의학
의학
의학
의학 박사학위
박사학위
박사학위 논문
박사학위
논문
논문
논문
The Efficacy of DNA Vaccine Using a
Sindbis Virus-based Vector Against
Nucleocapsid Protein of Hantaan virus
아
아
아
아 주
주
주 대
주
대
대 학
대
학
학
학 교
교 대
교
교
대
대
대 학
학
학
학 원
원
원
원
의
의
의
의 학
학
학
학 과
과
과
과
우
우
우
우 규
규
규
규 진
진
진
진
The Efficacy of DNA Vaccine Using a
Sindbis Virus-based Vector Against
Nucleocapsid Protein of Hantaan virus
by
Gyu Jin Woo
A Dissertation Submitted to The Graduate School of Ajou
University
in Partial Fulfillment of the Requirements for the Degree of
DOCTOR OF PHILOSOPHY
Supervised by
Wan Kee Kim, Ph.D.
Department of Medical Sciences
The Graduate School, Ajou University
우규진의
우규진의
우규진의
우규진의 의학
의학
의학
의학 박사학위
박사학위
박사학위
박사학위 논문을
논문을
논문을 인준함
논문을
인준함
인준함
인준함
.
심사위원장
심사위원장
심사위원장
심사위원장 신
신
신 호
신
호
호
호 준
준
준 인
준
인
인
인
심사위원
심사위원
심사위원
심사위원 김
김
김
김 완
완
완 기
완
기
기
기 인
인
인
인
심사위원
심사위원
심사위원
심사위원 박
박
박
박 만
만
만 훈
만
훈
훈
훈 인
인
인
인
심사위원
심사위원
심사위원
심사위원 박
박
박
박 선
선
선
선 인
인
인
인
심사위원
심사위원
심사위원
심사위원 장
장
장
장 영
영
영 주
영
주
주
주 인
인
인
인
아
아
아
아 주
주
주 대
주
대
대 학
대
학
학
학 교
교 대
교
교
대
대
대 학
학
학
학 원
원
원
원
2005
년
년
년
년 12 월
월
월 22 일
월
일
일
일
i - ABSTRACT -
The Efficacy of DNA Vaccine Using a Sindbis Virus-based Vector
Against Nucleocapsid Protein of Hantaan Virus
Hantaan virus (HTNV) is a causative agent of hemorrhagic fever with renal
syndrome (HFRS). More than 100,000 cases of HFRS are reported yearly, with a
mortality rate of between 2% and 10%. But, there is no effective and safe vaccine
against HFRS. Even though neutralizing antibodies against the HTNV have been proven
to be critical against viral infections, the cellular immune responses to HTNV are also
assumed to be important for viral clearance. This study has examined the cellular and
humoral immune responses against the HTNV nucleocapsid protein (NP) elicited by
virus infection or DNA vaccination to investigate the immunogenicity of NP.
To examine the cellular immune response against HTNV NP, C57BL/6 mice were
injected with HTNV intraperitoneal. The NP-specific CD8+ T cell response was
analyzed using a 51Cr-release assay, intracellular cytokine assay, enzyme-linked immunospot assay and tetramer binding assay against H-2Kb restricted CTL epitopes of NP (M6 and N1 peptide). Using these methods, it was found that HTNV infection
elicited a strong NP-specific CD8+ T cell response at 8 days after infection, and several different methods to check the NP-specific CD8+ T cell response showed a perfect correlation among analyses. To examine the humoral immune response against HTNV
NP, the NP-specific antibody response was analyzed using an enzyme linked
immunosorbent assay (ELISA). HTNV infection elicited the NP-specific humoral
ii
DNA vaccine has been shown to elicit both humoral and cellular immune
responses, and confer protection against some viral, bacterial and parasitic pathogens.
Therefore, DNA vaccine strategy was applied to HTNV in this study. Sindbis
virus-based expression vector was carefully designed and constructed, in order to induce the
transient high level expression of target gene. In the case of DNA vaccination by
plasmid encoding nucleocapsid gene, a single dose injection of 100㎍ of plasmid DNA
into quardriceps muscle of C57BL/6 mice induced a high level of humoral and cellular
immune response. The NP-specific antibody response was elicited 2~4 weeks after
immunization and maximized at 6~10 weeks and sustained for over 14 weeks.
NP-specific CD8+ T cell response reached its peak 2~3 weeks after immunization. Even though NP-specific CD8+T cell response after DNA vaccination was not strong as the
HTNV infection, but the pattern of response was similar to that of HTNV infection.
In a challenge test with the recombinant vaccinia virus expressing NP
(rVV-HTNV-N), the rVV-HTNV-N titers in DNA vaccinated mice were decreased about 100
fold compared control mice. Even though challenge with rVV-HTNV-N in HTNV
infected mice were perfectly protected, but DNA vaccination showed the partial
protection.
In conclusion, this study showed that (i) HTNV infection in C57BL/6 mice elicited
the strong NP-specific CD8+ T cell response at 8 days after infection, (ii) DNA
vaccination with plasmid encoding HTNV nucleocapsid gene also elicited the strong
NP-specific humoral and cellular immune responses, and (iii) DNA vaccination elicited
the partial protective immunity against challenge with the recombinant vaccinia virus
expressing NP. (iv) DNA vaccine expressing HTNV NP was shown to be a possible
iii
Key words: Hantaan virus, Nucleocapsid protein, DNA vaccine, Cellular immune
response, Sindbis virus-based expression vector, Challenge test, Recombinant vaccinia
iv
TABLE OF CONTENTS
ABSTRACT --- i
TABLE OF CONTENTS --- iv
LIST OF FIGURES --- vii
LIST OF TABLES --- ix
ABBREVIATION --- x
I. INTRODUCTION --- 1
A. Immune responses against virus infection --- 1
B. Vaccine development strategy and DNA vaccine --- 3
C. Enhancing the antigen expression --- 7
D. Sindbis virus vector --- 14
E. Hantaan virus --- 16
F. Objectives of this study --- 19
II. MATERIALS AND METHODS --- 20
A. Plasmid DNA and reagents --- 20
B. Mice, virus and cells --- 20
C. Cloning of HTNV nucleocapsid gene and identification of nucleocapsid protein --- 21
1. PCR amplification and sequence analysis --- 21
2. In vitro transcription and translation (TNT) --- 22
D. Construction of DNA vaccine vector --- 23
E. Construction of DNA vaccine encoding HTNV nucleocapsid gene --- 26
F. Establishment of the assay systems for immune responses against NP-- 26
v
2. Assay for cellular immune response --- 29
G. In vivo test --- 32
1. Infection of mice with live HTNV --- 32
2. Immunization of mice with DNA vaccines --- 34
H. Generation of recombinant vaccinia virus expressing HTNV NP (rVV-HTNV-N) --- 35
1. Generation of rVV-HTNV-N --- 35
2. Challenge studies using the recombinant vaccinia virus --- 39
III. RESULTS --- 40
A. Cloning of HTNV nucleocapsid gene and identification of nucleocapsid protein --- 40
B. Construction of DNA vaccine vectors --- 40
C. Construction of DNA vaccine vectors expressing HTNV NP --- 45
D. Establishment of the assay system for in vivo test --- 45
1. Assay of humoral immune response --- 47
2. Assay of cellular immune response --- 53
E. In vivo test --- 59
1. Immune responses after HTNV infection --- 59
2. Immune responses after DNA vaccination --- 64
F. Generation of recombinant vaccinia virus expressing HTNV NP (rVV-HTNV-N) --- 72
1. Selection and screening of recombinant virus plaques --- 72
2. Challenge studies using recombinant vaccinia virus --- 72
vi
V. CONCLUSION --- 83
REFERENCES --- 85
vii
LIST OF FIGURES
Fig. 1. Factors influencing efficacy of DNA vaccines. --- 12
Fig. 2. Construction of sindbis virus-based DNA vaccine vectors. --- 25
Fig. 3. Schematic presentation of the tetramer analysis to detect CD8+
T lymphocytes. --- 33
Fig. 4. Construction of a vaccinia transfer plasmid for the generation
of recombinant vaccinia virus. --- 37
Fig. 5. Cloning of HTNV nucleocapsid gene. --- 41
Fig. 6. Comparison of marker gene expression among the constructed
sindbis virus-based DNA vaccine vectors. --- 44
Fig. 7. Construction of plasmid encoding nucleocapsid gene and
confirmation of NP expression in vitro. --- 46
