저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Master's Thesis in Department of
Biomedical Sciences
Underlying mechanisms of
Ophiobolin A-induced paraptosis-like
cell death in glioma cells
Ajou University Graduate School
Major in Cancer Biology
Mi Ri Kwon
Underlying mechanisms of Ophiobolin A-
induced paraptosis-like cell death
in glioma cells
Kyeong Sook Choi, Advisor
I submit this thesis as the
Master's thesis in Department of
Biomedical Sciences.
August 2017
Ajou University Graduate School
Major in Cancer Biology
i
-ABSTRACT-
Underlying mechanisms of Ophiobolin A-induced
paraptosis-like cell death in glioma cells
Ophiobolin A (OP-A), a fungal sesterterpene from Bipolaris oryzae, has recently been shown to have anti-glioma activity. I found that OP-A induces paraptosis-like cell death accompanied by the endoplasmic reticulum (ER) dilation, accumulation of poly-ubiquitinated proteins in glioma cells and CHOP-mediated ER stress plays a critical role in this process. Inhibition of protein synthesis using cycloheximide (CHX) almost completely blocked the vacuolation and glioma cell death, suggesting a requirement of new protein synthesis for this death accompanied by vacuolation. Interestingly, thiol-based antioxidants, including N-acetylcysteine (NAC) and reduced glutathione (GSH) and N-(2-mercaptopropionyl)-glycine (NMPG), but not other non-thiol antioxidants, including Ascorbic acid and Tiron and MnTBAP (Mn(III)tetrakis (4-benzoic acid) porphyrin) very effectively blocked OP-A-induced vacuolation and cell death. In addition, notable generation of reactive oxygen species (ROS) by OP-A was not detected, suggesting that the anti-cancer effects of OP-A may be mediated though its ability to covalently modify free sulfhydryl groups on proteins causing protein misfolding, rather than through ROS generation. Collectively, our results indicate that ER stress due to disruption of thiol proteostasis may be responsible for the potent anti-glioma activity of OP-A.
Keywords: Ophiobolin A, Glioblastoma multiforme (GBM), endoplasmic
ii
TABLE OF CONTENTS
ABSTRACT
--- ⅰTABLE OF CONTENTS
--- ⅱLIST OF FIGURES
--- ⅳI. INTRODUCTION
--- 1II. MATERIALS AND METHODS
--- 41. Chemicals and antibodies --- 4
2. Cell culture --- 4
3. Measurement of cellular viability (Live/Dead assay) --- 5
4. Examination of the stable cell lines expressing the fluorescence specifically in the endoplasmic reticulum --- 5
5. Western blotting --- 6
6. Immunocytochemistry (ICC) --- 6
7. Gene silencing of CHOP (Knockdown experiments using siRNAs and shRNAs) --- 7
8. Measurement of reactive oxygen species (ROS) production --- 7
9. Statistical analysis --- 8
III. RESULTS
--- 91. Ophiobolin A induces paraptosis-like cell death in glioma cells via dilation of the ER --- 9
iii
2. OP-A induces ER stress and CHOP plays a critical role in OP-A-induced
paraptosis-like cell death --- 19
3. Thiol-antioxidants but not other ROS scavengers block OP-A-induced paraptosis-like cell death --- 28
4. Disruption of thiol homeostasis by A critically contributes to OP-A-mediated anti-cancer effects in various cancer cell lines --- 36
IV. DISCUSSION
--- 47V. REFERENCES
--- 56iv
LIST OF FIGURES
Figure 1. OP-A induces the cell death in various glioma cells
--- 11Figure 2. Ophiobolin A induces cytoplasmic vacuolation prior to cell
death in diverse glioma cells
--- 13Figure 3. Ophiobolin A triggers ER swelling in T98G cells
--- 14Figure 4. Apoptosis is not a major cell death mode induced by OP-A in
glioma cells
--- 15Figure 5. OP-A-induced cell death and vacuolation are not associated
with necroptotic cell death
--- 17Figure 6. Ophiobolin A induces ER stress by accumulation of
misfolded protein aggregates in glioma cells
--- 21Figure 7. CHX pretreatment effectively blocks the ER dilation and cell
death by OP-A. in T98G cells
--- 23Figure 8. OP-A-induced vacuolation and cell death were commonly
blocked by CHX in the tested other glioma cells
--- 24Figure 9. Upregulation of CHOP plays a critical role in Ophiobolin
A-induced ER dilation and cell death
--- 25v
Figure 11. Ophiobolin A-induced vacuolation and cell death is blocked
by thiol antioxidants, but not by other radical scavengers
--- 29Figure 12. Ophiobolin A does not generate ROS
--- 31Figure 13. Disruption of thiol homeostasis, but not ROS generation, is
important for OP-A-induced paraptosis-like cell death in various
glioma cells
--- 32Figure 14. Thiol antioxidants, but not other ROS scavengers, block
OP-A-induced accumulation of misfolded proteins and CHOP
--- 33Figure 15. Proposed mechanisms involved in OP-A-induced
paraptosis-like cell death
--- 35Figure 16. OP-A induces the cell death in various cancer cells
--- 38Figure 17. Ophiobolin A induces cytoplasmic vacuolation prior to cell
death in diverse cancer cells
--- 40Figure 18. OP-A-induced cell death and vacuolation have a different
effect by apoptosis inhibitor (z-VAD-fmk) depending on the cancer cell
types
--- 41Figure 19. OP-A-induced cell death and vacuolation have a different
effect by necroptosis inhibitor (Necrostatin-1) depending on the cancer
cell types
--- 43Figure 20. OP-A-induced vacuolation and cell death were commonly
blocked by CHX in the tested various cancer cells
--- 44vi
Figure 21. Ophiobolin A-induced vacuolation and cell death is blocked
by thiol antioxidants, but not by other radical scavengers in various
cancer cells
--- 45- 1 -
I. INTRODUCTION
Glioblastoma multiforme (GBM), also known as grade IV astrocytoma, is the most common primary malignant brain tumor (Furnari et al., 2007). Complete resection remains virtually impossible due to the invasive nature of GBM cells into the brain parenchyma (Furnari et al., 2007; Stupp et al., 2009). Despite the use of aggressive surgical resection, intensified radiation therapy and concomitant chemotherapy with temozolomide, GBM remains a fatal disease with an average survival time of 14.6 months following diagnosis (Stupp et al., 2009). This extremely unfavorable prognosis for GBM patients is due to tumor recurrence, arising in part from the tumor cells’ intrinsic and acquired resistance to apoptosis (Furnari et al., 2007; Stupp et al., 2009). Therefore, strategies to induce non-apoptotic cell death may offer an innovative therapeutic strategy to combat GBM, which are notoriously difficult to treat with existing medications.
Previously, a new type of non-apoptotic cell death, termed paraptosis, was reported to occur during the development of the nervous system, as well as in some cases of neurodegeneration (Sperandio, de Belle, Bredesen., 2000; Bredesen, Rao, Mehlen., 2006). Paraptotic cells in brain tissues were observed to be filled with small and large vacuoles (Pais, Danaila, Pais., 2013). Paraptosis is characterized by a process of vacuolation that begins with physical enlargement of the endoplasmic reticulum (ER) and/or mitochondria (Sperandio et al., 2000; Wang et al.,2004). Paraptosis does not involve the apoptotic characteristics of pyknosis, DNA fragmentation, or caspase activation, but it is known to require new protein synthesis (Sperandio et al.,2000). Although the molecular basis of paraptosis or paraptosis-like cell death is still clearly understood, imbalanced homeostasis of ions (e.g., Ca2+ (Yoon et al., 2012 & 2014) and K+ (Bury et al., 2013a)), generation
- 2 -
proteasomal inhibition (Yoon et al., 2010 & 2014) and disruption of sulfhydryl homeostasis (Lee et al., 2016; Kar et al., 2009; Singha et al., 2013) were proposed as its underlying mechanisms. Recently, paraptosis was reported to be induced by several natural products and synthetic chemicals with anti-cancer activities (Lee et al., 2016).
