Augmentation of Stress Induced Gastric
Mucosal Damages after Helicobacter
pylori Infection; Mechanisms and
Strategy for Prevention
Augmentation of Stress Induced Gastric
Mucosal Damages after Helicobacter
pylori Infection; Mechanisms and
Strategy for Prevention
. . 2 , , . . . , , , . . . Proteome , EMSA . , 1 . , , , . , .
Augmentation of stress induced gastric mucosal
damages after Helicobacter pylori infection;
Mechanisms and strategy for prevention
by
Tae Young Oh
A Dissertation Submitted to The Graduate School of Ajou University in Partial Fulfillment of the Requirements for the Degree of
DOCTOR OF PHILOSOPHY
Supervised by
Ki-Baik Hahm, M.D., Ph.D.
Department of Medical Sciences The Graduate School, Ajou University
ABSTRACT
-Augmentation of Stress Induced Gastric Mucosal
Damages after Helicobacter pylori Infection;
Mechanisms and Strategy for Prevention
Purpose: Among the several environmental factors which can influence the
outcome of Helicobacter pylori (H. pylori) infection, stress might be one of the prime effectors responsible for stress ulceration in the stomach. However, the exact underlying mechanisms to explain why gastric lesions are augmented after stress in the presence of H. pylori infection and how stress affects the outcomes of H. pylori infection are not clearly documented yet. This study was aimed to reveal how the stress does influence the severity of H. pylori-related gastric mucosal damage and if so, to dig out the underlying molecular mechanism, and to search the proper strategy to prevent the disaster.
Materials & Methods: In order to establish the H. pylori-associated gastritis
model, we infected the Sprague-Dawley rats with SS1 strain of H. pylori (VacA+, CagA+). After the six months of infection, the presence of H. pylori in the stomach was identified. The water immersion restraint stress (WIRS) was imposed on the half of rats at 24 weeks after infection. Rats were restrained in a wire-stress cage for 30, 120 and 480 min at 21 water immersion state. After sacrificing rats with ethyl ether inhalation, stomach was immediately
isolated for gross and microscopic lesion scores and parts of the stomach were preserved for the measurement of MDA, iNOS, IL-1 , IL-2, IL-6, IL-10, TNF- , and IFN- . RNase protection assays (RPA) were done for looking at the transcriptional changes of cytokines and chemokines. Electrophoretic mobility shift assay (EMSA) was done for measuring the DNA binding activity of NF- B. In
vitro experiments were coincided with animal study. Cultured gastric mucosal
cells were either administered with H. pylori or subject to receive heat shock (43 , 5 min). Using extracted proteins, polyacrylamide gel electrophoresis or 2-dimensional gel electrophoresis were done, transferred to nitrocellular paper, and blotted with several heat shock protein (HSP) antibodies to look at the change of HSP after H. pylori or stress and both. Animal experiments were repeated to observe the protection against WIRS by antioxidants.
Results: Stress itself acted as augmenting factor in H. pylori-associated gastritis,
based on the finding that bleeding rates and bleeding index were significantly increased after both H. pylori infection and stress compared to stress alone without H. pylori infection. Significantly higher levels of oxidative stress and Th1 type lymphocyte responses were observed in group with both stress and H.
pylori infection. Significantly elevated levels of TBA-RS and iNOS were
observed in both WIRS and H. pylori infected group compared to WIRS or H.
pylori infection alone group. HSP27 and HSP70 acted as protective protein and
phosphorylation of HSP90 critically contributed to augmented gastric mucosal damage. Antioxidant pretreatment significantly attenuated the stress-associated gastric mucosal lesions.
severity of H. pylori-associated gastric lesions. The presence of H. pylori contributed to significant deterioration of stress-associated gastric mucosal lesion. HSP dyregulation and oxidative stress might be the responsible mechanism for that. Antioxidants could be considerated as the strategy to prevent the augmentation of gastric lesion by H. pylori infection and stress.