Fig. 8. Selection of recombinant baculovirus expressing HTNV NP. --- 48
Fig. 9. Analysis of subcellular localization of NP expressed in insect cells. - 49
Fig. 10. Purification of recombinant NP expressed by baculovirus
viii
Fig. 11. Establishment anti-HTNV NP ELISA conditions. --- 52
Fig. 12. Screening of transformed E. coli that expressed H2-kb heavy
chain and mouse β2M.. --- 54
Fig. 13. Preparation of inclusion body in E. coli. --- 55
Fig. 14. Refolding and biotinylation of purified heavy chain and β2M. --- 56
Fig. 15. Generation of tetramer by mixing with biotinylated MHC complex
and PE-conjugated streptavidin. --- 58
Fig. 16. Immune responses in HTNV infected C57BL/6 mice. --- 62
Fig. 17. Comparison of antibody responses by DNA vaccine injection site. -- 65
Fig. 18. Comparison of antibody responses between pcDNA3-N
and CSHAC-N by injection dose. --- 66
Fig. 19. Cellular immune response after DNA vaccination in C57BL/6 mice. -68
Fig. 20. Humoral immune response after DNA vaccination in C57BL/6 mice.-71
Fig. 21. Isolation and confirmation of recombinant vaccinia virus expressing
HTNV NP. --- 74
ix
LIST OF TABLES
x
ABBREVIATION
Ab : Antibody
Ag : Antigen
ATCC : American Type Culture Collection
ATM : Anterior-tibialis muscle
BHK : Baby Hamster Kidney
β2M : β2-microglobulin
BrdU : 5-Bromo-2’-deoxyuridine
BSA : Bovine Serum Albumin
CD : Cluster of Differentiation
CMV : Cytomegalo Virus
CTL : Cytotoxic T Lymphocyte
DMEM: Dulbecco’s Modified Eagle Medium
ELISA : Enzyme linked immuno-sorbent assay
ELISPOT : Enzyme linked immuno-spot
FACS : Fluorescence Activated Cell Sorter
FBS : Fetal Bovine Serum
FITC : Fluorescence Isothiocynate
HFRS : Hemorrhagic Fever with Renal Syndrome
HTNV : Hantaan Virus
ICCS : Intracellular Cytokine Staining
INFγ : γ-Interferon
IPTG : Isopropyl-beta-D-thiogalactoside
MHC : Major Histocompatibility Complex
xi PAP : Peroxidase antiperoxidase
PBS : Phosphate buffered Saline
PCR : Polymerase Chain Reaction
PE : Polyerythrin
PFU : Plaque forming Unit
PMSF : Phenyl-methyl-sulfonyl-fluoride
QM : Quadriceps muscle
RIP : Radio Immuno Precipitation
SDS-PAGE : Sodiumdodesyl Sulfate Polyacrylamide Gel Electrophoresis
Sf9 : Spodoptera frugiperda clone 9
TK : Thymidine kinase
1
I. INTRODUCTION
A. Immune responses against virus infection
Viruses are obligatory intracellular microorganisms that replicate within cells,
often using the nucleic acid and protein synthetic machineries of the host. Many
viruses enter host cells by binding to physiologically important, normal cell surface
molecules. After entering cells, viruses can cause tissue injury and disease by any
of several mechanisms. Viral replication interferes with normal cellular protein
synthesis and function, leading to injury to and ultimately death of the infected cell.
Immunity against viral infections is mediated by a combination of humoral and
cellular immune mechanisms. Specific antibodies are important in defense against
viruses early in the course of infection. Neutralizing antiviral antibodies bind to
envelope or capsid protein and prevent viral attachment and entry into host cells.
Compliment activation may also participate in antibody-mediated viral immunity,
mainly by promoting phagocytosis and possibly by direct lysis of viruses with lipid
envelopes.
However, several points about the role of humoral immunity in protection
against viruses should be emphasized. First, antibodies may be effective against
viruses before the organisms enter cells, or may block spread from cell to cell, but
intracellular viruses are inaccessible to antibodies. Second, it has generally proved
2
Third, the neutralizing capacity of an antibody in vitro often shows little or no
correlation with its protective capacity in vivo. Taken together, these observations
suggest that although antibodies are an important component of immunity to
viruses, they may not be sufficient for eliminating many viral infections (Protzer
and Schaller, 2000).
The principal mechanism of specific immunity against established viral
infections is CTLs. The best defined virus-specific CTLs are CD8+ cells that recognize endogenously synthesized viral antigens in association with class I MHC
molecules on virtually any cell type (Hackett and Eisenlohr, 1990; Long and
Jacobson, 1989). A smaller but detectable proportion of virus-specific CTLs in
humans and mice consist of CD4+ CTLs that recognize viral antigens presented in association with class II MHC molecules. CD4+ CTLs can be effective only against infected cells that express class II molecules, whereas CD8+ CTLs have a much broader range of cellular reactivity. The full differentiation of CD8+ CTLs requires cytokines produced by CD4+ helper cells, which recognize endogenously synthesized or shed viral antigens in association with class II molecules. The
antiviral effects of CTLs are due to lysis of infected cells, stimulation of
intracellular enzymes that degrade viral genomes, and secretion of cytokines with
interferon activity.
However, viruses have evolved other mechanisms for evading host immunity.
Many viruses are capable of great antigenic variation, and large numbers of
3
virus becomes insusceptible to immunity generated in the population by previous
infections. And some viruses suppress immune responses by various mechanisms.
They may infect the cells of the immune system, impairing their function and
resulting in inhibition of specific immunity.
B. Vaccine development strategy and DNA vaccine
The birth of immunology as a science may be dated from Edward Jenner’s
successful vaccination against smallpox, which was reported in 1798. The
importance of prophylactic immunization against infectious diseases is best
illustrated by the fact that worldwide programs of vaccination have led to the
complete or near complete eradication of many of these diseases in developed
countries. Smallpox and polio are perhaps the two most impressive examples.
Current vaccines may be divided into two categories, ‘live’ and ‘dead’. Live
vaccines comprise traditional attenuated microbes, viral or bacterial, selected for
reduced pathogenicity with maintained immunogenicity, and ‘recombinant’
vaccines, in which foreign antigens are expressed from a replicating viral or
bacterial vector. “Dead” vaccines consist of killed whole pathogens, or soluble
pathogen proteins or protein subunits. Dead vaccines cannot efficiently enter the
MHC I pathway. Although safer, vaccines composed of inactivated pathogens or
immunogenic protein subunits may be less effective in inducing the cell-mediated
4
intracellularly replicating organisms. Live vaccines may be dangerous to pregnant
women or immunocomprimised hosts, can may be contaminated by potentially
harmful adventitious agents during production.
Since effective vaccination as a public health measure requires long-lasting
immunity, the ability of vaccines to stimulate memory T and B lymphocytes is an
important consideration in vaccine design. Thus, the demonstration over the last
decade that plasmid DNA vaccines can induce both humoral and cellular immune
responses in a variety of murine and primate disease models has engendered
considerable excitement in the vaccine community (Table 1).