Ophiobolin A (OP-A) is a sesterterpenoid produced by pathogenic fungi of the genus Bipolaris (Au, Chick, Leung., 2000). Many studies on OP-A have been reported in plants (Sugawara et al., 1987; Tipton, Paulsen, Betts., 1977; Cocucci et al., 1983), but not in mammalian cells. Recently, it was reported that OP-A can be used as an anti-cancer agent (Bury et al., 2013a & 2013b; Bhatia et al., 2016; Rodolfo et al., 2016; Morrison et al., 2017). OP-A inhibits the tumor growth both in apoptosis-sensitive and apoptosis-resistant cancer cells and also in cancer cells displaying various MDR (Multi Drug Resistant) phenotypes (Bury et al., 2013b). Additionally, OP-A demonstrates the potential as a selective drug for cancer stem cells by inhibition of K-ras4B activity through inactivation of calmodulin (Najumudeen et al., 2016). OP-A was shown to induce different cell death mechanisms depending upon the cancer cell origin (Bury et al., 2013a & 2013b; Morrison et al., 2017). In particular, OP-A has been considered a promising candidate for treatment of GBMs (Bury et al., 2013a; Najumudeen., 2016). In an orthotopic mouse GBM model, OP-A treatment revealed an increased mice survival as well as tumor growth reduction with the ability to cross the blood-brain-barrier (BBB) (Dasari et al., 2015). In addition, OP-A was shown to decrease BKCa channel activity and trigger paraptosis-like cell death in human GBM cells (Bury et al., 2013a) although the functional significance of BKCa channel inhibition in this cell death was not corroborated. As the action mechanism OP-A, Dasari et al. (2015) proposed that the reaction of the 1,4-dicarbonyl moiety of OP-A with the primary amines of intracellular proteins to lead covalent modification is important for its toxicity in glioma cells. In addition, Chidley et al. (2016) recently
- 3 -
identified phosphatidylethanolamin (PE) as the molecular target of OP-A and proposed that formation of PE-OP-A adducts directly causes the observed cytotoxicity of OP-A through membrane destabilization. These results suggest that OP-A may have the multiple targets.
In the present study, I explored the underlying mechanism by which OP-A kills glioma cells. I found that OP-A induces ER stress and ER dilation leading to paraptosis-like cell death in glioma cells. In this process, CHOP upregulation plays a critical role. My results show that OP-A treatment may provide a therapeutic strategy against gliomas via induction of paraptosis-like cell death mediated by proteostasis impairment.
- 4 -
II. MATERIALS AND METHODS
1. Chemicals and antibodies
All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) unless indicated otherwise. Tiron was from Fluka (SchIrte, Deutschland). Ophiobolin A (OP-A) was from Adipogen (San Diego, CA, USA). cycloheximide (CHX), MitoTracker-Red (MTR), calcein-acetoxymethyl ester (calcein-AM), ethidium homodimer-1 (EthD-1), and 4',6-diamidino-2-phenylindole (DAPI), acetyl ester (CM-H2DCF-DA) was from Molecular probe (Eugene, OR, USA). Caspase
inhibitors benzyloxy-carbonyl-Val-Ala-Asp-(OMe)-fluoromethyl ketone (z-VAD-fmk) was from R&D systems (Minneapolis, MN). Recombinant human TRAIL (the nontagged 19 kDa protein, amino acids 114-281) was from KOMA Biotech (Seoul, South Korea). The antibodies used in this study were anti-PARP, (Abcam, Cambridge, MA); caspase-3 (Stressgen, Ann Arbor, MI); α-tubulin, anti-ubiquitin, anti-ATF4 (Santa Cruz Biotechnology, Santa Cruz, CA); anti-phospho-eIF2α, anti-anti-phospho-eIF2α, anti-IRE1α, anti-PERK, anti-CHOP (Cell Signaling Technology, Beverly, MA); anti-COX II (Invitrogen, Carlsbad, CA); anti-PDI (Enzo Life Sciences, Farmingdale, NY, USA); rabbit IgG HRP, mouse IgG HRP, and goat IgG HRP (Molecular probe, Eugene, OR, USA).
2. Cell culture
Human glioma cell lines T98G, U373MG, U343, U87MG, A172, U251MG and U251N were cultured in low glucose Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and antibiotics (GIBCO-BRL, Life Technologies, NY, USA) and incubated in 5% CO2 at 37°C. MDA-MB 435S, MDA-MB 468, Huh-7 and HeLa cells were cultured in low
- 5 -
glucose DMEM supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics (GIBCO-BRL, Life Technologies, NY, USA). U2OS cells were cultured in high glucose DMEM supplemented with 10% FBS and 1% antibiotics.
3. Measurement of cellular viability (Live/Dead assay)
Cells were cultured in 24-well plates and treated as indicated. The test drugs were then removed and the cells were washed twice with PBS. For measurement of cellular viability, 2 μM calcein-AM, a green fluorescent indicator of the intracellular esterase activity of cells, and 4 μM EthD-1, a red fluorescent indicator of membrane damaged (dead) cells, were added to each well, and the plates were incubated for 5 min in 5% CO2 at 37°C. Cells were then observed under a fluorescence microscope (Axiovert 200M, Carl Zeiss, Germany) equipped with Zeiss filter sets #46 and #64HE. Viable cells, corresponding to those that exclusively exhibited green fluorescence, were counted in seven fields per well at 200 × magnification. Only exclusively green cells were counted as live because bicolored (green and red) cells cannot be unambiguously assigned to live or dead groups.
4. Examination of the stable cell lines expressing the fluorescence specifically in the endoplasmic reticulum
The stable T98G sublines expressing the fluorescence specifically in the ER were established by transfection of T98G cells with the pEYFP-ER vector (Clontech, Mountain View, CA, USA) and selection with fresh medium containing 500 µg/mL G418 (Calbiochem, Darmstadt, Germany).
- 6 -
5. Western blotting
Cells were washed in PBS and lysed in boiling sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer (62.5 mM Tris [pH 6.8], 1% SDS, 10% glycerol, and 5% β-mercaptoethanol). The lysates were boiled for 5 min, separated by SDS–PAGE, and transferred to an Immobilon membrane (Millipore, Bredford, MA, USA). After blocking nonspecific binding sites for 30 min using 5% skim milk, membranes were incubated for 2 h with specific antibodies. Membranes were then washed three times with TBST and incubated further for 1 h with horseradish peroxidaseconjugated antirabbit, -mouse or -goat antibody. Visualization of protein bands was accomplished using ECL (Advansta, Menlo Park, CA, USA). The representative results from at least three independent experiments are shown.
6. Immunocytochemistry (ICC)
After treatments, cells were fixed with acetone/methanol (1:1) for 5 min at -20°C, blocked in 5% BSA in PBS for 30 min, and incubated overnight at 4°C with primary antibody [anti-COX II (1:500, mouse, Invitrogen, Carlsbad, CA), anti-PDI (1:500, rabbit, Enzo Life Sciences, Farmingdale, NY, USA), anti-CHOP (1:250, mouse, Cell Signaling Technology, Beverly, MA), anti-ubiquitin (1:1000, mouse, Santa Cruz biotechnology, Santa Cruz, CA)] diluted in antibody diluent solution (GBI Labs, Mukilteo, WA 98275, USA). And then slides were washed three times in PBS, incubated for 1 h at room temperature with anti-rabbit or anti-mouse Alexa Fluor 488 or 594 (1:1000, Molecular probe, Eugene, OR, USA). After, DAPI was stained for 5 min diluted in PBS (1:2000) and Slides were mounted with ProLong Gold antifade mounting reagent (Molecular probe, Eugene, OR, USA). Cell staining was visualized with a fluorescence microscope using (Axiovert 200M, Carl Zeiss, Germany).
- 7 -
7. Gene silencing of CHOP (Knockdown experiments using siRNAs and shRNAs)
The siRNA duplexes used in this study were purchased from Invitrogen (Carlsbad, CA, USA) and have the following sequences: CHOP (NCBI accession no. NM_004083,50-GAGCUCUGAUUGACCGAAUGGUGAA-30). Negative Universal Control (Invitrogen, Carlsbad, CA) was used as the control. After annealing of the pairs of siRNA oligonucleotide, cells in 24-well plates were transfected with 10 nM siRNA oligonucleotides using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. For the knockdown experiments using CHOP-targeting shRNA, HEK293TN cells were transfected with the plasmid containing the non-targeting shRNA (SHC002V, Sigma-Aldrich, St Louis, MO) or the plasmid containing CHOP-targeting shRNA (TRCN0000364328, Sigma-Aldrich, St Louis, MO), together with pMD2.G (the envelope plasmid) and pPsAX2.0 plasmid (the packaging plasmid), using TransIT-2020 transfection reagents (Mirus Bio LLC, Madison, WI, USA) according to the manufacturer’s instructions. After 48 h of lentiviral particle production, T98G cells were infected with the filtered lentiviral medium (derived from HEK293TN cultures) supplemented with 10 μg/ml polybrene. To confirm successful siRNA- or shRNA-mediated knockdown, western blotting of the proteins of interest was performed.
