저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
The Mechanism and Effects of TNF-α on
Apoptosis in Auditory Cell Line
by
Hun Yi Park
Major in Medicine
Department of Medical Sciences
The Graduate School, Ajou University
The Mechanism and Effects of TNF-α on
Apoptosis in Auditory Cell Line
by
Hun Yi Park
A Dissertation Submitted to The Graduate School of
Ajou University in Partial Fulfillment of the Requirements
for the Degree of Ph.D. in Medicine
Supervised by
Keehyun Park, M.D., Ph.D.
Major in Medicine
Department of Medical Sciences
The Graduate School, Ajou University
This certifies that the dissertation
ofHun Yi Park is approved.
SUPERVISORY COMMITTEE
Keehyun Park
Yun-HoonChoung
Jae Young Choi
Chul-Ho Kim
Seon-Yong Jeong
The Graduate School, Ajou University
i
-ABSTRACT-
The Mechanism and Effects of TNF-α on Apoptosis in Auditory Cell Line
TNF-α is released in a variety of pathological states in the inner ear. Inducible NO synthase (iNOS) can be induced by cytokines and other inflammatory factors, and is generally thought to be associated with inflammation and other pathological processes in the cochlea. The purpose of the present study is to reveal that TNF-α could induce apoptosis in auditory cell line and to investigate the role of nitric oxide (NO) in TNF-α-induced auditory cell death.UB-OC1 cells and zebrafish were exposed to TNF-α. Flow cytometry, terminal deoxynucleotidyltransferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) assay, assay of mitochondrial membrane potential (MMP) and electron microscopy were used to show that TNF-α could induce apoptosis. Western blot was used to measure iNOS expression and Mitogen-activated protein kinase (MAPK) pathway.Flow cytometic analysis, TUNEL assay, MMP and electron microscopy all demonstrated that TNF-α could induce apoptosis in UB-OC1 cells. TNF-α significantly increased NO generation and iNOS expression. Pretreatment with iNOS blocker NG-methyl-L-arginine (NMA) attenuated TNF-α-induced cell death and caspase-3 activation. Also, TNF-α treatment increased p-p38 and pretreatment of NMA reduced this increased expression of p-p38. In conclusion, TNF-α can induce apoptosis in auditory cell line, and NO production in response to TNF-α is essential for apoptosis.
ii
TABLE OF CONTENTS
ABSTRACT……….……….…………...i TABLE OF CONTENTS……….……….ii LIST OF FIGURES……….……….………iv I. INTRODUCTION……….…….……….…..1II. MATERIAL AND METHODS……….…………3
1. Materials………..……….…….……3
2. Cell culture………...……..…..3
3. Cell viability assay………..…….……….…4
4. Measurement of apoptotic cells by flow cytometry…….……….…..4
5. Terminal deoxynucleotidyltransferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) assay……….…5
6. Assay of mitochondrial membrane potential (MMP)……….…..5
7. Measurement of nitrite……….……….……….…..6
8. Western blot analysis ...6
9. Effects on zebrafish model …………..………....7
10. Transmission electron microscopy (TEM) ..……..……….…...7
11. Scanning electron microscopy (SEM)………..……….8
12. Examination of neuromast in zebrafish...8
13. Statistical analyses……….……….…8
III. RESULTS………10
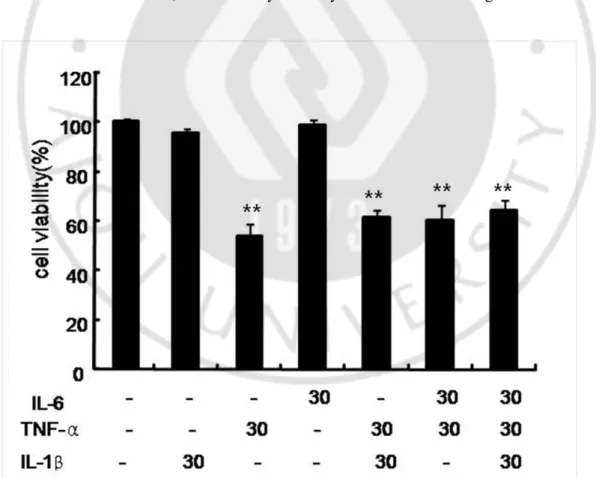
1. TNF-α decreased UB-OC1 cell viability…………..………10
2. TNF-α induced apoptosis in UB-OC1 cells………11
iii
4. TNF-α-induced ototoxicity in the zebrafish model……….…….…15
5. TNF-α-induced apoptosis in UB-OC1 cells was mediated by NO….17 IV. DISCUSSION……….…….…23
V. CONCLUSION……….……27
REFERENCES……….…….28
iv
LIST OF FIGURES
Fig. 1.Effect of proinflammatory cytokines on cytotoxicity in UB-OB1 cells ……..…10
Fig. 2.TNFα-induced apoptosis in UB-OC1 cells.……….………12
Fig. 3.Mitochondrial changes induced by TNF-α in UB-OC1 cells ………...…15
Fig. 4.Cytotoxicity induced by TNF-α in the zebrafishmodel ..………17
- 1 -
I. INTRODUCTION
Cochlear hair cells are mechanotransducers of the inner ear that are essential for hearing. Hair cell death commonly occurs following acoustic trauma or exposure to ototoxins, such as the aminoglycoside antibiotics and the antineoplastic agent cisplatin. In the process of hair cell damage, inflammatory cytokines play key roles in acute phase responses such as apoptosis and cellular stress responses. The inner ear can induce an inflammatory response to antigenor pathogens, and this response is temporally associated with degeneration of cochlear cells and eventually hearing loss (Keithleyet al., 1989; Ma CL et al., 2000).During inflammation, circulating leukocytes can penetrateinto the scala tympani from the spiral modiolar vein andits collecting venules (Harris et al., 1990). In spite of advantages of the prompt inflammatoryresponse to the organism as a whole, inflammation can damage the fine structures of the inner ear, leading to permanent hearing loss. Therefore,it would be beneficialto understand the factors andpathways that can initiate and maintain an inflammatoryresponse.