The historical basis for DNA vaccines rests on the observation that direct in
vitro and in vivo gene transfer of recombinant DNA by a variety of techniques
resulted in expression of protein. In the seminal study by Wolff et al of ‘‘plasmid
or naked’’ DNA vaccination in vivo, it was shown that direct intramuscular
inoculation of plasmid DNA encoding several different reporter genes could induce
protein expression within the muscle cells (Wolff et al., 1990). This study provided
a strong basis for the notion that purified/recombinant nucleic acids (‘‘naked
DNA’’) can be delivered in vivo and can direct protein expression. These
observations were further extended in a study by Tang et al. (Tang, DeVit, and
Johnston, 1992), who demonstrated that mice injected with plasmid DNA encoding
hGH could elicit antigen-specific antibody responses. Subsequently,
demonstrations by Ulmer et al. (Ulmer et al., 1993) and Robinson et al. (Robinson,
5
Table 1. Important considerations for vaccine design. Table 1. Important considerations for vaccine design. Table 1. Important considerations for vaccine design. Table 1. Important considerations for vaccine design.
* Adapted and modified from * Adapted and modified from
6
respectively, from influenza infection provided a remarkable example of how DNA
vaccination could mediate protective immunity. The mouse study further
documented that both antibody and CD8+ cytotoxic T-lymphocyte (CTL) responses were elicited, consistent with DNA vaccines stimulating both humoral and cellular
immunity.
DNA vaccination might provide several important advantages over current
vaccines. DNA vaccines mimic the effects of live attenuated vaccines in their
ability to induce major histocompatibility complex (MHC) class I restricted CD8+ T-cell responses, which may be advantageous compared with conventional
protein-based vaccines, while mitigating some of the safety concerns associated with live
vaccines. This advantage was caused by endogenous production of antigen,
especially for viral antigens where all the post translation modifications are similar
following DNA immunization as those present during infection. As a result, the
antigens are authentic with all the conformational epitopes required for protection
being expressed. A second advantage is that since the animal acts as a bioreactor,
there is no need for downstream processing of the vaccine after the plasmid is
purified. Antigen purification is often a laborious and expensive process. Thus,
DNA-based vaccination should be economical to produce. Since the antigen is
produced endogenously there is also no need for adjuvants or problems associated
with injection site reactions produced by adjuvants (Van Donkersgoed et al., 1999).
Because of the endogenous expression of antigens, plasmid-based vaccines induce
7
of many viral infections (Ulmer et al., 1998). Finally, since DNA vaccines are
simple to purify, and technologies are available for purification the risk of
extraneous contaminating agents, which are a major problem in conventional live
vaccines is eliminated (Evermann et al., 1994; Thornton, 1986).
Since the first reports regarding genetic (polynucleotide) immunization were
published (Cox, Zamb, and Babiuk, 1993; Tang, DeVit, and Johnston, 1992; Ulmer
et al., 1993), a lot of different reports of this technique have been published with
antigens from different bacteria, viruses, and parasites (Babiuk et al., 2000; Lewis
and Babiuk, 1999). Unfortunately, the majority of the most successful
demonstrations of the efficacy of DNA immunization have been performed in the
mouse. When similar approaches were used in humans or large animals the results
were not as encouraging. The reasons for the lower efficacy of DNA vaccines in
humans or large animals are not currently known, but it could be related to the
transfection efficiency.
C. Enhancing the antigen expression
In mouse cells, transfection appears to occur with much more efficiency than in
larger animal species. Thus, improving delivery of the plasmid to enhance cellular
transfection and subsequent expression of proteins will be a critical factor in
8 1. Plasmid vector
The simplest concept of a DNA based vaccine incorporates a promoter, the
gene of interest for use in the vaccine, and a backbone for delivery of the cassette
into cells. Manipulation of each of these different components of the plasmid has
been shown to alter the efficacy of DNA vaccination. Since it is generally believed
that the level of gene expression should correlate with the level of immunity, the
plasmid construct should be one that maximizes gene expression in vitro. It is
believed that this can be achieved in a number of ways. First, manipulation of the
plasmid to increase the level of gene expression (plasmid modification) on a per
cell basis and secondly by increasing the number of cells that are transfected
through improved delivery systems should achieve the desired goal (Fig. 1).
Although these two factors may be separated, a combination of approaches may be
the most effective in achieving the goal of optimal immunization.
It should be possible to optimize gene expression by modifying all of the
essential elements of the plasmid. These include the promoter, the gene, and the
plasmid backbone.
One bottleneck hindering protein synthesis from plasmid vectors is translation
of mRNA transcripts. The molecular mechanism for hindering protein synthesis
from mRNA is the presence of rare codons for tRNAs of low abundance that slow
translation. Rare tRNA codons can be changed to abundant tRNA codons through
site directed mutagensis of the gene coding region of the plasmid or creating
9
viral (Deml et al., 2001; Kotsopoulou et al., 2000; Vinner et al., 1999; zur Megede
et al., 2000) and non-viral gene products (Narum et al., 2001; Uchijima et al.,
1998).
All expression vectors used for vaccination must have adjuvant properties and
a transcriptional unit able to express the antigen-encoding gene at high levels.
Indeed, according to Galvin et al. (Galvin, Muller, and Khan, 2000), the level of
both humoral and cellular immunity induced by a DNA vaccine is directly
correlated to the promoter strength. In most studies on DNA immunization, viral
promoters, such as the Rous sarcoma virus (RSV) LTR promoter, the simian virus
40 (SV40) promotor and the IE promoter of HCMV, have been used to drive
antigen expression.
Even though efficient production of antigen by DNA vaccines has been
achieved with the above mentioned viral promoters, the safety of such promoters
has been questioned. In addition, lymphokines such as INFγ produced during
induction of immune responses may inhibit further transcription initiation by viral
promoters. This is particularly true for CpG-enhanced vectors. Therefore, several
alternative, tissue specific promoters have been investigated, with varying success.
Even if the optimized plasmid enters the cell and the nucleus, the quantity of
protein that is produced is generally very low. Thus, if the vaccines also encoded
some co-stimulatory molecules important for enhancing immunity, it should be
possible to improve the kinetics and magnitude of the immune response. This
10
incorporate genes into the plasmid which encode cytokines or co-stimulatory
molecules which would create a micro-environment conducive for both attraction
of antigen-presenting and responsive cells as well as in expansion of immune cells
(Krieg and Davis, 2001). Secondly, the plasmid backbone can also be constructed
to contain immune stimulatory CpG motifs (Krieg et al., 1998).
To enhance immune responses to DNA vaccines different co-stimulatory
molecules have been used. These include the addition of helper epitopes, antigen
targeting, co-stimulatory molecules and cytokines. Although the specific
requirements to activate the immune systems are currently unknown, several
co-stimulatory molecules have been shown to be important for induction of immune
responses such as B7-1 (CD80), B7-2 (CD86), CTLA4, ICAM1, and CD40L
(Deliyannis et al., 2000; Kim et al., 1999; Santra et al., 2000; Sin et al., 2001). The
functions of different co-stimulatory molecules such as CD28, CTLA4, CD80, and
CD86 in T cell co-stimulation are complex and distinct (Horspool et al., 1998) and
it is likely there effects will differ between antigens.
In addition to using co-stimulatory molecules to modulate or enhance immune
responses to DNA vaccines, the use of gene-encoded cytokines has also been
effective. Several different cytokines have been used in DNA vaccines with some
small effects. The most effective cytokines used to modulate and enhance immune
responses to DNA vaccines in mice have been IL-12 and GM-CSF (Iwasaki et al.,
1997; Moore et al., 2002; Okada et al., 1997). The complexity of the biological
11
to design optimal stimulation for DNA vaccines.
Recently, it has been shown that the innate immune system of vertebrates
recognize CpG motifs as "danger" signals leading to various signal transduction
events including the production of various cytokines involved in immune
activation. Thus, not only do CpG motifs stimulate innate immunity, but they also
have a profound effect on specific immunity. More specifically, the cytokine
profile induced by CpG motifs generally drives a balanced or preferentially a
Th1-biased immune response.
2. Delivery
Although it is difficult to quantitate the number of plasmids that enter cells or are
degraded before they enter the nucleus and initiate gene expression it is believed that
in excess of 90% of the DNA never gets into the cytoplasm and of this 10% less than
1% enter the nucleus where gene expression occurs (Barry et al., 1999; Boutorine
and Kostina, 1993). Thus, there have been numerous approaches used to enhance
plasmid uptake not only into the cell but also into the nucleus. The earlier approaches
focused on lipid based delivery systems or transfection agents since such transfection
agents could enhance transfection in vitro (Gregoriadis, Saffie, and de Souza, 1997).