Key Words: Helicobacter pylori, Stress, Water immersion restraint stress,
TABLE OF CONTENTS
TITLE PAGE --- 1 ABSTRACT --- 2 TABLE OF CONTENTS --- 5 LIST OF FIGURES --- 7 LIST OF TABLES --- 9 . INTRODUCTION --- 10. MATERIALS AND METHODS --- 15
A. Materials --- 15
1. Animals --- 15
B. Methods --- 15
1. H. pylori Culture --- 15
2. Infection with H. pylori --- 16
3. Water Immersion Resraint Stress --- 16
4. Gross and Histological Observation --- 17
5. TBA-reactive Substance --- 18
6. Cytokine Measurment --- 18
7. 2-D Gel Electrophoresis --- 19
8. Cell Culture on Heat Shock Induced Stress --- 19
9. Western Blotting --- 20
10. RNase Protection Assay --- 21
12. Stastical Assay --- 22 . RESULTS --- 23 . DISCUSSION --- 49 . CONCLUSION --- 56 BIBLIOGRAPHY --- 57 --- 69
LIST OF FIGURES
Fig. 1. Bleeding index of WIRS-induced gastric mucosa in rats with or without
H. pylori infection. --- 26
Fig. 2. Bleeding rates (%) of WIRS-induced gastric mucosa in rats with or without H. pylori infection. --- 27 Fig. 3. Microscopic observation of WIRS-induced gastric mucosa in rats with or
without H. pylori infection after stress loaded 480 min. --- 28 Fig. 4. Changes of TBA-RS on WIRS-induced gastropathy in rats with H. pylori
infection. --- 31 Fig. 5. (A) Changes of IFN- on WIRS-induced GMD in rats with H. pylori
infection. (B) Changes of TNF- on WIRS-induced GMD in rats H.
pylori infection. --- 33
Fig. 6. Change of proteomic profiles at 30 (A), 120 (B) and 480 (C) min on WIRS-induced GMD in H. pylori-infected rats. --- 34 Fig. 7. Detection of various HSPs expression in cultured AGS cells by Western
blot analysis. --- 36 Fig. 8. 2 D-gel electrophoresis. --- 37 Fig. 9. Activation of HSP90, HSP70, HSP60 and HSP27 in cultured AGS cells.
--- 38 Fig. 10. Representative macroscopic findings of the gastric mucosa of rats.
--- 42 Fig. 11. Microscopic observation of WIRS-induced GMD in H. pylori-infected
rats. --- 43 Fig. 12. Effect of antioxidant on HSPs expressions according to group.
--- 44 Fig. 13. Results of RNase protection assay (RPA) for cytokine (rCK-3).
--- 45
Fig. 14. Results of RNase protection assay (RPA) for apoptosis (rAPO). --- 46
Fig. 15. Effect of NF-κB by DA-9601 on WIRS-induced GMD in rats with H.
LIST OF TABLES
.