Tumor necrosis factor-alpha (TNF-α) is a proinflammatory cytokine capable of initiating apoptosis in a variety of cells. It is believed that TNF-α has the role in cell survival and apoptosis in the inner ear like in other kinds of cells. TNF-α has the ability to induceextrinsically activated apoptosis via the cell surface receptortumor necrosis factor receptor 1. Therefore, TNF-α inthe cochlea can induce apoptosis in epithelial or othercells depending on their expression of this receptor (Keithley et al., 2008). How cell survival and apoptosis occur in the inner ear has become a popular topic as new therapeutic agents are being introduced in attempts to prevent the initiation of apoptosis within the sensory receptor epithelia (Wang et al., 2007).In autoimmuneinner ear disease,blocking of the activity TNF-α hasbecome a promising therapeutic strategy due to its ability to recruit leukocytes to tissueswhere damage may occur(Cohen et al., 2005; Van Wijk et al., 2006).In the cochlea, TNF-α is released in a variety of pathological states such as vibration- and noise-induced hearing loss; it has also been associated with hearing loss following bacterial meningitis, autoimmune neurosensory hearing loss, and cisplatin ototoxicity (Aminopur et al., 2005; Chung et al., 2008; Fujika et al., 2006; So et al., 2007; Van Wijk et al., 2006;
- 2 -
Zouet al., 2005). TNF-α is secreted mainly by activated macrophages, monocytes, T cells, B cells and fibroblasts. In the cochlea, it is reported that spiral ligament fibrocytes have been shown to release chemokines including TNF-α when challenged with otitis media pathogens and are a likely source of TNF-α in vivo (Moon et al., 2006).Other inner ear structures have also demonstrated TNF-α production, including outer hair cells and supporting cells within the organ of Corti (So et al., 2007; Zou et al., 2005). TNF-α has also been reported to lead to the generation of reactive oxygen species (ROS) that function as second messengers in its signal transduction pathways including Mitogen-activated protein kinase (MAPK) pathway (Shen and Pervaiz, 2006).
As a free radical, nitric oxide (NO) is an important signaling molecule that participates in a variety of mammalian physiological events (Vanin, 1998).Many recent investigations have demonstrated that NO is important in the sensorineural hearing loss of bacterial meningitis, chronic otitis media and noise-induced hearing loss (Amaee et al., 1997; Heinrich et al., 2005; Jung et al., 2003).Three forms of NO synthase (NOS), including neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS), catalyze the formation of NO and are produced by separate NOS genes in mammals. iNOS is generally thought to be associated with inflammation and other pathological processes, such as ischemia–reperfusion injury of the cochlea (Tabuchi et al., 2001)However, the causal relationship between TNF-α and NO has not been reported in the cochlea.
The present study investigated whether TNF-α could induce apoptosis in auditory cell line and investigated the role of NO in TNF-α-induced cell death.
- 3 -
II. Materials and Methods
A. Materials
Recombinant murine TNF-α, interleukin (IL)-1β, and IL-6 were from R&D Systems (Minneapolis, MN). NO scavenger N-acetylcysteine (NAC) and iNOS blocker NG-methyl-L-arginine (NMA) were obtained from Sigma-Aldrich (St Louis, MO, USA). An anti-iNOS polyclonal antibody was from Upstate Biotechnology (Lake Placid, NY, USA). Antibodies against phospho-p53 (p-p53), phospho-extracellular signal-regulated kinase (p-ERK), phospho-c-jun N-terminal kinase (p-JNK), phospho-p38 mitogen-activated protein kinase (p-p38 MAPK) and cleaved caspase 3 were from Cell Signaling Technology (Beverly, MA, USA).
B. Cell culture
Hair cell lines have been used as important experimental tools to enhance our understanding of hair cell function. In case of UB-OC1 cell line, it has been known that the expression profiles of these molecular markers, analyzed by RT-PCR, immunolabeling, and electrophysiology, suggest that UB/OC-1 represents key, dynamic features of early hair cell differentiation (Rivolta and Holley, 2002).The cell line UB-OC1 was established from E13 Immortomouse organs of Corti, with the intention of representing early stages of hair cell differentiation. And, the establishment and characterization of the conditionally immortalized UB-OC1 cell line has been described (Rivolta et al., 1998).
UB-OC1 cells were maintained in high glucose Dulbecco’s modified Eagle’s medium (DMEM; GIBCO, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS;GIBCO) and 50 U/ml interferon (INF)-γ (Genzyme, Cambridge, MA., USA). For the experiments described below, UB-OC1 cells were cultured under permissive conditions: 33°C, 5% CO2 in DMEM supplemented with 10% FBS.
- 4 -
C. Cell viability assay
To determine cell viability, the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5
diphenyltetrazolium bromide) was performed. The MTT assay is based on the conversion of MTT into formazan crystals by living cells, which determines mitochondrial activity. Since for most cell populations the total mitochondrial activity is related to the number of viable cells, this assay is broadly used to measure the in vitro cytotoxic effects of drugs on cell lines or primary patient cells (van Meerloo J et al., 2011).
UB-OC1 cells were exposed to various concentrations of TNF-α, IL-1β, IL-6
(recombinant mouse TNF-α, IL-1β, IL-6; R&D Systems). Then,
3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; Sigma-Aldrich) was added to 40 μl of cell suspension for 4 h. After three washes with phosphate buffered saline (PBS, pH 7.4), the insoluble formazan product was dissolved in dimethyl sulfoxide (DMSO). The optical density (OD) of each culture well was measured using a microplate reader (Bio-Tek, Winooski, VT) at 560 nm.
D. Measurement of apoptotic cells by flow cytometry
The quantitative analysis of apoptotic cell death caused by TNF treatment was performed using the Annexin V- fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (BD Biosciences, San Diego, CA, USA) following the manufacturer’s protocol.
Annexins are a family of proteins with the ability to bind aminophospholipids with high affinity under the influence of calcium ions. Especially, Annexin V has a greater specificity for the membrane lipid phosphotidylserine (PS) than for other phospholipid species. In normal conditions, PS is confined almost exclusively to the inner face of the plasma membrane. However, this asymmetric distribution is lost early during apoptosis in a caspase-dependent manner. Exposure of PS on the outside of the cell membrane can be detected easily, therefore detection of PS on the outside of the cell membranecan be a
- 5 -
sensitive and early marker of apoptosis. A population of cells staining positive for annexin V and negative for propium iodide (PI) is strongly suggestive of an apoptotic mechanism of cell death (Muppidi J et al, 2004).
Briefly, UB-OC1 cells were plated at 1 x 105 cells/well in a 6-well plate, incubated for 16
h, and then treated with various concentrations of TNF-α. The cells were harvested, washed with cold PBS and subjected to Annexin V-FITC and propidium iodide staining in binding buffer at room temperature for 10 min in the dark. The stained cells were analyzed by fluorescence activated cell sorting (FACS Calibur; BD Biosciences, San Jose, CA, USA) using Win MDI 2.9 Software.