Although such approaches have had some success they are far from ideal. Therefore,
other approaches have been employed.
Gene gun delivery was one of the earlier approaches used to enhance gene
12
Fig. 1. Factors influencing efficacy of DNA vaccines. (Adapted and modified from Vaccine, vol.21, 649-658)
13
into the cell and it is for this reason that immune responses can be demonstrated
with significantly lower doses of DNA than if free DNA or DNA mixed with
transfection enhancing agents are used. Indeed, the magnitude of immune
responses induced by gene gun administration can be equivalent to needle delivery
with 100 fold less DNA. This is possibly related to both the efficiency of
transfection as well as the specific cells that are transfected in vivo. Gene gun
delivery, as opposed to other methods of delivery appear to be very efficient in
transfecting Langerhans cells or dendritic cells (Raz et al., 1994).
Polylactide-co-glycolide (PLG) microparticles have been used extensively in
delivery of protein-based vaccines (Singh and O'Hagan, 1999). They have proven
to be especially effective at targeting antigens to M cells (Jones et al., 1997).
Encapsulation of DNA in PLG particles is especially of interest since these
microparticles could then be used for oral delivery. Following oral delivery, PLGs
are taken up by M cells through the gastrointestinal tract and should induce
immune responses (Jones, Clegg, and Farrar, 1998).
Electroporation of cells in vitro has been one of the ways to increase DNA
uptake by cells (Banga and Prausnitz, 1998). Based on in vitro success, it seemed
that similar enhancement of DNA uptake should be possible to achieve in vivo, if
the correct electrical field could be achieved. A number of studies have shown that
gene transfer and expression could increase in skin and muscle following
14 D. Sindbis virus vector
Alphaviruses (family: Togaviridae) are enveloped viruses that possess a
single-strand, positive-sense RNA genome. The development of infectious clones for a
number of alphaviruses has led to the description of several powerful expression
systems. Expression systems based on a self-replicating RNA (replicon) that can be
packaged into viral particles were developed for Semliki Forest virus (SFV),
Venezuelan equine encephalitis (VEE) virus, and Sindbis virus (SINV)
(Bredenbeek et al., 1993; Geigenmuller-Gnirke et al., 1991; Liljestrom and Garoff,
1991; Pushko et al., 1997; Xiong et al., 1989). The SFV and SINV systems also
were engineered into DNA-based vectors (Berglund et al., 1998; DiCiommo and
Bremner, 1998; Dubensky et al., 1996; Hariharan et al., 1998; Kohno et al., 1998).
The DNA based alphavirus vectors drive transcription of the replicon RNA from a
RNA polymerase II-dependent promoter (cytomegalovirus early promoter). This
replicon RNA then becomes self-replicating and drives expression of a
heterologous gene of interest. The plasmids including a full-length human
cytomegalovirus (hCMV) promoter-driven expression cassette are able to produce
their replicase complex following cytoplasmic transport of the corresponding RNA.
The replicase produces a full-length RNA coding for itself as well as an abundant
sub-genomic mRNA coding for the heterologous protein. and high-level expression
of the heterologous antigens (DiCiommo and Bremner, 1998).
15
infectious diseases (Berglund et al., 1998; Colombage et al., 1998; Fleeton et al.,
1999; Hariharan et al., 1998; Hevey et al., 1998; London et al., 1992; Mossman et
al., 1996; Pugachev et al., 1995; Pushko et al., 1997; Tsuji et al., 1998; Zhou et al.,
1995).
Alphavirus DNA-based expression vectors have several potential advantages
as vaccine vectors: (1) transient, high-level, protein expression; (2) cytoplasmic
mRNA transcription, eliminating potential mRNA splicing events that may be
associated with nuclear transcription; (3) The RNA-self-amplifying property is of
considerable interest with respect to vaccine biosafety: as the vector is replicating
at the RNA level and not at the DNA level, the rate of foreign DNA present in vivo
and possessing ‘genome integration potential’ is controlled and does not increase
following vaccination (contrary to some attenuated or recombinant vaccines); (4)
Another unique feature of this vector is its suicidal nature. When transfected into
cells, it eventually leads to apoptosis of the transfected cells (Kohno et al., 1998;
Leitner et al., 2000), which is particularly important to alleviate the concerns of
potential integration and cell transformation generated by the use of conventional
DNA vaccines (Gurunathan, Klinman, and Seder, 2000).
Several groups have demonstrated the ability of suicidal DNA vaccines to
induce high-level humoral and cell-mediated immunity against a variety of
antigens, and the immunized animals developed more pronounced immune
responses than those received a conventional DNA vaccine encoding the same
16
Kirman et al., 2003; Leitner et al., 2000). In addition, a most recent report has
showed that suicidal DNA vaccines could break immunological tolerance by
activating innate antiviral pathways, whereas the conventional DNA vaccines
encoding the same antigen could not (Leitner et al., 2003). All these advantages
indicate that suicidal DNA vaccines are an attractive vaccine delivery vehicle and
an alternative strategy to conventional DNA vaccines.
For these reasons, I was interested in testing alphavirus-based expression
vectors as possible hantaanvirus vaccine vehicles. In this study, I compared the
immunogenicity and protective efficacy of different vaccine vectors expressing the
HTNV nucleocapsid gene in a mouse model.
E. Hantaan virus
Hantaan virus (HTNV) (genus Hantavirus, family Bunyaviridae) is the
causative agent of the most severe form of a rodent-borne disease known as
hemorrhagic fever with renal syndrome (HFRS).Other hantaviruses that are known
to cause HFRS include Seoulvirus (SEOV), which causes disease primarily in Asia,
and Dobravavirus (DOBV) and Puumala virus (PUUV), which cause disease in
Europe, Scandinavia, and western Russia (Peters, Simpson, and Levy, 1999). In
addition, Sin Nombre and Andes viruses have been associated with outbreaks of a
highly lethal disease, hantavirus pulmonary syndrome (HPS), in the Americas
17
by only one principal rodent host species, their distribution is generally limited to
the range of that host (Schmaljohn and Hjelle, 1997). HTNV, which is carried by
Apodemus agrarius, is found in Asia; DOBV, carried by Apodemus flavicollis, and
PUUV, carried by Clethrionomys glareolus, are found in Europe. SEOV is more
widely disseminated than any other recognized Hantavirus because its host, the
common urban rat (Rattus norvegicus), is found throughout the world. Since the
transmission of HTNV to human is via aerosols of contaminated excreta such as
urine, feces or saliva from infected mice, much interest have been focused on the
way how the virus is maintained and spread among rodent hosts. But suitable
animal models that mimic the HFRS are so far unavailable.
More than 100,000 cases of HFRS are reported annually, with a mortality rate
between 2% and 10% (Hjelle et al., 1995; Lee, 1989; Ruo et al., 1994). It is an
acutely prostrating febrile illness, in which about one third of the patients develop
hemorrhagic manifestations with 10% to 15% developing shock. In the typical
severe case, there is a sudden onset of high fever, headache, myalgia, and severe
malaise. The major pathological lesions in fatal cases are disseminated
hemorrhages and microscopic abnormalities in the kidneys.