INTRODUCTION
Stress ulcer is a highly prevalent clinical entity, especially encounted frequently in emergency department because urgent symptoms like severe dyspepsia and upper gastrointestinal bleeding bothered the patients. Usually stress ulcer is described as acute gastric mucosal lesion (AGML) since its underlying fundamental mechanisms are mostly based on the disruption of the balance between the mucosal defense system and the mucosal offense system. Structural elements of gastric mucosal defense include the mucus and epithelial cell barrier, and physiological elements of protection during the acute phase of the injury involve mucin production, bicarbonate ion secretion and gastric mucosal microcirculation.1 Experimentally stress-induced AGML is due, in large part, to either gastric acid hypersecretion or the tremendous and prompt reduction in mucosal blood flow.2,3 Other factors, which contribute to AGML (stress ulceration) are decreases in prostaglandin synthesis, cytokine liberation, and the disruption of cellular restitution and repair mechanisms.4
Since we cannot provoke AGML in human, to develop the appropriate experimental models is necessitated and the model of water immersion restraint stress (WIRS) is most popularly used in these investigations.5 This model appears to be very suitable especially in testing various factors affecting the formation and healing of gastric mucosal lesion (GML).6,7 An important role in damage and protection of this barrier is played by gastric microcirculation. Disturbance in blood perfusion of gastric mucosa result in the formation of
erosions and ulcers. This phenomenon typically occurs in experimental model of stress induced or ischemic gastric lesions.8
In clinical study, stress ulceration of the stomach was first described in burned patients in 1842.9 Gastric mucosal lesions develop in 91% of patients suffering head trauma.10 The incidence of stress ulceration is approximately 82% in general surgical patients admitted to an intensive care unit (ICU). Stress gastritis is usually presented as upper gastrointestinal bleeding in critically ill patients due to severe physiological stress.11 The syndrome of stress-related GML of the gastrointestinal tract was first described in 1971 by Lucas et al. who termed this 'stress-related erosive syndrome'.12 Harvey Cushing first described the association between CNS injury and gastroduodenal disease in 1932.13 Gastric mucosal damage generally results from an imbalance between aggressive factors (acid, pepsin, mucosal hypoperfusion, ischemic- reperfusion injury, intramucosal acid-base balance, systemic acidosis, free radicals, bile salts, Helicobacter pylori and NSAIDs) and defense factors (mucosal prostaglandins, mucous bicarbonate barrier, epithelial restitution and regeneration, mucossal blood flow and cell membrane and tight junctions).14 Stress ulcer also might be caused by disequlibrium between these two factors, but the exact underlyng mechanisms are inferred and not clarified clearly yet.
However, the discovery that Helicobacter pylori (H. pylori) is a major cause of peptic ulcer and even gastric cancer has tempted many to conclude that psychological factors are unimportant. But this is dichotomised thinking. There is solid evidence that psychological stress triggers many ulcers and impairs response to treatment, while H. pylori is inadequate as a mono-causal explanation as most
infected people do not develop ulcers. Simply H. pylori is inadequate as a sole explanation for various gastric disease including peptic ulcer disease and gastric cancer. H. pylori alone does not explain fully the epidemiological patterns of upper gastrointestinal disease.
By the way, psychological stress is not only empirically associated with ulcers, but is a very plausible risk factor for ulcer disease. As plausible explanations for this, people affected by stress may also smoke more, sleep less, and take more NSAIDs, thereby increasing their susceptibility to ulcer by mechanisms that are not related to gastric acidity. Compared to health people, patients with duodenal ulcers are particularly likely to response to laboratory stressors by secreting more acid. Yamamoto et al. published interesting finding suggestive of the relationship between H. pylori infection and stress-induced gastric mucosal injury.15 Moreover, several epidemiologic evidences stress the contribution of stress on H. pylori-associated gastric pathologies. Immediately after the Great Hanshin Earthquake in Kobe in 1995, the recurrence rate of peptic ulcer in patients infected with H. pylori was significantly higher than that in patients in whom H. pylori had been eradicated.16 The recurrence rate of duodenal ulcers was 93% in patients infected with H. pylori, but was zero in H.
pylori-eradicated patients.
Other very conclusive evidence showing the contribution of environmental stress on gastric carcinogenesis came from the report of Cho et al..17 They extend their investigation by comparing the incidence rate of stomach cancer among the same three ethnic groups in the state of Illinois. The incidence of stomach cancer was observed to be the lowest in whites (22.5/100,000),
intermediate in African (28.2/100,000), and highest (62.6/ 100,000) in immigrant Koreans. The highest rate of stomach cancer in immigrant Koreans indicates profound genetic-environmental interaction. According to the study by Van der Voort et al., the severity of gastric and duodenal mucosal injury in critically ill patients during mechanical ventilation is significantly correlated with the presence of H. pylori infection.18
The existence of H. pylori itself, however, is not enough for the development of peptic ulcer, because most infected people never develop ulcer diseases. Chronic use of NSAIDs is also considered to be an independent risk factors, but there are still many patients with peptic ulcer who do not have H.