E. Terminal deoxynucleotidyltransferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) assay
TUNEL assay is a method for detecting apoptotic cells that undergo extensive DNA degradation during the late stages of apoptosis. TUNEL involves end-labeling the broken ends of the double-stranded DNA with biotin-conjugated dUTP using the enzyme terminal deoxynucleotidyltransferase (TdT)(Gavrieli et al., 1992).
After the monolayers achieved 60 - 70% confluence, the cells were exposed to medium with TNF for 48 h. Thereafter, the cells were washed with PBS and fixed in 4% paraformaldehyde. The cells were then incubated with 50 µl of TUNEL reaction mixture at 37℃ for 60 min in a humid atmosphere. The stained cells were analyzed under a fluorescence microscope (Carl Zeiss, Oberkochen, Germany).
F. Assay of mitochondrial membrane potential (MMP)
The MMP of intact cells was measured by flow cytometry with the lipophilic cationic probe 5,5 V,6,6 V-tetrachloro-1,1 V,3,3 V-tetraethylbenzimidazolcarbocyanine iodide (JC-1; Molecular Probes, Eugene, OR, USA). JC-1 is a ratiometric, dual-emission fluorescent dye that is internalized and concentrated by respiring mitochondria, thereby reflecting changes
- 6 -
in MMP in living cells. Two excitation wavelengths, 527 nm (green) for the monomer form and 590 nm (red) for the J-aggregate form, exist. With normal mitochondrial function (high MMP), JC-1 spontaneously forms complexes known as J-aggregates and the red fluorescence product is predominant. However, when mitochondrial injury occurs, MMP is generally reduced and JC-1 remains in the monomeric form with an increase in green fluorescence; accordingly, quantification of red and green fluorescent signals reflects the degree of mitochondrial damage. The culture medium was briefly removed from adherent UB-OC1 cells, and the cells were rinsed with PBS. Cell monolayers were incubated with DMEM and 5 mg/ml JC-1 at 33°C for 20 min. The cells were subsequently washed twice with cold PBS and trypsinized. Cell pellets were then resuspended in 500 μl of PBS. The change in MMP was measured by flow cytometry.
G. Measurement of nitrite
Nitric oxide (NO) is rapidly oxidized in culture medium into nitrite, which accumulates in the sample and can be easily correlated with NO production. Therefore, nitrite production was determined by adding 50 μl of the Greiss reagent to 50 μl culture supernatant (Green et al., 1982). The absorbance was measured at 540 nm, and nitrite concentrations were calculated from a sodium nitrite standard curve.
H. Western blot analysis
Total proteins were extracted using RIPA (Sigma-Aldrich) following the manufacturer’s instructions. Protein concentrations were measured using the Bio-Rad DC protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Proteins were separated by electrophoresis on 10-15% sodium dodecyl sulfate polyacrylamide gels. An equal amount of protein (10 μg) was loaded in each lane. After electrophoresis, the proteins were transferred onto polyvinylidenedifluoride (PVDF) membranes and subsequently subjected to immunoblot analysis using appropriate antibodies. The amount of loading was further checked by
- 7 -
Western blotting of a housekeeping protein (β-actin, 1:1,000 dilution; Cell Signaling Technology). Protein bands on the blots were visualized using horseradish peroxidase-labeled anti-rabbit IgG and enhanced chemiluminescence detection kit (Amersham Pharmacia Biotech, Piscataway, NJ).
I. Effect on zebrafish model
We also evaluated TNF-α toxicity using zebrafish model, because they haveneuromasts in lateral line, which are sensory organ composed of neuroepithelial clusters of hair cells, supporting cells, and integrated neurons. Although these organs are specific to fish and amphibians, they are essentially identical in ultra-structure to the neuroepithelium of the mammalian inner ear (Nicolson, 2005; Whitfield, 2002; Whitfield et al., 2002).
To determine the general toxicity and the influence of TNF-α on embryos of zebrafish (Daniorerio) was tested. One female and two male sexually mature zebrafish were maintained for a 14-h light/10-h dark photoperiod. Several rectangular mesh wire boxes were placed at the bottom of the aquaria to collect the eggs the following morning. Then, the 4 dpfzebrafish were treated with 10 ng/ml or 30 ng/ml TNF-α for 24 h.
J. Transmission electron microscopy (TEM)
After fixing UB-OC1 cells and zebrafish embryos (4 days post-fertilization, dpf) with 4% glutaraldehyde in 0.1 M sodium cacodylate (pH 7.4) + 0.001% CaCl2 for 1 h on ice, UB-OC1 cells and zebrafish were washed in cacodylate solution, dehydrated through a graded ethanol series, and infiltrated and embedded in Spurr’s epoxy resin via propylene oxide. In case of zebrafish, the larvae were oriented to obtain cross sections in a rostral to caudal manner. Semi-thin sections (2 μm) were collected and stained with 1% toluidine blue. Ultrathin sections (90 nm) were cut on an Ultracut S microtome (Leica, Wetzlar, Germany), mounted on 200-mesh Athene thin bar-grids, and contrasted with uranyl acetate and lead citrate. Grids were examined using a model EM 902A transmission electron microscope
- 8 -
(Carl Zeiss).
K. Scanning electron microscopy (SEM)
After fixing zebrafish embryos (4 dpf) in 2.5% glutaraldehyde in PBS (pH 7.2) at 4°C overnight, embryos were washed three times (5 min per wash) in distilled water and dehydrated through a graded series of 25, 50, 70, 80, 95, and 100% ethanol solution for 10 min each. Samples were then desiccated through a graded series of 25, 50, 75, and 100% isoamyl acetate in ethanol solution for 10 min. They were then dried using a critical point dryer (CPD). Dried specimens were sputter-coated twice with carbon using a MED010 evaporator (Baltec, Hudson, NH) and observed using a JSM-6700F scanning electron microscope (JEOL, Tokyo, Japan) operating at 5 or 7 kV.
L. Examination of neuromast in zebrafish
Wild type zebrafish (Dania rerio) were maintained at 28.5°C on a 14 h light/10 h dark cycle. At 4 dfp, larvae were maintained at a density of about 50 per 100 mm2 in a petri dish in embryo media (174 mMNaCl, 21 mMKCl, 12 mM MgSO4, 18mM Ca(NO3)2 4H2O, 15 mM HEPES) in a tissue incubator at 28.5°C. Then, 4 dpfzebrafish larvae were exposed to 10 and 30 ng/ml TNF-α for 4 h. Hair cell lateral line neuromasts were labeled using 2 μM YO-PRO1 (Molecular Probes) for 1 h followed by three rinses. Hair cells within neuromasts of the supraorbital (SO1 and SO2), otic (O1), and occipital (OC1) lines were analyzed. These neuromasts were selected for imaging without the need to reposition the fish during image capture. The total number of hair cells of the SO1, SO2, O1, and OC1 neuromasts were counted in each animal for all experimental and control conditions.