HTNV is a spherical, enveloped virus with a genome consisting of three
segments of single-stranded, negative-sense RNA. The three segments are
designated as large (L), medium (M), and small segments (S) that encode RNA–
dependent RNA polymerase, two envelope glycoproteins (G1, G2) and
18
It is well known that neutralizing antibodies, especially to G1 and G2, play
major roles in protection against HTNV infection (Schmaljohn et al., 1990;
Yoshimatsu et al., 1993; Zhang, Takashima, and Hashimoto, 1988). Those
circulating antibodies are thought to prevent the primary infection of the virus in
host animals but do not contribute to the clearance of the virus that has already
multiplied in the host cells. Kariwa et al. found that HTNV could replicate or
survive for a certain period in adult mice, in spite of the presence of specific
antibodies (Kariwa et al., 1995). Although a high level of a neutralizing antibody
was present in suckling mice inoculated with HTNV, the virus persisted in the
animal for several weeks (Nakamura et al., 1985a). Furthermore it was reported
that neutralizing monoclonal antibody-escape mutants of HTNV were generated in
the presence of antibodies the G1, G2 proteins. Therefore, it seems that efficient
protection against HTNV infection could not be provided by neutralizing
antibodies alone, and thereby need the cellular immune system. The NP of HTNV
is highly immunogenic and genetically more conservative than the envelope
protein. And natural HTNV infections of rodents and humans result in the
induction of strong N-specific antibody and T cell responses (Khaiboullina and St
Jeor, 2002). Therefore the report showing that baculovirus recombinants
expressing only NP could protect animals from HTNV challenge is very interesting.
It suggests that the NP elicited a nonneutralizing, perhaps T cell-mediated,
protective immune response. Human CD4+ and CD8+ CTL epitopes on NP have been identified in SNV (Sin Nombre Virus) and in HTNV.
19 F. Objectives of this study
In this study, I want to know whether HTNV infection can be protected by NP-
specific immune response elicited by the safe and potent DNA vaccine vector.
Therefore, the present study was focused on the following; First, the establishment
of assay systems for cellular and humoral immune response to HTNV NP; Second,
confirmation of HTNV NP-specific immune response in H-2Kb mice infected with HTNV; Third, development of sindbis virus-based DNA vaccine vector expressing
HTNV NP; Fourth, confirmation of HTNV NP-specific immune response in mice
immunized with HTNV NP expressing DNA vaccine; Fifth, establishment of
HTNV surrogate challenge model system and investigation of protective efficacy
20
II. MATERIALS AND METHODS
A. Plasmid DNA and reagents
Mammalian expression vector, pcDNA3.1 (Invitrogen, Carlsbad, Calif, USA),
was used for control vector. Sindbis virus-based DNA vaccine vector was derived
from pSinRep5 (Invitrogen). The Escherichia coli strain TOP10 was used as the host
during the plasmids construction and selected with ampicillin. Plasmid DNA was
purified using endotoxin-free DNA purification kits (Qiagen, Valencia, CA, USA)
according to the manufacturer’s directions.
Liposome, GenePoter-1 (GeneTherapySystem, Italy), was used for in vitro
transfection for identification of target gene expression.
B. Mice, virus and cells
C57BL/6 mice were purchased from Charles River Laboratories (Wilmigton,
MA). The studies used 5~6 week old female mice. BHK, Sf9, Hela, Vero E6 and
143B TK(-) cells were obtained from American Type Culture Collection (ATCC ;
Monassas, VA). BHK and Vero E6 were cultured in Dulbecco’s Modified Eagle
Medium (DMEM), the Sf9 cell was in TC yeastolate and TC lactalbumin (Difco)
supplemented Grace insect cell media, and the 146B TK(-) cell was in alpha-MEM.
All media were supplemented with 10% FBS and 40㎍/ml gentamycin (Gibco-BRL).
21
incubator. The HTNV strain 76-118 was obtained from Dr. Lee (Lee, 1989). HTNV
expanded in Vero E6 cells monolayer through five times passage, a viral stock was
prepared from the culture medium as described (Elliott, Kiley, and McCormick,
1984). A titer of virus stock was determined by foci formation in Vero E6 cells
followed by staining with peroxidase-antiperoxidase (PAP) as previously described
(Tanishita et al., 1984).
Baculovirus (Wild type virus : Autographa californica nucleopolyhedrovirus)
and Vaccinia virus (Western Reverse strain) were obtained from American Type
Culture Collection (ATCC ; Monassas, VA). Baculovirus was cultured in Sf9 cells
grown in monolayer, viral stock was prepared from the culture medium. Titer of
virus stock was determined by plaque assay in Sf9 cells followed by agarose overlay.
Vaccinia virus was cultured in Hela cell, viral stock was prepared from the cell lysate.
Titer of virus stock was determined by plaque assay in Vero E6 cells followed by
agarose overlay.
C. Cloning of HTNV nucleocapsid gene and identification of its protein
1. PCR amplification and sequence analysis
Total cellular RNA was isolated from HTNV-infected Vero E6 cells using TRIsol
(Invitrogen, Carlsbad, Calif) by standard methods. cDNA was synthesized and
amplified using a One-step RT-PCR (Invitrogen, Carlsbad, Calif) with N-gene
22
GG, 3-HTNV-N: GCGAATTCTTAGAGTTTCAAAGGCTCTTGGTTGGA). The
PCR reaction mixture was incubated for 1 min at 94℃, 1 min at 50℃, 3.5 min at
72℃ and 30 cycles of amplification were performed with a DNA Thermal Cycler
(MJ Research; Massachusetts, USA).
Sequence analysis of nucleocapsid gene was performed with the ABI Prism
BigDye Terminator Cycle Sequencing Ready Reaction Kit and ABI Prism 310
Genetic Analyzer (Perkin-Elmer Cetus, Norwalk, CT).
2. In vitro transcription and translation (TNT)
In vitro TNT was accomplished with TNT Coupled Reticulocyte Lysate Systems
(Promega, Madison, WI, USA) to identify whether this cloning the HTNV
nucleocapside gene have functional open reading frame. Reaction mixture (DNA
template 1㎍, Rabbit Reticulocyte Lysate 25㎕, Reaction buffer 2㎕, T7 RNA
polymerase 1㎕, Amino acid mixture minus methionin (1 mM) 1㎕, 35S-methionin 0.3 mCi/ml, RNase inhibitor (RNasin : 40U/㎕) 1㎕, Nuclease free H2O to final
volume 50㎕) was incubated at 30℃ for 90 minutes.
TNT products were analyzed by SDS-PAGE and autoradiography. After mixed 5
㎕ of TNT product and 20㎕ of SDS sample buffer, heat at 100℃ for 5 minutes to
denature the proteins. SDS-PAGE was carried out at a 30 mA in the 10% separating
gel. After electrophoresis, gel was dried under a vaccum and exposed on X-ray film
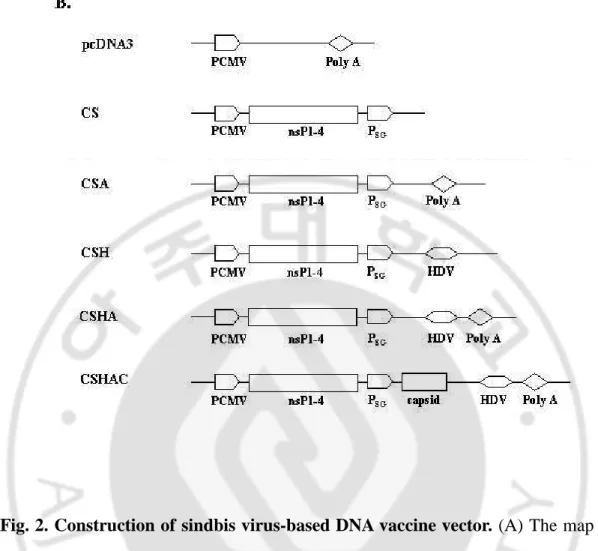
23 D. Construction of DNA vaccine vectors
Sindbis virus-based DNA vaccine vector was derived from pSinRep5
(Invitrogen). SP6 promoter, prokaryotic promoter, was replaced with human CMV
promoter. CMV promoter was inserted at Sac1 site before SP6 promoter, and 5’ of
sindbis genome was linked directly to the CMV promoter by in vivo homologous
recombination with PCR product, which was reacted with 5’CMVsin
(5’-AGTGAACCGATTGACGGCGTAGTACACACTATTGAATCA-3’) and 3’CMVsin
(5’-GCCGTCAATCGGTTCACTAAACCAGCTCTGCTTATATAG-3’) primer.