8. Measurement of reactive oxygen species (ROS) production
CM-H2DCF-DA was used to measure the intracellular generation of hydrogen
peroxide (H2O2). Briefly, 3 × 105 T98G cells were plated in 6-well plates and
allowed to attach overnight. Cells were incubated with or without OP-A and then incubated with 5 μM of H2DCF-DA for 30 min in the dark at 37°C. After washing
- 8 -
with Hank’s Buffered Salt Solution (HBSS) containing Ca2+ and Mg2+, cells were
further processed for fluorescence activated cell sorting (FACS) analysis using a FACScan flow cytometer system (BD Biosciences, San Jose, CA). Data were analyzed using WinMDI 2.8 software (BD Biosciences, San Jose, CA).
9. Statistical analysis
Statistical analysis was performed with GraphPad Prism (GraphPad Software Inc., Sandiego, CA, USA) was used. Unless otherwise specified, each experiment was repeated at least three times. Normality of data was assessed by Kolmogorov-Smirnov testes and equal variance using Bartlett's test. For normal distribution, statistical differences were determined using an analysis of variance (ANOVA) followed by Bonferroni multiple comparison test. If the data were not normally distributed, Kruskal-Wallis test was performed followed by Dunn’s test. p < 0.001 was considered statistically significant.
- 9 -
III. RESULTS
1. Ophiobolin A induces paraptosis-like cell death in glioma cells via dilation of the ER
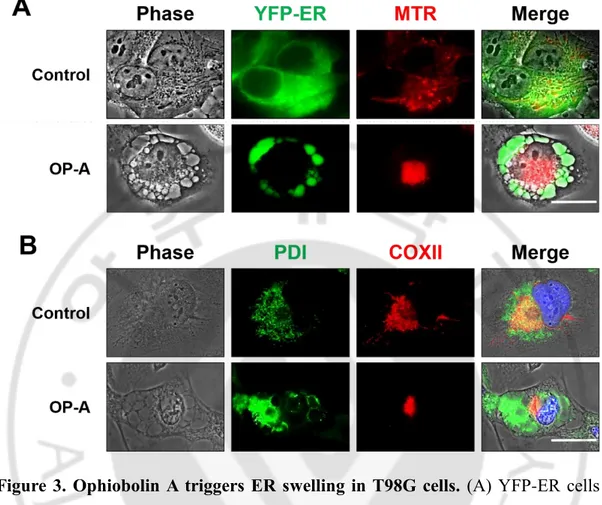
To investigate the underlying mechanism of OP-A-induced glioma cell death, I first examined the effect of various concentrations of OP-A on the viability of various glioma cells, including T98G, U373MG, U343, U251MG, U251N, A172 and U87MG cells. OP-A treatment dose-dependently reduced the viability of these cells, as assessed using calcein-AM and EthD-1 (Figure 1A and 1B). The sensitivity of glioma cells to OP-A was slightly different and A172 cells demonstrated the highest sensitivity against OP-A. Notably, OP-A-induced cell death was accompanied by a marked vacuolation (Figure 2). To identify the origin of OP-A-induced vacuolation, I examined the morphologies of the endoplasmic reticulum (ER) and mitochondria employing YFP-ER cells (T98G sublines that express the fluorescence specifically in the ER) and Mito-Tracker Red (red-fluorescent dye that specifically stains mitochondria). While the reticular patterns of ER and filamentous or elongated mitochondria were observed in untreated YFP-ER cells, the ER green fluorescence was detected within the vacuoles and aggregated mitochondria were observed beside the nuclei in YFP-ER cells treated with 2 µM OP-A for 12 h (Figure 3A). Immunocytochemistry of PDI, an ER resident protein, and COXII, a mitochondrial protein, showed that PDI was mainly expressed at the periphery of the extensively dilated vacuoles in the cytosol and COXII was expressed as aggregated forms beside the nuclei in T98G cells treated with 2 µM OP-A for 12 h (Figure 3B). Electron microscopy showed that ER cisternae were distended and mitochondria were shortened by treatment with 2 µM OP-A for 6 h
- 10 -
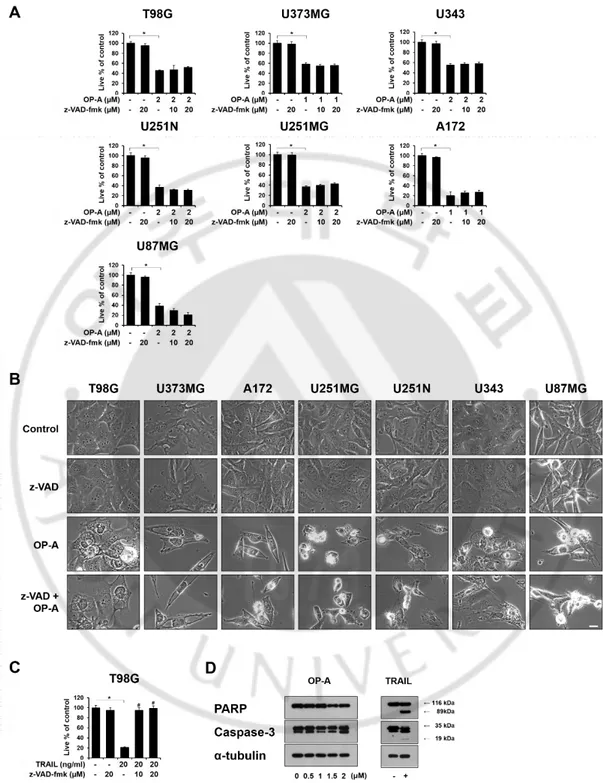
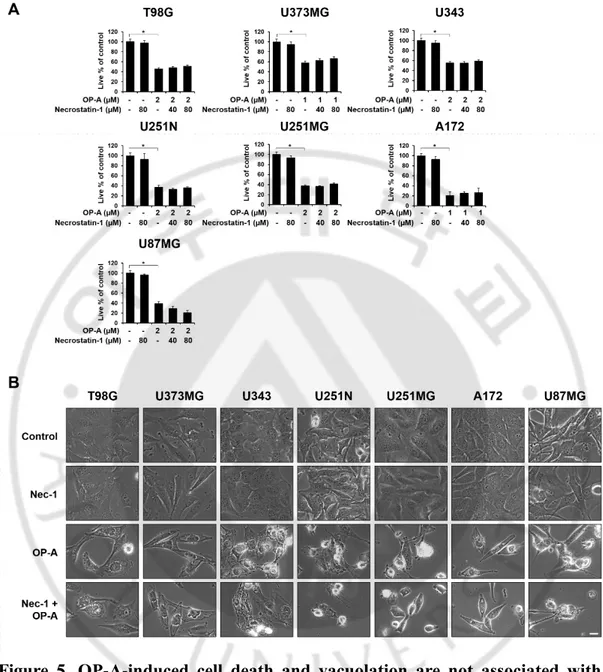
in T98G cells (data not shown). At 12 h, further expansion and fusion of swollen ER were observed and a dramatic dilation of the perinuclear space was also detected. At time points beyond 12 h, fusion of the dilated ER was further progressed until the most of the cellular space was almost fully occupied by expanded ER-derived vacuoles. These results indicate that OP-A-induced vacuolation is mainly derived from the dilation of the ER. OP-A-induced vacuolation and cell death in T98G cells were not affected by pretreatment with z-VAD-fmk, a pan-caspase inhibitor (Figure 4A and 4B). In contrast, TRAIL-induced cell death in T98G cells were almost completely blocked by z-VAD-fmk pretreatment (Figure 4C). In addition, while treatment of T98G cells with TRAIL induced the cleavage of caspase-3 and PARP, a substrate of caspase-3, OP-A treatment did not. Additionally, both csaspase-3 and PARP were cleaved by TRAIL, but not by OP-A treatment (Figure 4D). Furthermore, OP-A induced cell death as well as vacuolation was not affected by pretreatment with necrostatin-1, a specific inhibitor of necroptosis (Figure 5A and 5B). Collectively, these results suggest that OP-A kills glioma cells via induction of paraptosis-like cell death.