- 9 -
The Student’s t test and one-way ANOVA were used for the statistical analyses of the data. All statistical analyses were conducted using SPSS 12.0 statistical software (SPSS, Chicago, IL). Parameters of the data from three independent experiments are expressed as the mean ± SD. A p < 05 was considered statistically significant. (*p < 0.05; **p < 0.01; ***p < 0.001).
- 10 -
III. RESULTS
A. TNF-α decreased UB-OC1 cell viability
To confirm the direct roles of proinflammatory cytokines in the cytotoxicity in UB-OC1 cells, the MMP assay was performed. UB-OC1cells were exogenously treated with TNF-α, IL-1β, and IL-6, alone or in combination, for 48 h. The exogenous treatment with IL-1β and IL-6 alone or in combination did not significantly affect the viability of UB-OC1 cells. However, treatment with 30 ng/ml concentration of TNF-α significantly decreased cell viability (Fig. 1). These results indicate that TNF-α plays a key role in the cytotoxicity in UB-OC1 cells. Thereafter, we induced cytotoxicity in UB-OC1 cells using TNF-α alone.
- 11 -
exogenously treated with 30 ng/ml concentrations of each TNF-α, IL-1β, and IL-6 alone or in combination for 48 h. Cell viability was measured by MTT assay. *p< 0.05 and ** p < 0.01 by one-way ANOVA, compared with the media-only control group (n = 5).
B. TNF-α induced apoptosis in UB-OC1 cells
To examine whether TNF-α-induced cell death was attributable to apoptosis, the cells were exposed to 10 ng/ml or 30 ng/ml TNF-α for 48 h. Apoptosis was detected by TUNEL assay and flow cytometic analysis. The TUNEL assay showed that TNF-α increased the number of TUNEL-positive cells (Fig. 2A). To quantify the effect of TNF-α on apoptosis in UB-OC1 cells, cytometry was used. The annexin V-FITC and propidium iodide staining were used to analyzethe percentage of apoptotic cells. The number of apoptotic cellswas counted as late apoptotic cells shown in the upper right quadrant(UR) and early apoptotic cells as shown in the lower rightquadrant (LR) of the histograms. We found that TNF-α increased the number of early apoptotic cells (Fig. 2B, 2C).
- 12 -
Fig. 2.TNFα-induced apoptosis in UB-OC1 cells.A. TUNEL assay. The cells were then incubated with 50 μl of TUNEL reaction mixture (TdT and fluoresccin––dUTP) and stained with Hoechst 33258 (5 µg/ml). The stained cells were observed under a fluorescence microscope. The analysis by TUNEL assay confirmed that TNF-α induced TUNEL-positive
- 13 -
cells. Scale bar = 50 µm. B, C. To quantify TNF-α-induced apoptosis, we used flow cytometry. Cells were seeded into six-well plates at 1×105 cells/ml and incubated overnight.
10 ng/ml or 30 ng/ml TNF-α for 48 h.Annexin V-FITC and propidium iodide staining were used to analyze the percentage of apoptotic cells treated with TNF-α. These results indicate that TNF-αpromoted apoptotic cell death in concentration dependent manner. The data represent the mean ± SD of three independent experiments (* p < 0.05, ** p < 0.01).
C. TNF-α induced mitochondrial changes in UB-OC1 cells
We examined the effect of TNF-α in UB-OC1 cells to determine whether the loss of MMP could play a role in TNF-α-induced apoptosis. JC-1 is dual-emission fluorescent dye that is internalized and concentrated by respiring mitochondria, thereby reflecting changes in MMP in living cells. Two excitation wavelengths, 527 nm (green) for the monomer form and 590 nm (red) for the J-aggregate form, exist. With normal mitochondrial function (high MMP), JC-1 spontaneously forms complexes known as J-aggregates and the red fluorescence product is predominant. While mitochondrial injury occurs, MMP is generally reduced and JC-1 remains in the monomeric form with an increase in green fluorescence. TNF-α increased green cell fluorescence, indicating a loss of MMP and mitochondrial damage (Fig. 3A).
Ultrastructural analysis of TNF-α-treated UB-OC1 cells revealed morphological changes such as mitochondrial fission (arrow) indicating apoptosis-associated cytochrome c release (Fig. 3B). Compared to control, the number of small, discrete, round shape mitochondria increased significantly after TNF-α treatment.
- 15 -
Fig.3.Mitochondrial changes induced by TNF-α in UB-OC1 cells.A. MMP in TNF-α-treated UB-OC1 cells. The cells were treated with TNF-α and stained with JC-1 for visualization under the fluorescent microscope. The MMP change was objectively measured using flow cytometry. The data represent the mean ± SD of three independent experiments (* p < 0.05, ** p < 0.01). B. Electron microscopy images showing ultrastructural and morphological features of apoptosis in UB-OC1 cells exposed to TNF-α. Ultrastructural analysis of TNF-α-treated UB-OC1 cells revealed morphological changes such as mitochondrial fission indicating apoptosis-associated cytochrome c release (magnification x 12000). (Bar: 0.5 μm)
D. TNF-α-induced ototoxicity in the zebrafish model
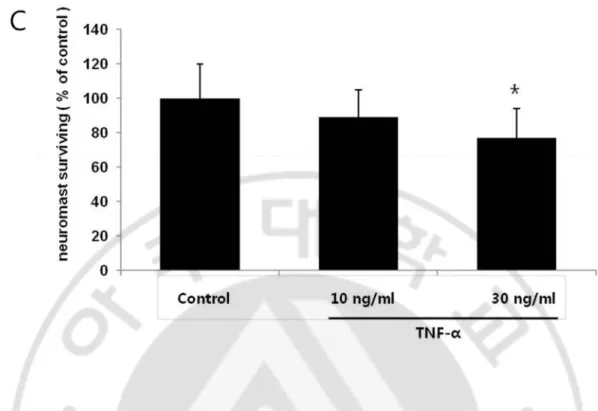
To further confirm that TNF-α could cause the morphologic changes in vivo, we usedzebrafish model. TNF-α was toxic to the neuromasts of zebrafish because the number and gross morphology of the hair cells in the neuromasts were adversely affected. In SEM and TEM, exposure to 30ng/ml TNF-α resulted in severe morphological damage such as loss or cutting of the kinocilium (Fig. 4A). Compared to control neuromasts, neuromasts from 30ng/ml TNF-α treated zebrafish showed marked damage (loss of kinocilium) to the hair cells in the neuromasts. Fig. 4B shows the expected distribution of neuromasts in live 4 dpfzebrafish as detected by staining with YO-PRO1. TNF-α treatment resulted in a significant loss of YO-PRO1 staining of neuromasts. To quantify neuromast changes after treatment with TNF-α, hair cells from four neuromasts (SO1, SO2, O1, and OC1) were counted. Treatment with 30ng/ml TNF-α significantly decreased the number of hair cells of neuromasts (Fig. 4C).