Hepatitis D virus ribozyme sequence for self-cleaving RNA sequence was inserted at
Not1 site (pCMVSinHDV: CSH) and bovine growth hormone polyadenylation signal
for transcripton termination was inserted at Xho1 site (pCMVSinHDVPolyA:
CSHA). Futhermore, sindbis virus capside gene was inserted after subgenomic
promoter at Xba1 site (pCMVSinCHDVPolyA: CSHAC). Because of sindbis virus
capside protein have self-cleavage site at C-terminal and acts as translational
enhancer (Fig. 2).
LacZ gene was inserted in multi cloning site as reporter gene for in vitro
expression test. BHK cells grown in 6-well plate were transfected with 2㎍ of
plasmid DNA with GenePoter 1 (GeneTheraphySystem). After 48 h, expression
products were assayed with β-gal assay kit (Invitrogen) according to manufacturer’s
24
25
Fig. 2. Construction of sindbis virus-based DNA vaccine vector. (A) The map of
original sindbis virus vector. (B) Schematic presentation of modified sindbis virus
vectors. PCMV, HDV and Poly A indicates CMV promoter, ribozyme sequence of
Hepatitis delta virus and bovine growth hormone polyadenylation signal. nsP1-4, PSG,
and capsid, indicates nonstructural protein 1-4, subgenomic promoter and capsid
26
E. Construction of DNA vaccine vectors encoding HTNV nucleocapsid gene
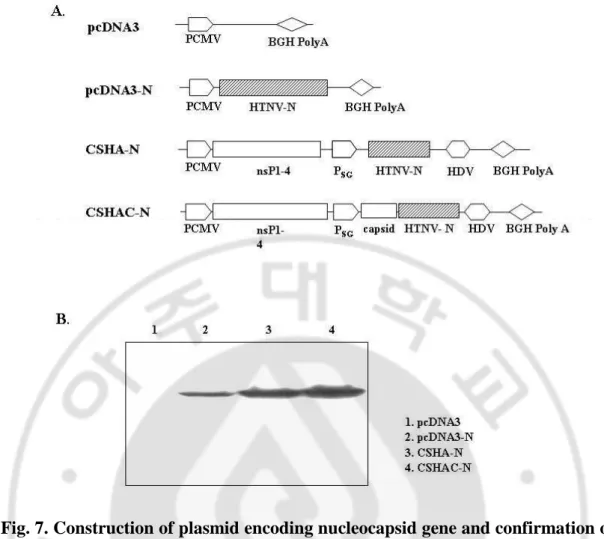
pcDNA3-N was constructed by cloning the cDNA of HTNV nucleocapsid gene
into EcoRI site of pcDNA3.1. CSHA-N was constructed by insertion of HTNV NP
gene under the transcription control of sindbis virus subgenomic promoter, and
CSHAC-N constructed by fusion of NP gene with sindbis virus capsid gene.
pcDNA3-N, CSHA-N, CSHAC-N plasmids were transfected in BHK cell for in
vitro expression test. After 48 h, expression products were assayed by Western blot
analysis with HTNV NP-specific monoclonal antibody (NA8B).
F. Establishment of the assay systems for immune responses against HTNV NP
1. Assay for humoral immune response
After DNA vaccination or HTNV infection in mouse, humoral immune response
was detected by anti-HTNV NP ELISA. In order to estabilish anti-HTNV NP ELISA
the recombinant HTNV NP was expressed and purified using baculovirus expression
system.
(A) Generation of recombinant baculovirus expressing HTNV NP
Nucleocapsid gene encoding His-tagged NP were amplified from pcDNA3-N by
PCR using specific primers (5-HTNV-N: GCGAATTCATGGCAACTATGGAGG
AATTACAGAAG, 3-HIS-N: GCGAATTCTTAATGGTGATGGTGATGATGGAGT
27
(Clontech, USA). Cotransfection with baculovirus genomic DNA into Sf9 cells were
performed as recommended by Clonetech.
Recombinant baculovirus carring the nucleocapsid gene were selected by plaque
assay and confirmed by Western blotting with anti-NP antibody. Finally, a plaque
isolate was amplified to yield high-titer virus stocks by infection of successively
larger numbers of Sf9 cells.
(B) Analysis of subcellular localization of recombinant HTNV NP expressed in insect cells
Analysis of subcellular localization of recombinant HTNV NP expressed in
insect cells was done to set up the optimized purification method. Sf-9 cell were
grown on tissue culture flask and infected by recombinant baculoviruses. Cells were
harvested at 24 h post-infection, cytoplasmic fraction and nucleus fraction was
separated by 1% NP40 treatment and centrifugation. Nuclear fraction was separated
to high-salt extract and insoluble fraction by 0.5 M NaCl treatment. And subcellular
localization of recombinant HTNV NP was analyzed by SDS-PAGE with each
fraction.
(C) Purification of HTNV NP
Sf9 cells were infected with recombinant baculovirus at a multiplicity of
infection of 10. The infected cells were cultured for 96 h and then collected by
28
(8 M urea, 0.1 M NaH2PO4, 0.01 M Tris-Cl, pH 8.0) for 1 h at room temperature.
After centrifugation, the supernatant was applied to Ni2+-NTA agarose resin (Qiagen) equilibrated with a binding buffer (8 M urea, 0.1 M NaH2PO4, 0.01 M Tris-Cl, pH
8.0) at room temperature. The column was then washed with a 10-column volume
wash buffer (8 M urea, 20 mM imidazole, 0.1 M NaH2PO4, 0.01 M Tris-Cl, pH 6.3).
The recombinant HTNV NP was finally eluted with elution buffer (8 M urea, 250
mM imidazole, 0.1 M NaH2PO4, 0.01 M Tris-Cl, pH 4.5). The purified protein was
analyzed by SDS-PAGE.
(D) Anti-HTNV NP ELISA
ELISA was performed to determine specific antibodies to the NP of HTNV.
Flat-bottomed 96-well plates (Nunc, Denmark) were coated with 0.1㎍/well of purified
NP in 0.05 M carbonate buffer (pH 9.6) at 4℃ for 16 h. The plate was blocked with
10% FBS in PBS for 2 h at 37℃. After washing with PBS containing 0.05%
Tween-20 (PBS-T), the plate was incubated at 37℃ for 1 h with sera from the immunized
mice using 50 fold diluted in PBS containing 2% BSA. The plate was washed and
incubated at 37℃ for 1 h with horseradish peroxidase-conjugated polyclonal goat
anti-mouse IgG (Sigma) diluted in 1:5,000. After washing with PBS-T, TMB
solution (KPL) was added and the reaction was left at room temperature for
approximately 30 min. The reaction was stopped by the addition of 0.5 N H2SO4 and
the optical density at 450 nm was measured. The reciprocal end-point titres of the
29 2. Assay for cellular immune response
(A) Enzyme linked immunospot (ELISPOT) assay
Spleen cells were isolated and IFN-γ ELISPOT assays were performed at 8 days after HTNV infection, or every week following DNA vaccination. MultiScreen-IP
plates (Millipore, Bedford, MA) were coated with 100㎕of anti-mouse IFN-γ Ab (5
㎍/ml in PBS) and were incubated at 4℃ overnight. The plates were then blocked
with PBS-10% FBS for 2 h at room temperature. Splenocyte (1~5 X 105cells) isolated from the spleens were added to wells with 5㎍/ml M6 (HTNV-NP221-228:
SVIGFLAL), N1 (HTNV-NP328-335: LGAFFSIL) peptides (Chang et al., 2001;
Park et al., 2000) and incubated at 37℃ for 24 h. After incubation, the cells were
removed and biotinylated anti-mouse IFN-γ Ab was added (2㎍/ml in PBS, 5% FBS, 0.05% Tween20) and kept for 2 h at room temperature. Then streptavidin-HRPO
(BD-Pharmingen) was added and kept for 2 h at RT. Finally, the plates were treated
with 3-amino-9-ethyl-carbazole (10 mg of AEC in 0.1 M Sodium acetate buffer (pH
5.0)) peroxidase substrate at room temperature for 10 min. The reaction was stopped
under running distilled water. The numbers of spots were counted using an ELISPOT
reader (AID ELISpot reader system).