- 11 -
Figure 1. OP-A induces the cell death in various glioma cells. (A) Cellular
viability was assessed using calcein-AM and EthD-1 after treatment with the indicated concentration of OP-A for 24 h. The percentage of live cells was normalized to that of untreated control cells (100%). Data represent the means ± SD. One-way ANOVA and Bonferroni’s post hoc test. * p < 0.05, ** p < 0.01 vs. untreated control. (B) The values of IC50 (the concentration of drug that is required
to the viability of treated cell for 24 h to 50%) after the viability assay using Live and Dead assay were assessed.
- 13 -
Figure 2. Ophiobolin A induces cytoplasmic vacuolation prior to cell death in diverse glioma cells. Cells treated with the indicated concentrations of OP-A for
- 14 -
Figure 3. Ophiobolin A triggers ER swelling in T98G cells. (A) YFP-ER cells
treated with 2 μM OP-A for 12 h were stained with 100 nM mitotracker-red (MTR) and observed under the phase-contrast and fluorescence microscope. Bar 20 μm. (B) T98G cells were treated with 2 μM OP-A for 12 h, fixed, and subjected for immunocytochemistry of COX II and PDI. Bar 20 μm.
- 15 -
Figure 4. Apoptosis is not a major cell death mode induced by OP-A in glioma cells. (A, B) Various glioma cells pretreated with 20 µM z-VAD-fmk for 30 min
- 16 -
and further treated with the indicated concentrations of OP-A for 24 h. (A) Cellular viability was assessed using calcein-AM and EthD-1. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p < 0.05 vs. untreated control. (B) Treated cells were observed under the phase-contrast microscope. Bar 10 μm. (C) Cellular viability was assessed using calcein-AM and EthD-1. As a positive control of apoptosis, the effect of 20 ng/ml TRAIL was compared. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p < 0.05 vs. untreated control, # p <0.05 vs TRAIL treatment. (D) T98G cells were treated with the indicated concentrations of OP-A for 12 h or 20 ng/ml TRAIL for 24 h. Cell extracts were prepared from the treated cells and Western blotting of was performed using anti-caspase 3 and anti-PARP antibody. α-tubulin was used as a loading control in Western blots.
- 17 -
Figure 5. OP-A-induced cell death and vacuolation are not associated with necroptotic cell death. (A, B) Cells were pretreated with necroptosis inhibitors
(Necrostatin-1) for 30 min and further treated with the indicated concentrations of OP-A for 24 h. (A) Cellular viability was assessed using calcein-AM and EthD-1 to detect live and dead cells, respectively. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p <0.05 vs
- 18 -
untreated control. (B) Treated cells were observed under a phase-contrast microscope. Bars, 10 μm.
- 19 -
2. OP-A induces ER stress and CHOP plays a critical role in OP-A-induced paraptosis-like cell death
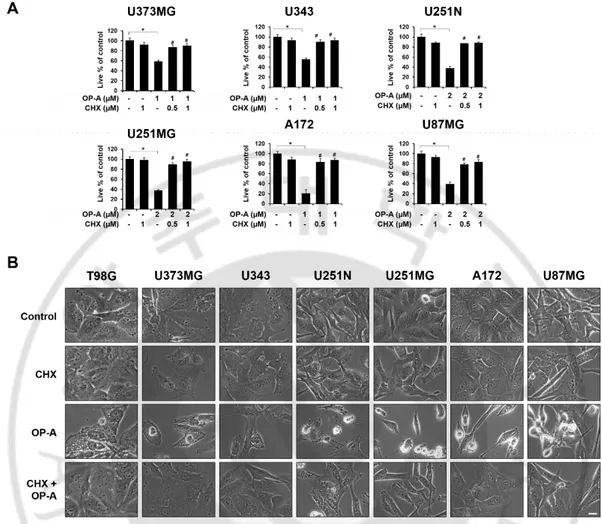
Previously, ER dilation was shown to be associated with the ER stress due to accumulation of misfolded proteins in the ER lumen (Lee et al., 2016; Mimnaugh et al., 2006). Therefore, I first examined whether OP-A affects the expression of the proteins related to ER stress. I found that OP-A treatment for 12 h dose-dependently upregulated the protein levels of Grp78, IRE1α, ATF4, and CHOP as well as the phosphorylation levels of PERK and eIF2α in T98G and U373MG cells (Figure 6A). Time-course experiment showed that 2 µM OP-A treatment progressively increased the protein levels of GRP78 and IRE1α (Figure 6A). In addition, while the phosphorylation levels of PERK and eIF2α as well as ATF4 protein levels were increased with a peak at 6 h of OP-A treatment. In contrast, CHOP protein levels were upregulated from 6 h of OP-A treatment and fairly sustained thereafter (Figure 6A). In addition, I found that 2 µM OP-A treatment dose- and time-dependently accumulated poly-ubiquitinated proteins. Immunocytochemistry also revealed that treatment with 2 µM OP-A progressively accumulated the poly-ubiquitinated protein aggregates (Figure 6B), showing the disruption of proteostasis. Paraptosis or paraptosis-like cell death is known to require de novo protein synthesis (Sperandio et al., 2000). When I tested whether blocking of the protein synthesis affects OP-A-induced cellular responses, protein synthesis blocker cycloheximide (CHX) pretreatment almost completely inhibited OP-A-induced accumulation of poly-ubiquitinated proteins and CHOP (Figure 6C) as well as ER-derived vacuolation and cell death (Figure 7A and 7B) in T98G cells. And pretreatment with Tauroursodeoxycholic acid (TUDCA), a chemical chaperone, slightly OP-A-induced ER-derived vacuolation and partially, but significantly, attenuated OP-A-induced cell death (Figure 7C and 7D). In addition,
- 20 -
OP-A-induced vacuolation and cell death were commonly and effectively blocked by CHX in the tested other glioma cell lines (Figure 8A and 8B) suggesting that CHX, which arrests de novo protein synthesis, reduces the burden on homeostatic protein-folding mechanisms and significantly delayed OP-A-induced cell death response. Taken together, these results suggest that OP-A induces ER stress and dilation via accumulation of misfolded proteins, leading to paraptosis-like cell death in glioma cells.
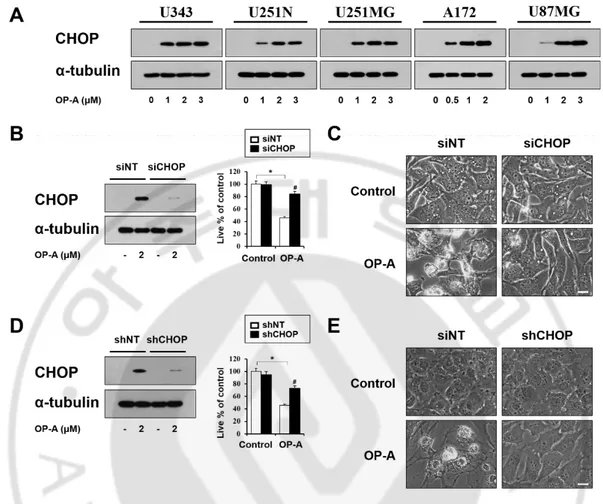
Among the proteins related to the unfolded protein response, CHOP is involved in making the cell death decision associated with ER stress (Rutkowski, Kaufman., 2007). In addition, since OP-A-mediated CHOP induction was commonly observed in not only T98G and U373 MG cells (Figure 6A) but also in the tested other glioma cells (Figure 9A), I further asked whether OP-A-induced cytotoxicity is associated with CHOP induction. I found that the gene silencing of CHOP employing siRNA effectively inhibited OP-A-induced cell death (Figure 9B) and vacuolation (Figure 9C). Similar results were obtained using CHOP shRNA (Figure 9D and 9E). In addition, inhibition of OP-A-induced ER-derived vacuolation by CHOP knockdown was also confirmed by the fluorescence microscopy using YFP-ER cells (Figure 10A) and immunocytochemistry of PDI and CHOP (Figure 10B). Collectively, these results suggest that CHOP upregulation plays a crucial role in OP-A-induced paraptosis-like cell death in glioma cells.