- 17 -
Fig. 4.Cytotoxicity induced by TNF-α in the zebrafish model.TNF-α was toxic to the neuromasts of zebrafish. A. In SEM (magnification x 7000) and TEM (magnification x 4000), exposure to TNF-α resulted in severe morphological damage such as loss or cutting of the kinocilium (Bar: 2μm). B. The 4 dpfzebrafish were treated with 10 ng/ml or 30 ng/ml TNF-α for 24 h. The fish were then stained with YO-PRO1. Treatment with 30 ng/ml TNF-α significantly decreased the number of hair cells of the neuromasts. C. To quantify changes in neuromasts after treatment with TNF-α, hair cells from four neuromasts (SO1, SO2, O1, and OC1) were counted. Hair cell survival was calculated as a percentage of the control group. Data bars represent mean hair cell survival (n=5 fish) ± SD.
E. TNF-α-induced apoptosis in UB-OC1 cells was mediated by NO
We first examined the contribution of NO to cytokine-induced apoptosis in UB-OC1 cells. TNF-α significantly increased NO generation in a dose dependent manner (Fig. 5A). TNF-α stimulated the expression of iNOS, suggesting that NO is involved in TNF-α-induced cell death (Fig. 5B). To clarify that cellular NO change is important in TNF-α-induced cell death,
- 18 -
we examined the effect of NAC, a scavenger of NO, on TNF-α-induced cell death by annexin V-FITC and propidium iodide staining. Pretreatment with NAC prevented TNF-α-induced cell death (Fig. 5C). Next, to determine whether this NO increase is responsible for TNF-α-induced cell death, the cells were pretreated with NMA (iNOS inhibitor). Following 24 h of exposure to TNF-α in the presence or absence of NMA, apoptotic cell death was evaluated by annexin V-FITC and propidium iodide staining. Pretreatment with NMA attenuated TNF-α-induced cell death (Fig. 5D). We also examined Western blot analysis of caspase-3 in TNF-α-induced apoptotic cell death. As shown in the Fig. 5E, cleaved caspase-3 was increased by TNF-α. However, pretreatment of NMA decreased caspase-3 activation.
In the next series of experiments, we investigated the role of MAPK subfamilies in TNF-α-induced cell death. Cells were exposed to 30 ng/ml TNF-α for 1 h in the absence or presence of NMA, and Western blot analysis demonstrated that TNF-α treatment increased expression levels of phosphorylated p38 (p-p38) and p-ERK, and pretreatment of NMA reduced this increased expression level of p-p38 and p-ERK. However, the change in the expression levels of p-JNK and p-p53 were not significant (Fig. 5F). These data suggest that NO-mediated TNF-α-induced apoptosis might be associated with activation of the p38 and ERK pathway in UB-OC1 cells.
These collective results indicate that NO production in response to TNF-α is essential for TNF-α–induced apoptosis in UB-OC 1 cells.
- 21 -
Fig. 5.Nitric oxide mediates cytokine-induced apoptosis.A. UB-OC1 cells were treated with TNF-α for 24h. TNF-α increased NO generation in a dose-dependent manner. The data represent the mean ± SD of five independent experiments (* p < 0.05, ** p < 0.01). B. UB-OC1 cells were left untreated or exposed to TNF-α for 24 h. Cell extracts were analyzed
- 22 -
by Western blotting for iNOS and actin. Blots shown are representative of n=3. C. UB-OC1 cells were left untreated or exposed to 30 ng/ml TNF-α for 24 h in the absence or 1 h pretreatment with 2.5 mM NAC. Apoptotic cell death was determined by Annexin V-FITC and propidium iodide staining. The data represent the mean ± SD of five independent experiments (* p < 0.05, ** p < 0.01). D. UB-OC1 cells were left untreated or exposed to 30 ng/ml TNF-α for 24h in the absence or 1 h pretreatment of 50 μM NMA. Apoptotic cell death was determined by Annexin V-FITC and propidium iodide staining. The data represent the mean ± SD of five independent experiments (* p < 0.05, ** p < 0.01). E. UB-OC1 cells were left untreated or exposed to 30 ng/ml TNF-α for 24h in the absence or 1 h pretreatment of 50 μM NMA. Cell extracts were analyzed by Western blotting for cleaved caspase 3 and actin. Blots shown are representative of n=3. F. UB-OC1 cells were left untreated or exposed to 30 ng/ml TNF-α for 24h in the absence or 1 h pretreatment of 50 μM NMA. Cell extracts were analyzed by Western blotting. Blots shown are representative of n=3.
- 23 -
IV. Discussion
Exposure of several cell types to the inflammation-related cytokine TNF-α has been shown to induce apoptosis (Rath and Aggarwal, 1999; Udo et al., 2001). In the pathogenesis of many types of sensorineural hearing loss (SNHL), inflammation has been implicated as a central theme. However, the inner ear appears to have evolved mechanisms for the protection from inflammatory response, such as the blood-labyrinthine barrier, which limit immune responses, in order to prevent damage to the sensitive sensory cells and neurons. For example, previous studies have found none or only a few resident leukocytes within the cochlea (Hirose and Liberman, 2003; Satoh, 1997; Takahashi and Harris, 1988). On the other hand, spiral ligament fibrocytes, outer hair cells and supporting cells within the organ of Corti have been reported to release chemokines and are a likely source of TNF-α in vivo( Moon et al., 2006; So et al., 2007; Zou et al., 2005). Taking these results into consideration, it could be postulated that cytokine-induced apoptosis is very important in the pathogenesis of SNHL. Specifically, TNF-α has been known to be associated with inner ear trauma, such as high levels of noise and vibration (Fujioka et al., 2006; Zou et al., 2005). In these settings, TNF-α produced by inner ear structures including spiral ligament fibrocytes, outer hair cells and supporting cells within the organ of Corti is pro-inflammatory and associated with apoptosis. It is suggested that apoptosis is the principal pathway of hearing loss and cell death in various kinds of hearing loss and that TNF-α can contribute to this process of apoptosis.