(B) Intracellular cytokine staining (ICCS) assay
30
h at 37℃ in RPMI containing 10% FBS, 2㎍/ml of brefeldin A (Sigma) and 50㎍/ml
peptides (M6 or N1). The cells were then washed and stained with PE-labelled
anti-CD8 Ab at 4℃ for 20 min. Then the cells were washed in PBS and fixed with 4%
paraformaldehyde in PBS for 20 min. After washing, the cells were permeated with
0.5% saponin (Sigma) in PBS for 10 min. After washing, the cells were then stained
with FITC-labeled anti-mouse IFN-γ Mab for 30 min. The cell samples were analyzed with a FACSCalibur (Becton Dickinson), and the data analysis was
conducted with CellQuest software (Becton Dickinson).
(C) 51Cr release assay
Antigen-specific CTL function was measured by standard 51Cr release assay. Splenocytes were cultured for 5 days in RPMI1640 medium supplemented with 10㎍
/ml M6 or N1 peptide. Peptide-pulsed target cells were prepared by incubating EL-4
cells with synthetic peptide (10㎍/ml) for 4 h in a CO2 incubator, then washed
extensively with a PBS buffer to eliminate unbound peptides. Subsequently, the cells
were incubated with 100 µCi 51Cr for 4 h at 37℃. They were brought into contact with each other by centrifugation for 2 min and incubated for 4 h in 96-well
round-bottom plates. The specific lysis was calculated as follows: (Experimental release –
spontaneous release) / (100% release – spontaneous release) X 100. All assays were
performed in triplicate.
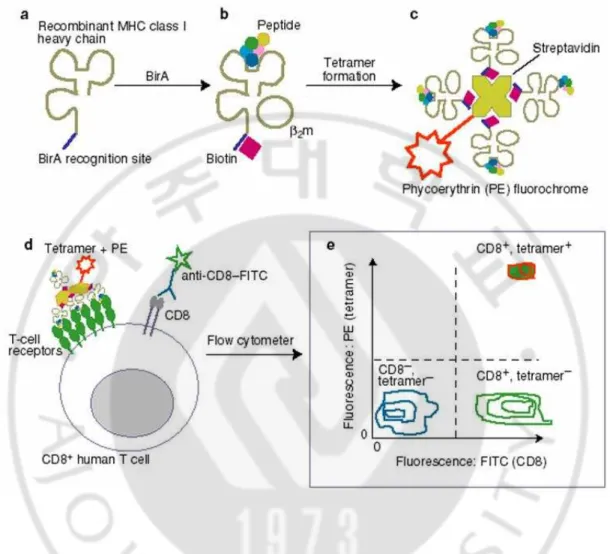
31 (1) Construction of peptide-MHC tetramers
MHC class 1 – peptide tetramers were produced as described by Altman et al.
(Altman et al., 1996; Ogg et al., 1998). Extracellular domain of the H2-kb heavy chain containing a biotinylation site was amplified by PCR and cloned into the
pET15b vector (Promega). Mouse β2M cDNA was similarly amplified and cloned
into the pET11a (Promega) vector. E. coli BL21(DE3) were transformed with these
plasmids, and colonies were selected by SDS-PAGE following expression of their
protein. For large production of proteins, selected clones were grown and induced by
addition of 1mM IPTG to the growth media at 0.5~1 O.D. (at 660 nM). After grown
for 3 h, the cells were harvested by centrifugation and resuspened pellet in solution
buffer (50 mM Tris-HCl, 25% sucrose, 1 mM NaEDTA, 0.1% sodium azide, 10 mM
DTT, pH 8.0). Preparation of inclusion body was done by sonication on ice, added
lysozyme (0.5 mg/ml) for 30 min at 37℃ and then MgCl2 (1.25 mM) and DNase I
(25㎕/ml) for 30 min at 37℃. After centrifugation at 12,000 rpm, 20 min, 4℃, the
pellets were washed with lysis buffer (50 mM Tris-HCl, 1% Triton X-100, 0.1%
sodium deoxycholate, 100 mM NaCl, 0.1% sodium azide, 10 mM DTT, pH 8.0).
Inclusion body was harvested by centrifugation at 12,000rpm, 30min, 4℃, and
resolved in 8 M urea at room temperature
Purified heavy chain and β2M were refolded in refolding buffer (100 mM
Tris-HCl, 400 mM L-Arg-Tris-HCl, 2 mM NaEDTA, 0.5 mM oxidized Glutathione, 5 mM
32
refolding buffer, and 65 ㎍/ml of β2M, 90 ㎍/ml of heavy chain were added with
stir. Exchanged the buffer with washing buffer (20 mM Tris-HCl, 50 mM NaCl, pH
8.0), and concentrated with ultra-filtration membrane (MWCO=10,000, Amicon) and
spin-filteration tube (Centricon YM-10, Amicon).
Biotinylation was done in solution mixture [Solution A (bicin buffer: 0.5 M bicin,
pH 8.3) + Solution B (100 mM ATP, 100 mM MgOAc, 200 µM biotin) + extra
d-biotin + Biotin-protein ligase (BirA) + protease inhibitor (Pepstatidin, Leupeptin)]
with peptide-MHC complex, and incubated overnight at room temperature.
Biotinylated–MHC complexes were purified with FPLC (superdex 200HR gel
filteration column). Complex peak fractions were pooled and concentrated with
centricon-10, and tetramers were generated by adding phycoerythrin conjugated
streptavidin (BD Pharmingen) over the 15 h to a final molar ratio of 1:4.
(2) Staining and sorting of T cells using peptide-MHC tetramers
Sample analysis was performed using 1~2 X 106 cells in 30㎕ of 1%FBS/PBS. Anti-CD8-FITC, and relevant tetramer-PE (final concentration of 20㎍ MHC/ml)
were added, and the staining was conducted for 4 h at 4℃. The cells were then
analyzed on a FACScalibur using CellQuest software (Becton Dickinson) (Fig. 3).
G. In vivo test
33
Fig. 3. Schematic presentation of the tetramer analysis to detect T lymphocytes. (Adapted from Expert Rev Mol Med)
34
C57BL/6 mice inoculated with 1 X 105 FFU/Head of cell-cultured HTNV via intraperitonial administration. Mouse sera were collected by retro-orbital puncture
each day in order to check the NP-specific humoral immune response. After eight
days, splenocyte was isolated by ficoll density gradient and the NP-specific cellular
immune response assayed. All experiments with infectious viruses were conducted in
the BSL3 containment facilities.
2. Immunization of mice with DNA vaccines
(A) Comparison of antibody response by injection sites
Mice were anesthetized with sodium pentobarbital anesthesia (75 mg/kg,
Intraperitonially). While they asleep, the mice were received bilateral injections in
the anterior-tibialis muscles (ATM) or quadriceps muscles (QM) of 50㎍
pcDNA3 or pcDNA3-N. All intra-muscular injections into the mouse ATM or QM
were through the skin using an insulin syringe, which comes with a pre-attached
29G1/2 needle. Mouse sera were collected by retro-orbital puncture at 2 week
intervals. The serum was recovered by centrifugation 10 min. at 4,000 g and
anti-HTNV NP was measured by ELISA methods.
(B) Comparison of antibody response between pcDNA3-N and CSHAC-N by injection dose
35
Anesthetized mice were received injection in the QM. pcDNA3-N was injected
with 25, 12.5, 6.25㎍/head, and CSHAC-N was injected with 12.5, 6.25, 3.1㎍
/head. Mouse sera were collected at 2 week intervals and the anti-HTNV NP was
measured by ELISA described previously.
(C) The humoral and cellular immune responses after DNA vaccination
For DNA vaccination, mice were injected with pcDNA3-N or CSHAC-N into
both quadriceps muscles with 50㎍DNA each. Empty pcDNA3 was used as a
negative control. Three days before vaccination, mice were injected with 0.25%
bupivacane into both quadriceps muscles. After vaccination mouse sera were
collected by retro-orbital puncture at two week intervals for antibody titration. Mice
were sacrificed every week after immunization for spleen isolation.