pylori infection or use NSAIDs.19-21 These findings strongly suggest that other factors, such as stress, diet or smoking, may contribute to the pathogenesis of peptic ulcer.22 An association between peptic ulcer and physical stresses such as trauma or burn is well established.23,24 There is also revelent evidence that psychological stress causes ulcers and impairs the patient's response to the treatment.25,26 Additive effects of H. pylori infection on stress-induced gastric ulcer were also ssuggested, because the prevalence of H. pylori infection in ulcer patients after the earthquake was much higher than in ulcer-free controls living in same area.16
Based on these backgrounds that the simple presence of H. pylori in the stomach might predispose to augmented response after stress, we performed the current study to 1) reveal whether the H. pylori infection could affect the severity of AGML after stress in animal model, 2) what is underlying mechanism for the augmented response after the both H. pylori infection and
stress, and 3) to provide the effective strategy to prevent the disaster of H.
pylori infection on the stomach after stress. The following experiments including
animal experiment and in vivo experiment including 2D-gel electrophoresis, Western blot, RNase protection assay, and histopathological evaluations were done.
.
MATERIALS and METHODS
A. Materials
1. Animals
Six week-old specific-pathogen-free (SPF) Sprague-Dawley rats (male, Charles River Japan) were used for experiments. They were fed on sterilized commercial pellet diets (Biogenomics Co., Seoul, Korea) and given sterile water
ad libitum. A total of 200 rats were maintained. They were housed in an
air-conditioned biohazard room designed for infectious animals, with a 12 h L:12 h D cycle.
B. Methods
1. H. pylori Culture
H. pylori (SS1 strain) known to fulfil the Lausanne criteria27 were kindly provided in a frozen state by Prof. Adrian Lee.28 Bacteria with the typical S shape, Gram-negative rods with modified Gram stain, and positive oxidase, urease and catalase were all presumed to be H. pylori. They were stored in liquid nitrogen and infected in the mice after cultivation. For the liquid culture, 6 days before inoculation 150 of H. pylori isolates were inoculated into Brain Heart Infusion (BHI) broth supplemented with 10% sheep blood, 5% horse serum, and Skirrow's supplement in an anaerobic jar with the micro-aerophilic
gas-generating kit CampyPack-Plus (Becton Dickinson, USA) at 37 and 5% O2/10% CO2/85% N2 for 5 days. Cultures used for dosing the mice were grown
under micro-aerophilic conditions for 4 days in brain heart infusion broth (Difco, CA) supplemented with 10% foetal calf serum and 5% sheep whole blood in a shaking incubator.
2. Infection with H. pylori
The rats were inoculated with a bacterial suspension, a single intragastric dose of a 1 ×107 CFU/mL, obtained from 4-day liquid cultures of SS1 strain. The rats were dosed three times for a 3 day period with 100 of bacterial suspension (approximately 1×106 CFU) by oro-gastric tube. They received three inoculations over a period of 3 days. Control rats were given same amout of normal saline and were housed in isolators in order to prevent risk of infection.
3. Water Immersion Restraint Stress
Twenty four weeks after inoculation of H. pylori, gastric mucosal lesions induced by WIRS in rats. The animals were deprived of food, but allowed free access to water 24 h before insult of WIRS. Rats were places in strain cage and immersed in water (21 ) for 30, 120 or 480 min. Animals were sacrificed immediately after the end of water immersion. Stomachs were removed and opened along the greater curvature, followed by rinsing with phosphate-buffered saline. For RNA and protein extraction, the stomachs were dipped into RNA later solution and quickly frozen in liquid nitrogen and stored at 80 until extraction. For, histological examination, gastric tissue was fixed in 10% buffered
formalin. We divided the animals into five groups, that is, control group only with WIRS, H. pylori infection group with WIRS, two other groups orally administered DA-9601 6 or 20 mg/kg, and final group pretreated with -tocopherol 40 mg/kg.