In the present study, we showed apoptotic cell death induced by TNF-α on molecular and morphological basis in UB-OC1 cells and zebrafish. This means that TNF-α alone could induce apoptotic cell death in the cochlea. Though these results were mainly from in vitro study, they could be supportive data for many previous studies revealing the relationship between TNF-α and the inner ear damage in hearing loss. We have chosen hair cell lines because they have provided important experimental tools to enhance our understanding of hair cell function. In case of UB-OC1 cell line, it has been known that the expression profiles of these molecular markers, analyzed by RT-PCR, immunolabeling, and electrophysiology, suggest that UB/OC-1 represents key, dynamic features of early hair cell
- 24 -
differentiation (Rivolta and Holley, 2002). We also have chosen zebrafish because neuromasts in lateral line of zebrafish are sensory organ composed of neuroepithelial clusters of hair cells, supporting cells, and integrated neurons. Although these organs are specific to fish and amphibians, they are essentially identical in ultra-structure to the neuroepithelium of the mammalian inner ear (Nicolson, 2005; Whitfield, 2002; Whitfield et al., 2002).
It has been well established that NO plays an important role in physiology of the inner ear, such as mediating neurotransmission and regulating blood flow (Takumidaand Anniko, 2002). Meanwhile, increasing evidences suggest that excessive NO production might well play an essential role in pathological damage of the cochlea, such as following inoculation of lipopolysaccharide, gentamycin, and cisplatin into the ear. The production of an inducible form of iNOS may be closely related to this phenomenon (Takumida et al., 1999; Takumida et al., 2000; Watanabe et al., 2000).
In the present study, the exposure of UB-OC1 cells to TNF-α led to iNOS and NO production. Furthermore, treatment with NAC (NO scarvenger) or NMA (iNOS blocker) significantly reduced TNF-α-induced apoptosis. These results suggest that TNF-α induced apoptosis by the expression of functional iNOS in UB-OC1 cells. The role of NO on TNF-α-induced apoptosis in UB-OC1 cell could be diverse. Firstly, NO that is trigged by iNOS reacts with free oxygen radicals, leading to the formation of the most harmful peroxynitrite anion (ONOO−), and this anion leads to lipid peroxidation, cellular damage, and apoptosis. Secondly, NO could play an important role in TNF-α-induced apoptosis by activating signaling molecules related to apoptosis. In UB-OC1 cells, TNF-α-induced apoptosis was almost completely abrogated by the NOS blocker NMA, indicating a strong dependence on NO for cytokine-induced UB-OC1 cell death. Although it cannot be excluded that NO also may induce necrotic cell death of UB-OC1 cells, the finding that NO caused activation of caspase-3 indicates that NO specifically causes activation of the classic apoptosis pathway in UB-OC1 cells.
The molecular signaling mechanisms by which TNF-α contributes to cochlear cell apoptosis is poorly understood. Tumor necrosis factor receptor 1 (TNFR1) is the membrane bound receptor for TNF-α and is expressed constitutively on most cell types, including those cells in the organ of Corti (Dinh et al., 2008; Palladino et al., 2003). TNFR1 can initiate
- 25 -
signaling cascades leading to transcription factor activation and/or cell death. The results of the present study showed that exposure of TNF-α to UB-OC1 cells led to iNOS expression and NO production. This suggests that TNF-α can induce functional iNOS by way of TNFR1 signals in UB-OC1 cells. It has been reported that iNOS expression is linked to activation of intracellular signaling proteins such as ERKs, JNKs, p38 MAPK, or PI3 K/AKT/p70S6 K (Cruz et al., 1999; Jang et al., 2004; Salhet al., 1998). Interestingly, in the present study, TNF-α induced phosphorylation of p-38 and p-ERK in UB-OC1 cells,suggesting the specificity of TNF-α to activate p-38 and p-ERK in UB-OC1 cells. These results differ from some previous findings in other cochlear cell line. Nam reported that IL-1β induced iNOS expression and NO production in House Ear Institute-organ of Corti1 (HEI-OC1) cells (Nam, 2006). In his study, IL-1β treatment induced phosphorylation of ERKs, JNKs, and p70S6 K. However, IL-1β did not induced phosphorylation of p-38 in HEI-OC1 cells. In the present study, TNF-α induced phosphorylation of ERKs and p-38. This difference may be due to different cytokines or different cell lines. In the present study, we examined the effect of IL-1β on cell viability. As you can see in Fig. 1, TNF-α, not IL-1β has significant effect on cell viability in UB-OC1 cells. In the present study, the pretreatment of iNOS inhibitor suppressed phosphorylation of ERKs and p-38, not JNKs. It could be postulated that NO induced by iNOS in turn could play a role in TNF-α-induced apoptosis by activating signaling molecules. It is reported that NO can induce apoptosis via activation of the p38 MAPK pathway (Guner et al., 2009). And, NO-induced activation of p38 MAPK appears to mediate neuronal loss in some models of ischemic brain damage (Chen et al., 2999).It is known that high levels of NO can induce energy depletion-induced necrosis. While, if energy levels are maintained, NO can induce apoptosis, via oxidant activation of: p53, p38 MAPK pathway or endoplasmic reticulum stress (Brown, 2010). High levels of NO can also trigger mitochondrial permeability transition. And, sustained permeability transition generally leads to necrotic cell death, as in this state the mitochondria can rapidly hydrolyse mitochondrial and cytosolic ATP. However, transient permeability transition can lead to apoptosis, as the mitochondria swell, rupturing the outer membrane, leading to release of cytochrome c, which triggers caspase activation, if ATP is maintained (Brown, 2010). In regard to mitochondrial change in TNF-α-induced apoptosis, loss of MMP and mitochondrial fission were found. Mitochondria play an important role in the progression of apoptosis through the release of pro-apoptotic factors, such as cytochrome c, from the mitochondrial intermembrane space. During this process,
- 26 -
mitochondrial networks are dramatically reorganised from long filamentous interconnected tubules into small punctate spheres. Also, it is known that mitochondrial fission can be induced by NO and can be blocked by NOS inhibitors (Barsoum et al., 2006).
In the present study, TNF-α induced apoptosis with NO production and iNOS expression in UB-OC1 cells. And, the pretreatment of iNOS blocker attenuated cell death and caspase-3 activation. Regarding signaling pathway, the pretreatment of iNOS blocker reduced phosphorylation of p-38 induced by TNF-α. We think that NO production by iNOS enhanced by TNF-α plays a crucial role in TNF-α-induced apoptosis in UB-OC1 cells. Moriyama et al. reported TNF-α induced NO production and NOS inhibitor suppressed NO production in cultured spiral ligament fibrocytes (Moriyama et al., 2007). They claimed that TNF-α may act on spiral ligament fibrocytes to produce NO, and this NO could damage the cochlear tissue. We believe that TNF-α can act on cochlear hair cells to produce NO, and this NO could damage the cochlear tissue.