H. Generation of recombinant vaccinia virus expressing HTNV NP
(rVV-HTNV-N)
In order to establish the surrogate challenge system, the recombinant vaccinia
virus expressing HTNV NP was selected as follows.
1. Generation of rVV-HTNV-N
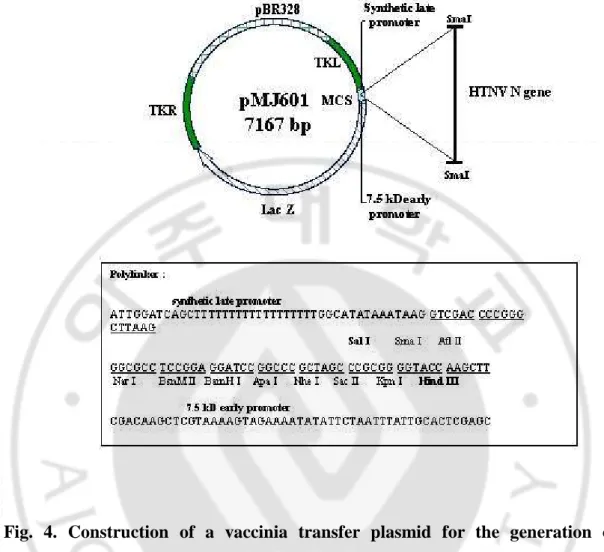
(A) Cloning of HTNV nucleocapsid gene into vaccinia transfer vector and transfection
36
fragment (NEB) and cloned into the Sal1 site of pMJ601 vector under the control of
the synthetic late promoter (Fig. 4). This construct, along with a wild type vaccinia
virus (strain Western Reverse), was transfected into BHK cells using a GenePoter 1
(GeneTherapySystem, Italy). Two days after transfection, the cell was harvested with
a disposable scraper and transfer to a conical centrifuge tube. After aspirating the
medium, cells were resuspened and disrupted in 0.5ml MEM by three cycles of
freeze-thawing.
(B) Selection and screening of recombinant virus plaques
The recombinat vaccinia virus was selected by β-gal staining and TK(-) selection.
Crude recombinant virus contained in the lysate was infected in confluent
monolayers of TK(-) 143B cell. Allow the virus to adsorb for 60 min, then remove
the inoculum and add a 1:1 mixture of pre-warmed 2X alpha-MEM containing 100
㎍/ml BrdU with an equal volume of 2%LMP agarose (plaque medium). The next
day, 2 ml of media containing 0.13% X-gal (6.4㎕/ml of 20% X-gal in dimethyl-
-formamide) were added to the agar overlay. One or two days after, blue-colored
plaque was harvested by sterile Pasteur pipet. The agarose plug was transfered to a
microfuge tube containing 0.5 ml alpha-MEM. Plaque isolates were freeze-thawed 3
times, another round of plaque purification was performed using this freeze-thaw
sample.
37
Fig. 4. Construction of a vaccinia transfer plasmid for the generation of recombinant vaccinia virus. DNA coding for NP of HTNV was subcloned into the
38
The presence of recombinant virus in isolated plaques was confirmed by RIP. The
isolated virus was infected to the confluent monolayered BHK cells for 60 min, then
inoculum was removed and DMEM containing 2% FBS was added. The next day,
culture media were replaced with methionine free media (GibcoBRL). After
starvation for 1 h in 37℃ CO2 incubator, culture media were replaced with media
containing 35S-methionine (25 µCi/ml). About 2 h later, cells were harvested and extracted with RIPA extraction buffer (20 mM Tris (pH 7.4), 150 mM NaCl, 2 mM
EDTA, 1% NP40, 1 mM PMSF, 0.1% Sodium deoxycholate) for 15 min. at 4℃.
Anti-HTNV NP monoclonal antibody was added to the supernatent after
centrifugation (12,000 rpm for 3 min.). After the mixture was rotated for overnight at
4℃, 50㎕ Protein A-sepharose 4B (1.5g/30ml) was added, and then rotated for 1 h
at 4℃. Wash with RIPA buffer 4 times, and the pellet was boiled with SDS-PAGE
sample buffer for 5 min. SDS-PAGE was done with 12% gel. Gel was dried after
amplified for 20 min with amplify sol. (Amersham), and then exposured to X-ray
film.
(D) Amplification of a recombinant vaccinia virus
A plaque isolate was amplified by infection of successively larger numbers of
cells. Sonicated plaque isolates were infected to confluent monolayed 143B TK(-)
cell in T-25 flask, and overlayed with selection media. After incubation for 2 days or
until cytopathic effect is obvious, cells were harvested and resuspended in 0.5 ml
39
were diluted with 1.75 ml of media and infected to the confluent monolayered HeLa
cell in T-175 culture flask. Selection media was not required at this step. After 2~3
days, the cell was harvested with cell scraper. Pellet by centrifugation for 5 min at
1,800 X g was resuspended in 2ml of media. Disrupt the cells by three cycles of
freeze-thawing. The supernatant of cell lysate was deposit in -80℃ deepfreezer, and
determine the titer of virus stock by plaque assay in 143B TK(-) cell.
2. Challenge studies using the recombinant vaccinia virus
The virus challenge experiment was performed four weeks after immunization.
Mice were challenged with 1 X 106 PFU/head of rVV-HTNV-N via intraperitonial administration. After five days, the ovaries were harvested and homogenized using a
pellet pestle motor (Kontes, Vineland, NJ), and the sample was adjusted to a final
volume of 200㎕. The titer of rVV-HTNV-N in the ovaries was analyzed using
human 143B TK(-) cells. Thus, TK(-) 143B cells, plated at 5 X 105 cells/well in 6 well-plate before 18 h, were infected with rVV-HTNV-N in ovary homogenization at
various dilutions for 1 h with rocking. Cell were cultured for two days after washing
in alpha-MEM containing 5% FBS, 20㎍/ml BrdU. Plaque was revealed in
40
III. RESULTS
A. Cloning of HTNV nucleocapsid gene and identification of its protein
The cDNA representing the nucleocapsid gene derived from of the 76-118 strain
of HTNV was synthesized by reverse transcription of viral RNA. After PCR
amplification, about 1.3kb HTNV nucleocapsid gene was identified (Fig. 5B). The
cDNA was cloned into EcoRI site in T7 promoter-based expression plasmid,
pBluescript II SK (pBSK) (Stratagene: Canada), to create pBSK-N (Fig. 5A, 5C).
Sequence analysis of nucleocapsid gene was performed with the ABI Prism BigDye
Terminator Cycle Sequencing Ready Reaction Kit and confirmed the sequence
identity.
To identify whether the nucleocapsid gene has functional open reading frame, in
vitro TNT was accomplished with TNT Coupled Reticulocyte Lysate Systems
(Promega). About 47kDa of HTNV NP was identified by SDS-PAGE analysis and
autoradiograpy (Fig. 5D).
B. Construction of DNA vaccine vectors
Sindbis virus-based DNA vaccine vector was derived from pSinRep5
(Invitrogen). SP6 promoter, prokaryotic promoter, was replaced with human CMV
promoter. And HDV ribozyme sequence, BGH polyadenylation signal, and sindbis
virus capsid gene were inserted serially. LacZ gene was inserted into multi cloning
41 A.
Fig. 5. Cloning of HTNV nucleocapsid gene. (A) Plasmid for HTNV nucleocapsid
gene cloning. DNA coding for NP of HTNV was subcloned into the EcoRI site of
42
one-step RT-PCR with nucleocapsid gene specific primer as described in Materials
and Methods. (C) The pattern of EcoRI enzyme digestion of pBSK and pBSK-N. (D)
In vitro transcription and translation. In vitro TNT was accomplished with TNT
Coupled Reticulocyte Lysate Systems (Promega). Products were analyzed by