4. Gross and Histological Observation
Infected rats were killed by cervical dislocation at 24 weeks after H. pylori inoculation. The stomach was placed in 10% buffered formalin and embedded in paraffin, and 4 sections were cut. In order to determine the presence of H.
pylori and for pathological findings, haematoxylin and eosin (H&E) staining and
Warthin-Silver stainings were employed on formalin-fixed sections. According to the criteria of Yamamoto et al. (Table 1), bleeding index was evaluated on formalin-fixed section and the bleeding rate (%) was calculated.15
Table 1. Criteria of bleeding index
Bleeding index Criteria
0 no bleeding at all
1 mild bleeding
(small amounts of coagula in the stomach) 2 moderate bleeding
(intermediate between 1 and 3 points)
3
severe bleeding
(contents of the stomach were filled with blood in duding coagula)
5. TBA-reactive Substance
The concentration of thiobarbituric acid-reactive substance (TBA-RS), an index of lipid peroxidation, was measured in the gastric mucosal homogenates using the method of Ohkawa et al..2 9 The level of TBA-RS in the mucosal homogenate was expressed as nM of malondialdehyde per mg of protein using 1,1,3,3-tetramethoxypropane as a standard.
6. Cytokine Measurement
The concentration of IFN- and TNF- in the gastric tissue of rat was measured by enzyme-linked immunosorbent assay (ELISA). First, 2 ml of phosphate-buffered saline (PBS) containing 0.1% sodium dodesylsulfate (SDS) and 0.1% Tween 20 was added to each frozen specimen. The specimen was
then homogenized for 90 sec in cold water. After overnight storage at 20 , it was centrifuged at 400 g for 15 min at 4 . The supernatant was analysed with a Biotrak Rat Cytokine ELISA System (Amersham International, Buckinghamshire, England). Results were expressed in terms of concentration per mg of protein. SPECTRAmax 250 (Molecular Devices, Sunnyvale, CA, USA) was used as the ELISA reader, and protein was measured using Bicinchoninic acid (BCA) Protein Assay Reagents (Pierce Chemical, Rockford, IL, USA).
7. 2-Dimensional Gel Electrophoresis
Cultures grown in liquid media to mid-log phase were pelleted, washed once in 1 ml 0.9% NaCl, and frozen at 80 . Cell pellets were then treated with urea-SDS sample buffer, including DNase and RNase, according to procedures of the manufacturer (Genomic Solution, ESA), and 2-D gel analysis was performed as described.30 The ESA isoelectric focusing system with pH 3-10 ampholines was used to generate gradients ranging approximately from pH 4.5 to 6.5. Alternatively, the Amersham IPGphor system was used with 18 cm IPG strip gels generating a nonlinear pH gradient of 3 to 10. The second dimension SDS-PAGE gel 11.5% Duracryl separated proteins over the range of molecular weight from 10 to 80 kD. Proteins were stained with Silver stain or Coomassie blue.
8. Cell Culture on Heat Shock Induced Stress
Human gastric adenocarcinoma (AGS) cells were purchased from American Type Culture Collection (ATCC, Rockville, MD). They were cultured for
fluorescence microscopy on glass coverslips or for micro injection studies on etched grid cover slips marked by numbers and letters (Bellco, Vineland, NJ) in Ham's F12 medium (Gibco BRL, Gaithersburg, MD) supplemented with L-glutamine, with 10% fetal calf serum (FCS, HyClone, Logan, UT), antibiotics and antimycotics in a humidified 5% CO2 atmosphere. The H. pylori were
washed with PBS (0.01 M NaH2PO4/ Na2HPO4, 145 mM NaCl, pH 7.2) and
harvested in F12 medium without FCS and antibiotics/antimyotics. Bacterial concentration was adjusted to optical density 0.5 using wave length of 565 nm. AGS cells were transferred to medium without FCS, antibiotics and antimycotics about 15 min before bacteria were added (1 ml H. pylori / 2 ml media) and the mixture was incubated at 37 for time period indicated usually 2 h. Cells were induced heat stress 43 for 30 min.