- 21 -
Figure 6. Ophiobolin A induces ER stress by accumulation of misfolded protein aggregates in glioma cells. (A) Cell extracts were prepared from the cells
treated with 2 µM OP-A for the indicated time points or indicated concentrations of OP-A for 12 h. Western blotting of the indicated proteins was performed. α-tubulin was used as a loading control in Western blots. (B) T98G cells treated with 2 µM OP-A for 20 h were fixed, immunostained using anti-ubiquitin antibody (red) and subjected to immunocytochemistry. (C) T98G cells were untreated or pretreated with 1 µM CHX and further treated with 2 µM OP-A for 12 h. Western blotting of
- 22 -
- 23 -
Figure 7. CHX pretreatment effectively blocks the ER dilation and cell death by OP-A in T98G cells. (A, C) YFP-ER cells pre-treated with or without 2 μM
CHX (A) or 20 μM TUDCA (C) and further treated with 2 μM OP-A for 12 h. Cells were observed under the phase-contrast and fluorescence microscopy. (B, D) T98G cells were pretreated with the indicated concentrations of CHX (B) or TUDCA (D) and further treated with 2 µM OP-A for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p < 0.05 vs. untreated control, # p <0.05 vs OP-A treatment.
- 24 -
Figure 8. OP-A-induced vacuolation and cell death were commonly blocked by CHX in the tested other glioma cells. (A, B) Cells untreated or pretreated with 2
μM CHX for 30 min and further treated with the indicated concentrations of OP-A for 24 h. (A) Cellular viability was assessed using calcein-AM and EthD-1 to detect live and dead cells, respectively. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p < 0.05 vs. untreated control, # p <0.05 vs OP-A treatment. (B) Treated cells were observed under a phase-contrast microscope. Bars, 10 μm.
- 25 -
Figure 9. Upregulation of CHOP plays a critical role in Ophiobolin A-induced ER dilation and cell death. (A) Cell extracts were prepared from the cells treated
with the indicated concentrations of OP-A for 12 h. Western blotting of the indicated proteins was performed. α-tubulin was used as a loading control in Western blots. (B, C) T98G cells transfected with the non-targeting siRNA (siNT) or CHOP siRNA were further treated with 2 μM OP-A for 24 h. (B) Cellular viability was assessed using calcein-AM and EthD-1. Knockdown of CHOP was confirmed by Western blotting. α-tubulin expression was analyzed to confirm equal loading of the protein samples. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p < 0.05 vs. siNT untreated control, # p <0.05 vs siNT OP-A treatment. (C) Treated cells were observed under the phase-contrast microscope. Bars, 10 μm. (D, E) T98G cells were incubated with
- 26 -
the lentivirus containing non-targeting (NT) shRNA or a CHOP-targeting shRNA (CHOP shRNA) and further treated with 2 μM OP-A for 24 h. (D) Cellular viability was assessed using calcein-AM and EthD-1. shRNA-mediated knockdown of CHOP was confirmed by Western blotting. To confirm successful siRNA- or shRNA-mediated knockdown, Western blotting of the proteins of interest was performed. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p < 0.05 vs. siNT untreated control, # p <0.05 vs siNT OP-A treatment. (E) Treated cells were observed under the phase-contrast microscope. Bars, 10 μm.
- 27 -
Figure 10. CHOP is critically involved in OP-A-induced ER dilation. (A)
YFP-ER cells were transfected with siNT or CHOP siRNA and further treated with 2 μM OP-A for 8 h. Phase-contrast and fluorescence microscopy was performed. Bar 20 μm. (B) T98G cells transfected with siNT or CHOP siRNA were further treated with 2 μM OP-A for 8 h. Cells were subjected to immunocytochemistry of PDI and CHOP. Representative pictures of cells are shown. Bar 20 μm.
- 28 -
3. Thiol-antioxidants but not other ROS scavengers block OP-A-induced paraptosis-like cell death
Previously, our lab reported that generation of reactive oxygen species (ROS) play a critical role in the paraptosis induced by curcumin (Yoon et al., 2014). In an attempt to decipher the underlying mechanism of OP-A-induced paraptosis-like cell death, I tested the possible involvement of ROS in this cell death. Interestingly, experiment using various antioxidants showed that OP-A-induced cell death as well as ER-derived vacuolation were commonly and effectively blocked by pretreatment with various thiol-based antioxidants, including N-α–acetyl-L-cysteine (NAC), GSH, and N-mercapto-propionyl-glycine (NMPG), but not by pretreatment with other non-thiol ROS scavengers, including ascorbic acid (Vitamin C), MnTBAP (Mn(III)tetrakis (4-benzoic acid) porphyrin) (superoxide dismutase (SOD) mimetic), and Tiron (a radical scavenger) (Figure 11A – 11C). Measurement of ROS levels using DCF-DA by flow cytometry and fluorescence microscopy revealed that OP-A did not increase ROS levels, in contrast to H2O2
(Figure 12A and 12B). In addition, OP-A-induced vacuolation and cell death were effectively blocked by NAC, but not by Tiron, in tested other glioma cells (Figure 13A and 13B), indicating that OP-A-induced cell death is ROS-independent. When I examined the effects of thiol antioxidants on induced ER stress, OP-A-induced accumulation of poly-ubiquitinated proteins and CHOP was also effectively inhibited by thiol antioxidants, but not by non-thiol antioxidant (Figure 14), suggesting thiol antioxidant blocks all the OP-A-induced cellular responses.
- 29 -
Figure 11. Ophiobolin A-induced vacuolation and cell death is blocked by thiol antioxidants, but not by other radical scavengers. (A) T98G cells were
pretreated with the indicated concentrations of various antioxidants and further treated with 2 µM OP-A for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p < 0.05 vs. untreated control, # p <0.05 vs OP-A treatment. (B, C) T98G (B) or YFP-ER (C) cells were pretreated with 1 mM NAC, 1 mM GSH, 0.2 mM NMPG, 0.25 mM ascorbic acid, 200 µM MnTBAP, or 1 mM Tiron and further treated with 2 µM OP-A for 24 h (B) or 8 h (C). Cells
- 30 -
were observed under the phase-contrast microscope (B) or fluorescence microscopy (C). Bar 10 μm (B) or 20 μm (C).
- 31 -
Figure 12. Ophiobolin A does not generate ROS. (A) T98G cells were treated
with 1.5 µM OP-A for the indicated time points and then incubated with DCF-DA. Cells were subjected to flow cytometry. As a positive control to generate ROS, T98G cells treated with 10 mM H2O2 for 30 min were processed for the flow
cytometry.
(B) T98G cells treated with 10 mM H2O2 for 10 h or 1.5 µM OP-A for 10 h were
- 32 -
Figure 13. Disruption of thiol homeostasis, but not ROS generation, is important for OP-A-induced paraptosis-like cell death in various glioma cells.
(A) Diverse glioma cells were pretreated with the indicated concentrations of various antioxidants and further treated with the indicated concentrations of OP-A for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p < 0.05 vs. untreated control, # p <0.05 vs OP-A treatment. (B) Glioma Cells were untreated or pretreated with with 1 mM NAC or 1 mM Tiron and further treated with 1 μM or 2 μM OP-A for 24 h. Cells were observed under the phase-contrast microscope. Bar 10 μm.
- 33 -
Figure 14. Thiol antioxidants, but not other ROS scavengers, block OP-A-induced accumulation of misfolded proteins and CHOP. T98G cells were
pretreated with 1 mM NAC, 1 mM GSH, 0.2 mM NMPG, 0.25 mM ascorbic acid, 200 M MnTBAP, or 1 mM Tiron and further treated with 2 M OP-A for 12 h. Cell extracts were prepared for Western blotting of CHOP, ubiquitin, and -tubulin.
- 35 -
Figure 15. Proposed mechanisms involved in OP-A-induced paraptosis-like cell death. Michael adduction formation between OP-A and cysteine residues on
the proteins could disrupt the formation of correct disulfide bond in the protein folding. The resultant alterations in their structure and function may cause accumulation of misfolded proteins to induce ER stress and CHOP expression, ultimately leading to paraptosis-like cell death.
