomarker changes precede symptom onset of mild cognitive impairment.
Neurology, 81, 1753-8.
53. Moody, D. M., W. R. Brown, V. R. Challa, H. S. Ghazi-Birry & D. M. Reboussin (1997) Cerebral microvascular alterations in aging, leukoaraiosis, and Alzheimer's disease. Ann N Y Acad Sci, 826, 103-16.
54. O'Neill, L. A. J., D. Golenbock & A. G. Bowie (2013) The history of Toll-like receptors [mdash] redefining innate immunity. Nat Rev Immunol, 13, 453-460. 55. Okun, E., K. J. Griffioen, J. D. Lathia, S.-C. Tang, M. P. Mattson & T. V.
Arumugam (2009) Toll-like receptors in neurodegeneration. Brain research
reviews, 59, 278-292.
56. Okun, E., K. J. Griffioen & M. P. Mattson (2011) Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci, 34, 269-81.
57. Ono, K. & J. Han (2000) The p38 signal transduction pathway activation and function. Cellular signalling, 12, 1-13.
58. Pantoni, L., J. H. Garcia & J. A. Gutierrez (1996) Cerebral white matter is highly vulnerable to ischemia. Stroke, 27, 1641-6.
59. Raasch, J., N. Zeller, G. van Loo, D. Merkler, A. Mildner, D. Erny, K. P. Knobeloch, J. R. Bethea, A. Waisman, M. Knust, D. Del Turco, T. Deller, T.