Recently, anti-TNF-α therapy has changed the progress of several immune-related diseases, such as rheumatoid arthritis, psoriasis or Crohn’s disease. In terms of SNHL, there are some reports on the beneficial effects of the anti-TNF-α therapy in immune-mediated hearing loss (Rahmanet al., 2001; Van Wijk et al., 2006;Wang et al., 2003). Furthermore, in cisplatin exposed rats, intraperitoneal injections of the anti-TNF-α receptor antibody Etanercept reduced concentrations of inflammatory cytokine protein and mRNA in the serum and cochleae (Van Wijk et al., 2006). Based on these results and the result of the current study, it is possible that anti-TNF-α therapy could be beneficial to inner ear damage in acute SNHL.
- 27 -
V. Conclusion
In conclusion, the present study provides evidence that NO contributes to TNF-α-induced apoptosis.NO-mediated TNFα-induced apoptosis is associated with activation of the p38 and ERK pathways in UB-OC1 cells. These results might add to understanding of the role of NO in cytokine-induced apoptosis in the inner ear. Furthermore, these results support that anti-TNF-α therapy could be beneficial in inner ear damage.
- 28 -
REFERENCES
1. Amaee FR, Comis SD, Osborne MP, Drew S, Tarlow MJ. Possible involvement of nitric oxide in the sensorineural hearing loss of bacterial meningitis.ActaOtol 117:329–336, 1997
2. Aminpour S, Tinling SP, Brodie HA. Role of tumor necrosis factor-alpha in sensorineural hearing loss after bacterial meningitis. OtolNeurotol 26(4):602–609, 2005
3. Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waqqoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide induced mitochondrial fission is regulated by dynamin-related GTPases in neurons.EMBO J 256:3900–3911, 2006
4. Brown GC. Nitric oxide and neuronal death. Nitric Oxide 23:153-165, 2010
5. Chen M, Sun HY, Li SJ, Das M, Kong JM, GaoTM. Nitric Oxide as an Upstream Signal of p38 Mediates Hypoxia/Reoxygenation-Induced Neuronal Death. Neurosignals 17:162–168, 2009
6. Chung WH, Boo SH, Chung MK, Lee HS, Cho YS, Hong SH. Proapoptotic effects if NF-kappaB on cisplatin-induced cell death in auditory cell line.ActaOtolaryngol 128(10):1063–1070, 2008
7. Cohen S, Shoup A, Weisman MH, Harris J. Etanercept treatmentfor autoimmune inner ear disease: results of a pilot placebocontrolledstudy. OtolNeurotol 26:903-907, 2005
8. Cruz MT, Duarte CB, Goncalo M, Carvalho AP, Lopes MC. Involvement of JAK2 and MAPK on type II nitric oxide synthase expression in skin-derived dendritic cells.Am J Physiol 277:1050–1057, 1999
9. Dinh CT, Haake SM, Chen S, Hoang K, Nong E, Eshraghi AA, Balkany TJ, Van De Water TR. Dexamethasone protects organ of corti explants against tumor necrosis factor-alpha-induced loss of auditory hair cells and alters the expression levels of apoptosis-related genes. Neuroscience 157:405–413, 2008
- 29 -
Proinflammatory cytokines expression in noise-induced damaged cochlea. J Neurosci 83(4):575–583, 2006
11. Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation.J Cell Biol119:493-501, 1992
12. Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal Biochem 126:131–138, 1982
13. Guner YS, Ochoa CJ, Wang J, Zhang X, Steinhauser S, Stephenson L, Grishin A, Upperman JS. Peroxynitrite-induced p38 MAPK pro-apoptotic signaling in enterocytes. BiochemBiophys Res Commun 384: 221-225, 2009
14. Harris JP, Fukuda S, Keithley EM. Spiral modiolar vein: its importancein inner ear inflammation.ActaOtolaryngol110:357-365, 1990
15. Heinrich UR, Selivanova O, Feltens R, Brieger J, Mann W. Endothelial nitric oxide synthase upregulation in the guinea pig organ of Corti after acute noise trauma. Brain Res 1047:85-96, 2005
16. Hirose K, Liberman MC. Lateral wall histopathology and endocochlear potential in the noise-damaged mouse cochlea.JARO4:339–352, 2003
17. Jang BC, Kim DH, Park JW, Kwon TK, Kim SP, Song DK, Bae JH, Mun KC, Baek WK, Suh MH, Hla T, Suh SI. Induction of cyclooxygenase-2 in macrophages by catalase: Role of NF-κB and PI3K signaling pathways. BiochemBiophys Res Commun 316:398–406, 2004
18. Jung TT, Llaurado RJ, Nam BH, Park SK, Kim PD, John EO. Effects of nitric oxide on morphology of isolated cochlear outer hair cells: possible involvement in sensorineural hearing loss.OtolNeurotol 24:682–685, 2003
19. Keithley EM, Woolf NK, Harris JP. Development of morphologicaland physiological changes in the cochlea induced by cytomegalovirus.Laryngoscope 99:409-414, 1989
20. Keithley EM, Wang X, Barkdull GC. Tumor Necrosis Factor -α Can Induce Recruitment ofInflammatory Cells to the Cochlea.OtolNeurotol 29:854-859, 2008 21. Ma CL, Billings PB, Harris JP, Keithley EM. Characterization ofan experimentally
induced inner ear immune response.Laryngoscope110:451-456, 2000
- 30 -
fibrocytes release chemokines in response to otitis media pathogens. ActaOtolaryngol 126(6): 564–569, 2006
23. Moriyama M, Yoshida K, Ichimiya I, Suzuki M. Nitric oxide production from cultured spiral ligament fibrocytes: effects of corticosteroids.ActaOtolaryngol 127:676-681, 2007
24. Muppidi J, Porter M, Siegel RM. Measurement of apoptosis and other forms of cell death.CurrProtocImmunol59:3.17.1-3.17.36, 2004
25. Nam SI. Interleukin-1β up-regulates inducible nitric oxide by way of phosphoinositide 3-kinase-dependent in a cochlear cell model. Laryngoscope 116(12):2166-2170, 2006
26. Nicolson T. The genetics of hearing and balance in zebrafish. Annu Rev Genet 39:9-22, 2005
27. Palladino MA, Bahjat FR, Theodorakis EA, Theodorakis EA, Moldawer LL. Anti-TNF-α therapies: the next generation. Nat Rev 2:736–746, 2003
28. Rahman MU, Poe DS, Choi HK. Etanercept therapy for immune-mediated
cochleovestibular disorders: preliminary results in a pilot study.