- 36 -
4. Disruption of thiol homeostasis by OP-A critically contributes to OP-A-mediated anti-cancer effects in various cancer cell lines
Next, I examined whether inhibition of OP-A-induced cell death by thiol antioxidants occurs also in various cancer cell lines, but not is restricted in glioma cells. Recently, Morrison et al. (2017) have shown that OP-A induces different mechanisms of cell death in mammalian cells depending on the cancer cell origin. When I examined the effects of OP-A on various cancer cells, including MDA-MB 468 and MDA-MB 435S breast cancer cells, Huh-7 and SNU-449 hepatocellular carcinoma cells (HCC), and U20S osteosarcoma cells, and HeLa cervical cancer cells, OP-A dose-dependently reduced their viabilities with a slight difference in its cytotoxicity (Figure 16A and 16B). When I examined the effect of different doses of OP-A on cellular morphologies, treatment with 3 µM OP-A for 24 h induced cellular vacuolation and cell death in HeLa, U2OS, Huh-7, and SNU-449 cells (Figure 17). Treatment with 2 µM OP-A induced a marked blebbing and swelling in MDA-MB468 and MDA-MB 435S cells together with cellular vacuolation. In an attempt to understand OP-A-mediated cell death mechanism, I tested the effects of z-VAD-fmk (an inhibitor of apoptosis), necrostatin-1 (an inhibitor of necroptosis), and CHX (an inhibitor of paraptosis). I found that OP-A-induced cell death was not affected by z-VAD-fmk in MDA-MB 435S, Huh-7, SNU-449, and HeLa cells. However, it was very effectively blocked by z-VAD-fmk in MDA-MB 468 cells and slightly but significantly inhibited in U2OS cells (Figure 18A and 18B). OP-A-induced cell death was not altered by necrostatin-1 in MDA-MB 468, Huh-7, and HeLa cells (Figure 19A and 19B). In contrast, it was effectively blocked in MDA-MB 435S and SNU-449 cells and partially blocked in U2OS cells. OP-A-induced cell death was effectively blocked by CHX in all the tested cancer cells (Figure 20A and 20B). These results suggest that paraptosis may be a major contributor to OP-A-mediated toxicity in HeLa and Huh-7 cells, whereas both
- 37 -
paraptosis and necroptosis contribute to it in SNU-449 and MDA-MB 435S cells. In addition, both paraptosis and apoptosis are involved in MDA-MB 468 treated with OP-A, mixed cell death including paraptosis, necroptosis, and apoptosis occurs in OP-A-treated U2OS cells. Collectively, these results suggest that depending on the cancer cell types, possibly depending on the genetic backgrounds, OP-A may induce different cell death modes but paraptosis may be an important death mechanism to explain OP-A-mediated toxicity in various cancer cells.
Finally, I investigated whether the dependency of OP-A-mediated cytotoxicity on cellular thiol modulation in these cancer cells. Very interestingly, NAC pretreatment, but not Tiron pretreatment, potently blocked OP-A-induced cell death commonly in all the tested cancer cells (Figure 21A and 21B). These results suggest that disruption of thiol homeostasis may be the key underlying mechanisms involved in the anti-tumor effect of OP-A in various cancer cells, irrespective the ultimate cell death mode(s) induced by OP-A.
- 38 -
Figure 16. OP-A induces the cell death in various cancer cells. (A) Various
cancer cells were treated with OP-A at the indicated concentrations for 24 h. Cellular viability was assessed using calcein-AM and EthD-1 to detect live and dead cells, respectively. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p <0.05, ** p<0.01 vs untreated control. (B) The values of IC50 (the concentration of drug that is required to the
viability of treated cell for 24 h to 50%) after the viability assay using Live and Dead assay were assessed.
- 40 -
Figure 17. Ophiobolin A induces cytoplasmic vacuolation prior to cell death in diverse cancer cells. Cells treated with the indicated concentrations of OP-A for 24
- 41 -
Figure 18. OP-A-induced cell death and vacuolation have a different effect by apoptosis inhibitor (z-VAD-fmk) depending on the cancer cell types. (A, B)
Cells were pretreated with apoptosis inhibitors (z-VAD-fmk) for 30 min and further treated with the indicated concentrations of OP-A for 24 h. (A) Cellular viability was assessed using calcein-AM and EthD-1 to detect live and dead cells, respectively. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p <0.05 vs untreated control, # p <0.05
- 42 -
vs OP-A treatment. (B) Treated cells were observed under a phase-contrast microscope. Bars, 10 μm.
- 43 -
Figure 19. OP-A-induced cell death and vacuolation have a different effect by necroptosis inhibitor (Necrostatin-1) depending on the cancer cell types. (A, B)
Cells were pretreated with necroptosis inhibitors (Necrostatin-1) for 30 min and further treated with the indicated concentrations of OP-A for 24 h. (A) Cellular viability was assessed using calcein-AM and EthD-1 to detect live and dead cells, respectively. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p <0.05 vs untreated control, # p <0.05 vs OP-A treatment. (B) Treated cells were observed under a phase-contrast microscope. Bars, 10 μm.
- 44 -
Figure 20. OP-A-induced vacuolation and cell death were commonly blocked by CHX in the tested various cancer cells. (A, B) Cells untreated or pretreated
with paraptosis inhibitor (CHX) for 30 min and further treated with the indicated concentrations of OP-A for 24 h. (A) Cellular viability was assessed using calcein-AM and EthD-1 to detect live and dead cells, respectively. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p < 0.05 vs. untreated control, # p <0.05 vs OP-A treatment. (B) Treated cells were observed under a phase-contrast microscope. Bars, 10 μm.
- 45 -
Figure 21. Ophiobolin A-induced vacuolation and cell death is blocked by thiol antioxidants, but not by other radical scavengers in various cancer cells. (A)
- 46 -
Diverse cancer cells were pretreated with the indicated concentrations of NAC, Tiron and further treated with the indicated concentrations of OP-A for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. Statistical significance was determined using one-way ANOVA followed by Bonferroni’s post hoc tests. * p < 0.05 vs. untreated control, # p <0.05 vs OP-A treatment. (B) Cells were untreated or pretreated with with 1 mM NAC or 1 mM Tiron and further treated with 1 μM or 2 μM OP-A for 24 h. Cells were observed under the phase-contrast microscope. Bar 10 μm.
- 47 -
IV. DISCUSSION
GBM, the most common adult primary brain cancer, remains the deadliest of all forms of brain tumors despite the many clinical trials that have attempted to improve the dismal outcomes. GBM is characterized by resistance to apoptosis, which is largely responsible for the low effectiveness of the classical chemotherapeutic approaches based on apoptosis induction in cancer cells (Krakastad, Chekenya., 2010). For treating apoptosis-resistant tumors, the induction of non-apoptotic cell death could be an option.
Recently, natural products including curcumin (Yoon et al., 2010 & 2012), celastrol (Yoon et al., 2014), and paclitaxel (Rutkowski, Kaufman., 2007) reportedly induce paraptosis or paraptosis-like cell death in resistant malignant cancer cells, although the molecular basis of this cell death mode is not fully understood. Therefore, identification of the agents to induce paraptosis or paraptosis-like cell death and understating of their underlying mechanisms may provide an alternative therapeutic strategy for overcoming innate and acquired resistance to the current proapoptotic anticancer therapies. OP-A, a natural compound made by a fungus in order to attach plant cells, was found to have significant activity against apoptosis-resistant GBM cells through induction of paraptosis-like cell death (Bury et al., 2013a), but its underlying mechanism is largely unknown. In this study, I confirmed that OP-A induces paraptosis-like cell death in glioma cells based on the following morphological and biochemical characteristics: (a) OP-A induces progressive vacuolation prior to glioma cell death mainly via swelling and fusion of the ER without apoptotic features, such as formation of apoptotic bodies and the dependency on caspases (Figure 1-5). (b) OP-A-induced ER-derived vacuolation and cell death are effectively inhibited by CHX (Figure 7 and 8). (c) OP-A-induced ER-derived dilation and cell death is closely linked to ER stress (Figure 6 and 7).
- 48 -
In the kingdoms of plant, fungi and Protista vacuoles are common intracellular organelles to maintain their life (Marty., 1999; Kilonsky, Herman, Emr., 1990; Plattner., 2015). But it is not normal to have vacuoles in most animal cells. Formation of large vacuoles (cytoplasmic vacuolization or vacuolation) in mammalian cells spontaneously or after exposure to bacterial or viral pathogens or various natural and artificial low-molecular-light compounds (Henics, Wheatley., 1999; Aki, Nara, Uemura., 2012; Chen et al., 2013). Irreversible vacuolization in mammalian cells indicates cytopathological or cytotoxic conditions leading to cell death. Moreover, irreversible vacuolation is induced by various natural and synthetic compounds of different chemical structure (Rogers-Cotrone et al., 2010; Solano et al., 2013; Grandin et al., 2012; Korsnes et al., 2011; Zhang et al., 2010). Sometimes irreversible vacuolization by several inducers causes known types of caspase-independent cell death including paraptosis or paraptosis-like cell death, necroptosis (Sperandio et al., 2004; Christofferson, Yuan., 2010). In addition, irreversible vacuolation can affect the ER and mitochondria and Golgi apparatus. The most striking feature of paraptosis is a vacuolization derived from the ER and/or mitochondria. When I performed ICC to observe mitochondrial changes following OP-A treatment, mitochondrial aggregation rather than its dilation was detected (Figure 3). EM (electron microscopy) analysis showed the mitochondrial fragmentation but not megamitochondria formation due to fusion among swollen mitochondria, which was shown in curcumin- or celastrol-induced paraptosis (Yoon et al., 2012 & 2014). Therefore, OP-A-induced vacuolation may be derived from the dilation of the ER, but not mitochondria.
ER is composed of a series of continuous membranous structures including the rough ER, smooth ER in eukaryotic cells (Park, Blackstone., 2010). Each domain has different functions. For example, the rough ER comprise sheets and associated with polyribosomes for protein synthesis. Smooth ER is composed of tubules and associated with lipid synthesis and delivery, establishing contact with other
- 49 -
organelles (Park, Blackstone., 2010). When I observed OP-A-induced cellular morphological changes by EM, ER expansion due to the fusion of swollen ER was progressed following OP-A treatment. Although free ribosomes were not discernably detected in our electron microscopic observation, I think the rough ER, rather than the smooth ER, has expanded possibly because of OP-A-induced disruption of proteostatic balance. Similar to our results, Kar et al. (2009) showed that 15d-PGJ2 induces cancer cell death accompanied by ER-derived vacuolation. They argued that the origins of 15d-PGJ2-induced vacuoles might be rough ER from their electron microscopy. To further clarify the origins of ER-derived vacuoles, immunocytochemistry of Climp63, an ER sheet marker protein, and Reep-5, an ER tubule marker protein were performed. Our lab observed that Climp63 is present in Eeyarestain 1 (another paraptosis inducer)-induced ER vacuoles.
As a cause of ER-derived vacuolation, Mimnaugh et al. (2006) proposed the accumulation of misfolded and ubiquitinated proteins within the ER lumen. They suggest that buildup of misfolded proteins within the ER lumen could induce an osmotic force, leading to an influx of water from the cytoplasm to the ER lumenal space.
The ER is the subcellular organelle in which secretory and membrane proteins are folded, stabilized by disulfide bonds, post-translationally modified, oligomerized, and ultimately exported. This process is tightly monitored by ER quality control mechanisms that sense any disruption and retain unfolded protein in the ER, triggering ER stress.The unfolded protein response (UPR) is triggered by accumulating misfolded proteins in the ER lumen and attempts to restore homeostasis. This is a general notion that ER stress-induced cell death is triggered by terminal UPR when early UPR fails to restore ER functions (Sano, Reed., 2013). I found that OP-A treatment progressively accumulated the aggregates of poly-ubiquitinated proteins and the protein levels of GRP78 and IRE1αin T98G and
- 50 -
U373MG cells. The phosphorylation levels of PERK and eIF2αas well as ATF4 expression increased with a peak at 6 h and then decreased by OP-A treatment (Figure 6). In contrast, CHOP protein levels were also upregulated from 6 h of 2 M OP-A treatment and but fairly sustained afterwards, suggesting that ER stress may contribute to OP-A cytotoxicity in these glioma cells. I also found that CHOP knockdown effectively attenuated OP-A-induced ER-derived vacuolation and subsequent cell death, suggesting a crucial role of CHOP in OP-A-induced paraptosis-like cell death glioma cells (Figure 9 and 10). Han et al. (2013) recently reported that ATF4- and CHOP-mediated transcriptional regulation increases proteins synthesis, critically contributing to ER-stress-induced cell death. Therefore, further work is warranted to determine whether the induction of CHOP observed in the present work contributes to ER dilation through the transcriptional control of specific protein(s) or a global increase in protein synthesis.
Previously, our lab reported that ROS play a key role in paraptosis induced by curcumin, DMC (dimethylated curcumin), celastrol. Therefore, I investigated whether ROS are also importantly involved in OP-A induced cell death. However, measurement of ROS using DCF-DA revealed that ROS levels were not increased by Ophiobolin A (Figure 12). In addition, when I tested the effects of various antioxidants, various thiol antioxidants, including NAC, GSH, and NMPG, but not other ROS scavengers, including MnTBAP, ascorbic acid and tiron, very effectively blocked Ophiobolin A-induced vacuolation and cell death (Figure 11). In addition, I found that ER stress markers, such as accumulation of poly-ubiquitinated proteins as well as upregulation of CHOP, induced by Ophiobolin A was effectively inhibited by thiol antioxidants, but not by other ROS scavengers (Figure 14). Taken together, these results indicate that Ophiobolin A induces paraptosis-associated cell death via disruption of sulfhydryl homeostasis rather than ROS generation. Similar to OP-A, both 15d-PGJ2 (Kar et al., 2009) and manumycin A (Singha et al., 2013) were shown to induce cancer cell death
- 51 -
accompanied by ER-derived vacuolation. Like our results, the cytoplasmic vacuolation and cell death induced by 15d-PGJ2 or manumycin A were effectively blocked by thiol antioxidants, but not by other ROS scavengers (Kar et al., 2009). Since these compounds also commonly harbor sulfhydryl (-SH)-reactive, , β-unsaturated carbonyl group(s), their covalent modification of sulfhydryl groups and a resultant protein misfolding were proposed to be the cause of ER-derived vacuolation (Kar et al., 2009; Oliva et al., 2003). Xinhe et al. (2006) suggested that disruption of disulfide bonding by thiol-Michael adduct formation lead to protein misfolding and ER stress. As the action mechanism OP-A in animal cells, Dasari et al. (2015) proposed that the reaction of the 1,4-dicarbonyl moiety of OP-A with the primary amines of intracellular proteins to lead covalent modification is important for its toxicity. Chidley et al. (2016) recently identified phosphatidylethanolamin (PE) as the molecular target of OP-A and proposed that formation of PE-OP-A adducts directly causes the observed cytotoxicity of OP-A through membrane destabilization. Based on these reports, I guessed that OP-A reacts with cysteinyl thiols to form Michael adduct. And I suggest that the ability of OP-A to covalently modify free sulfhydryl groups on proteins may cause protein misfolding and their accumulation, leading to paraptosis-like cell death. Structurally, the highly electrophilic character of the , β-unsaturated ketone substructure of OP-A allows it to react with the thiol groups of NAC or GSH to form a covalent Michael adduct, such as NAC-OP-A and GSH-OP-A conjugate, respectively (data not shown). I further investigated the effect of the interaction between OP-A and NAC on OP-A-mediated cytotoxicity. I preincubated the mixture of 2 M OP-A and different doses of NAC for different time points at room temperature in the tube to allow the chemical adduct formation before its treatment to T98G cells for 24 h. I found that increased preincubation time of the mixture of OP-A and NAC diminished the cytotoxic effect of OP-A and lower doses of NAC were required to alleviate the cytotoxicity of OP-A at the prolonged preincubation (data not shown). Furthermore,
- 52 -
OP-A-induced cell death is blocked by various thiol antioxidants, but not by other ROS scavengers, not only in many GBM cells but also in many cancer cells of different tissue origins (Figure 11, 13 and 21). These results strongly suggest that NAC blocked OP-A cytotoxicity by eliminating its ability to form Michael adducts, particularly with their nucleophilic thiol groups. Disulfide bonds are formed in the ER through a series of exchange reactions between cysteinyl proteins and ER thiol-disulfide oxidoreductases (Oka, Bulleid., 2013; Vlashi et al., 2010). Disulfide bonds are also formed during the folding process of secretory proteins. The potential of OP-A to covalently modify the free sulfhydryl groups of proteins leading to protein adduct formation could disrupt the formation of correct disulfide bond and cause protein misfolding. Therefore, this disruption of sulfhydryl homeostasis by OP-A may lead to the accumulation of misfolded and subsequently ubiquitinated proteins. This could then trigger cytoplasmic vacuolation derived from ER dilation and subsequent cell death.
Paraptosis requires transcription and de novo protein synthesis (Sperandio et al., 2000 &2004). In this study, OP-A induced vacuolation and cell death were completely inhibited by the protein synthesis blocker, CHX (Figure 7A and 7B). Probably, since continued protein synthesis may aggravate accumulation of misfolded proteins by adduct formation with OP-A, CHX pretreatment might protect cancer cells from OP-A-induced ER-derived vacuolation and death by reducing the overall protein load within the ER. In addition, newly synthesized proteins may be more sensitive than mature proteins for covalent modification by OP-A.