OtolNeurotol22(5):619-624, 2001
29. Rath PC, Aggarwal BB. TNF-induced signaling in apoptosis. J ClinImmunol 19:350–364, 1999
30. Rivolta MN, Grix N, Lawlor P, Ashmore JF, Jagger DJ, Holley MC. Auditory hair cell precursors immortalized from the mammalian inner ear.ProcBiolSci 265:1595–1603, 1998
31. Rivolta MN, Holley MC. Cell lines in inner ear research. J Neurobiol53(2):306-318, 2002
32. Salh B, Wagey R, Marotta A, Tao JS, Pelech S. Activation of phosphatidylinositol 3-kinase, protein kinase B, and p70 S6 kinases in lipopolysaccharide-stimulated Raw 264.7 cells: differential effects of rapamycin, Ly294002, and wortmannin on nitric oxide production. J Immunol161:6947–6954, 1998
33. Satoh H. Anti-glomerular basement membrane antibody-induced inflammation in rat cochlear plexus. ActaOtolaryngol117:80–86, 1997
34. Shen HM, Pervaiz S. TNF receptor superfamily-induced cell death: redox-dependent execution.Faseb J 20:1589–1598, 2006
- 31 -
Moon SK, Lim DJ, Park R. Cisplatin cytotoxicity of auditory cells requires secretions of proinflammatory cytokines via activation of ERK and NF-κB.J Assoc Res Otolaryngol 8:338–355, 2007
36. Tabuchi K, Tsuji S, Asaka Y, Hara A, Kusakari J. Ischemia–reperfusion injury of the cochlea: effects of an iron chelator and nitric oxide synthase inhibitors.Hear Res 160:31–36, 2001
37. Takahashi M, Harris JP. Anatomic distribution and localization of immunocompetent cells in normal mouse endolymphatic sac.ActaOtolaryngol 106:409–416, 1998
38. Takumida M, Popa R, Anniko M. Free radicals in the guinea pig inner ear following gentamicin exposure.J Otorhinolaryngol 61:63–70, 1999
39. Takumida M, Anniko M, Popa R, Zhang DM. Lipopolysaccharide-induced expression of inducible nitric oxide synthase in the guinea pig organ of Corti.Hear Res 140:91–98, 2000
40. Takumida M, Anniko M. Nitric oxide in the inner ear.CurrOpinNeurol 15:11–15, 2002
41. Udo KM, Pereda-Fernandez C, Manderscheid M, Pfeilschifter J. Dexamethasone inhibits TNF-α-induced apoptosis and IAP protein down regulation in MCF-7 cells. Br J Pharmacol133: 467–476, 2001
42. Vanin AF. Biological role of nitric oxide: history, modern state, and perspectives for research.Biochemistry 63:731–733, 1998
43. vanMeerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Methods MolBiol 731: 237-245, 2011
44. vanWijk F, Staecker H, Keithley E, Lefebvre PP. Local perfusionof the tumor necrosis factor alpha blocker infliximab to the innerear improves autoimmune neurosensory hearing loss. AudiolNeurootol11:357-365, 2006
45. Wang J, Ruel J, Ladrech S, Bonny C, van de Water TR, Puel JL. Inhibition of the c-Jun-N-terminal kinase-mediated mitochondrial cell death pathway restores auditory function in sound exposed animals. MolPharmacol 71(3):654–666, 2007 46. Wang X, Truong T, Billings PB, Harris JP, Keithley EM. Blockage of
immune-mediated inner ear damage by etanercept.OtolNeurotol 24(1):52-57, 2003 47. Watanabe K, Hess A, Bloch W, Michel O. Expression of inducible nitric oxide
- 32 -
cisplatin.Anticancer Drugs 11:29–32, 2000
48. Whitfield TT. Zebrafish as a model for hearing and deafness. J Neurobiol 53(2):157-171, 2002
49. Whitfield TT, Riley BB, Chiang MY, Phillips B. Development of the zebrafish inner ear. DevDyn223(4):427-458, 2002
50. Zou J, Pyykkö I, Sutinen P, Toppila E. Vibration induced hearing loss in guinea pig cochlea: expression of TNF-α and VEGF. Hear Res 202(1–2):13–20, 2005
- 33 - - 국문요약 -
종 양 괴사 인자 가청 각세 포주 의세 포 자멸 사 에미 치는 영향 및기 전
아주대학교대학원의학과이비인후과 박헌이 (지도교수: 박기현) 염증반응은우리몸의중요한방어기전이지만, 염증반응과정중에정상조직에해를미칠수있다. 소음성난청, 이독성난청등여러병적조건하에서내이에서염증반응이발생함이보고되었으며, 이로인하여청각세포의손상이유발되어청력저하를일으키게된다. 이러한이유로인해염증반응을유발하거나유지시킬수있는요인과경로를이해하는것은 난청을치료하는데많은도움이될것이다. 이에본연구에서는염증성사이토카인이청각세포에미치는영향과기전에대해알아보고 자하였다. 연구재료로청각세포주(UB-OC1 cells)와제브라피시(zebrafish)를이용하였으며, 염증성사이토카인에노출시킨후, 세포자멸사가일어나는지확인하였다. UB-OC1 cells 에서종양괴사인자에의해세포사멸이유발됨을확인하였다. 종양괴사인자에의한세포자멸사가유발되었는지확인하기위하여유세포분석기(Flow cytometry), TUNEL assay, 미토콘드리아막전위측정(assay of mitochondrial membrane potential)을하였으며, 전자현미경을이용하여세포자멸사를형태학적으로확인하였다. 또한, 제브라피시에서측선의유모세포가종양괴사인자에의해손상됨을확인하였다. 종양괴사인자에의한청각세포의세포자멸사에서산화질소(Nitric oxide)의역할을알아보기위해서산화질소와산화질소합성효소(nitric oxide synthase)의발현을측정하였으며, 종양괴사인자가산화질소와산화질소합성효소의발현을증가시킴을확인하였다. 또한, 산화질소합성효소를억제하였을때, 종양괴사인자에의한세포자멸사가억압됨을확인하였다.- 34 -
이러한결과들은종양괴사인자가청각세포주에서세포자멸사를유발하며,
종양괴사인자에의한산화질소의발생이세포자멸사에서중요한역할을한다는것을알려 준다.