Very recently, Morrison et al. (2017) investigated the effects of OP-A on various cancer cell lines with different tissue origins and they showed that OP-A induces apoptosis or necrosis depending on the cell line analyzed. I also analyzed the death modes induced by OP-A in 6 different cancer cell lines using z-VAD-fmk (an apoptosis inhibitor), necrostatin-1 (a necroptosis inhibitor), and cycloheximide
- 53 -
(a paraptosis inhibitor). I found that OP-A-induced vacuolation and cell death in HeLa, Huh-7 cells were effectively protected by CHX, but not by either z-VAD-fmk or necrostatin-1, suggesting that paraptosis or paraptosis-like cell death may be a major cell death in these cancer cells. In contrast, OP-A-induced vacuolation and cell death in MDA-MB 435S, SNU-449, and U2OS cells were effectively blocked by necrostatin-1, compared to z-VAD-fmk or cycloheximide, suggesting that OP-A demonstrates cytotoxicity via induction of necroptosis as a main cell death mode. OP-A-induced cell death in MDA-MB 468 and U2OS cells significantly attenuated by z-VAD-fmk, but not by necrostatin-1 or cycloheximide. In addition, notable vacuolation was not induced by OP-A in MDA-MB 468 cells, suggesting that apoptosis may be a major cell death mode in these cancer cells (Figure 18-20). Collectively, these results suggest that depending on the cell types and the used OP-A concentrations, the contribution of the respective cell death mode may be different. The final cell death mode induced by OP-A may be determined by the genotypes of cancer cells. However, OP-A progressively induced cellular vacuolation in these cells, albeit not in every cell, suggesting that paraptosis-like cell death critically contribute to OP-A-induced cytotoxicity in these cancer cells.
It is interesting to speculate how OP-A induces paraptosis-like cell death commonly in glioma cells as a main cell death mode. OP-A induced glioma cell death was not affected by z-VAD-fmk (an apoptosis inhibitor) or necrostatin-1 (a necroptosis inhibitor), differently from their effects on other types of cancer cells. These results suggest that glioma cells may be more vulnerable to induction of OP-A-induced paraptosis-like cell death, whereas OP-A induces mixed types of cell death modes in other cancer cells. In addition, lower OP-A concentrations were required to effectively kill glioma cells in the present study, compared to the effects of OP-A on other cancer cell types in the results by Morrison et al. (2017). These results suggest that glioma cells may be more sensitive to the anti-cancer effects of OP-A than cancer cells originated from other tissues.
- 54 -
Notably, OP-A-induced cell death in all the tested cells was completely blocked by NAC (Figure 11 and Figure 21). Therefore, disruption of thiol homeostasis due to the activity of OP-A to react with thiol-containing proteins and a resultant induction of ER stress may be a common mechanism to explain the anti-cancer effect of OP-A, which is not restricted to GBM cells. It is also intriguing to speculate whether OP-A specifically or non-specifically forms adduct with intracellular thiol-containing proteins. Recent studies have reported dysregulated potassium channel expression in human cancer (Huang X, Jan LY., 2014). For example, overexpression of the voltage-gated potassium channel Kv1.1 marks a subgroup of medulloblastoma (Taylor et al., 2012); elevated Kv1.3 expression is detected in a number of human malignancies including breast, colon, and prostate cancer (Comes et al., 2013); high Kv11.1 (HERG) expression marks both solid and blood cancer (Pillozzi et al., 2002; Jehle et al., 2011); and Overexpression of a specific splice isoform of the BK channel correlates with the malignancy grade of glioma (Liu et al., 2002). Huang X, Jan LY. (2014) summarized the overexpression of the specific types of potassium channel in cancer. They showed that many potassium channel genes are overexpressed in glioma cells. Especially, the KCNA5, channel gene expression is inversely correlated with tumor malignancy and clinical aggressiveness in glioma (Preussat et al., 2003) and lymphoma (Bielanska et al., 2009). Moreover, Previously, Bury et al. (2013a) proposed that the decrease in BK channel activity may be involved in Ophiobolin A-induced paraptosis-like cell death in glioblastoma cells, although its functional significance was not examined yet. Therefore, we cannot exclude the possibility that the cysteinyl residues of BKC channels, which are overexpressed in glioma cells, can be much more easily targeted by OP-A, contributing to effective induction of paraptosis-like cell death.
The present study clearly showed that CHOP plays a critical role in the context of ER dilation. However, it is not clear how the activity of CHOP was not affected by OP-A without the adduct formation. CHOP is a prominent resident of
- 55 -
the ER and intracellular thiols such as cysteine, homocysteine, and glutathione play critical roles in the regulation of ER protein synthesis and folding (Kumar et al., 2006). Interestingly, CHOP has no cysteine in its protein structure, suggesting that CHOP can be still active, because it is not targeted by OP-A to form adduct leading to its inactivation. Vinay et al. (2012) reported that free L-cysteine, but not D-cysteine and homoD-cysteine and glutathione, changes the global conformation in response to the cellular redox state and/or ER stress. Therefore, it remains to be clarified whether sequestration of cysteinyl thiols by OP-A to form adduct may affect the activity of CHOP, which is relatively upregulated at later phase of ER stress, by modulating its conformation.
In summary, the activity of OP-A to covalently modify free sulfhydryl groups on proteins may cause accumulation of misfolded proteins to induce ER stress and ER dilation, ultimately leading tor paraptosis-like cell death (Figure 15). Therefore, the ability of OP-A to disrupt sulfhydryl proteostasis may provide an effective therapeutic strategy against human glioblastoma, a deadliest malignancy which are resistant to various anti-cancer treatments.
- 56 -
V. REFERENCES
1. Andrey V. Shubin, Ilya V. Demidyuk, Alexey A. Komissarov, Lola M. Rafieva, Sergey V. Kostrov: Cytoplasmic vacuolization in cell death and survival. Oncotarget 7(34): 55863– 55889, 2016
2. Aki T, Nara A, Uemura K: Cytoplasmic vacuolization during exposure to drugs and other substances. Cell Biol Toxicol 28: 125–131, 2012
3. Au TK, Chick WS, Leung PC: The biology of ophiobolins. Life Sci 67: 733–742, 2000 4. Bhatia DR, Dhar P, Mutalik V, Deshmukh SK, Verekar SA, Desai DC, Kshirsagar R, Thiagarajan P, Agarwal V: Anticancer activity of ophiobolin A, isolated from the endophytic fungus Bipolaris setariae. Nat Prod Res 39(12): 1455-1458, 2016
5. Bielanska J, Hernández-Losa J, Pérez-Verdaguer M, Moline T, Somoza R, Ramón Y Cajal S, Condom E, Ferreres JC, Felipe A: Voltage-dependent potassium channels Kv1.3 and Kv1.5 in human cancer. Curr Cancer Drug Targets 9(8): 904-914, 2009
6. Bredesen DE, Rao RV, Mehlen P: Cell death in the nervous system. Nature 443: 796– 802, 2006
7. Bury M, Girault A, Mégalizzi V, Spiegl-Kreinecker S, Mathieu V, Berger W, Evidente A, Kornienko A, Gailly P, Vandier C, Kiss R: Ophiobolin A induces paraptosis-like cell death in human glioblastoma cells by decreasing BKCa channel activity. Cell Death Dis 4: e561, 2013a
8. Bury M, Novo-Uzal E, Andolfi A, Cimini S, Wauthoz N, Heffeter P, Lallemand B, Avolio F, Delporte C, Cimmino A, Dubois J, Van AntIrpen P, Zonno MC, Vurro M, Poumay Y, Berger W, Evidente A, De Gara L, Kiss R, Locato V: Ophiobolin A, a sesterterpenoid fungal phytotoxin, displays higher in vitro growth-inhibitor effects in mammalian than in plant cells and displays in vivo antitumor activity. Int J Oncol 43(2): 575-585, 2013b
9.Chen R, Duan CY, Chen SK, Zhang CY, He T, Li H, Liu YP, Dai RY: The suppressive role of p38 MAPK in cellular vacuole formation. J Cell Biochem 114: 1789–1799, 2013 10. Chidley C, Trauger SA, Birsoy K, O'Shea EK: The anticancer natural product ophiobolin A induces cytotoxicity by covalent modification of phosphatidylethanolamine.