9. Western Blotting
Each frozen gastric mucosa was homogenized in ice-cold 20 mM Tris-HCl buffer, pH 7.5 containing 2 mM EDTA, 0.5 mM EGTA, 300 mM sucrose, 2 mM phenylmethyl sulfonyl fluoride (PMSF) with a tissue tearer at 4C. Protein concentrations were determined by Bio-rad reagent (Bio-rad, Hercules, CA) using bovine serum albumin as standard. Ten microgram of protein from the homogenates was denatured in the sample buffer and subjected to electrophoresis on an 8 % Tris-glycine gel and transferred onto polyvinylidene difluoride membranes (PVDF). The blots were pretreated with Tris-buffered saline containing 5 % nonfat dry milk, 1 % albumin and 0.1 % Tween 20 and then incubated overnight at 4 with antibodies for COX-1 (SC1754, Santacruz
Biotech, Santa Cruz, CA), COX-2 (SC1756, Santacruz Biotech, Santa Cruz, CA) and 3,3'-diaminobenzidine reagents. Filters were washed three times and incubated with a horseradish peroxidase-conjugated secondary antibody against goat IgG, developed using a commercial enhanced chemiluminosense system (ECL), and exposed to films.
10. RNase Protection Assay (RPA)
The RPA is a highly sensitive and specific method for the detection and quantitation of mRNA species. Gene expressions of IFN- , TNF- , GM-CSF, TGF- 1, TGF- 3, TGF- 2, LT, TNF, MIF, IFN were examined by RNase protection assay with two housekeeping gene products, L32 and GAPDH in ethanol-induced GMD rat using rck-3 Multi-probe Template set (BD, PharMingen, San Diego, CA). Total RNA was extracted from the gastric mucosa by the Trizol method (Life technologies, Gibco BRL) according to the manufacturer's protocol and quantified spectrophotometrically by absorption at 260 nm and 280 nm. Total RNA (20 mg) was labeled with [3 2P] UTP (Amersham Pharmacia Biotech, Buckinghamshire, UK), using T7 RNA polymerase according to the manufacturer's description, and hybridized at 42 overnight with 1105 cpm of riboprobe, and then digested with RNase A cocktail. The reaction products were resolved on 5 % acrylamide gel and analyzed after 72 h by autoradiography.
11. Electrophoretic Mobility Shift Assay (EMSA)
and V. Nuclear proteins (10 mg) were incubated for 30 min at 25 with 20 pg of 32P-labeled oligonucleotides containing the NF- B binding site (GATCGAGGGGGACTTTCCCAGC) and 1 g of poly dI-dC in 5 ml of a solution consisting of 20 mM HEPES, 4 mM MgCl2, 50 mM CaCl2, 1 mM
EDTA, 1 mM DTT, and 4% glycerol. The mixtures were loaded onto a non-denaturing 6% polyacrylamide gel with 0.25 × TBE electrophoresis buffer. After electrophoresis, gels were dried and exposed to the radiography film for 18 h at 70 with intensifying screens. Supershift assays were also performed using rabbit antibodies against four kinds of Rel protein; p50, p65, p52, c-Rel to determine the Rel protein composition of oxygen-derived free radicals-activated NF- B dimers in nuclear extracts of esophageal mucosa of GERD. Each anti-p50, -p65, -p52, -c-Rel antibody (Santa Cruz, CA) was mixed with the
NF-B probe at the start of the 30 min-incubation.
12. Statistical Assay
Results are expressed as the mean±S.E.M.. The data were analysed by one-way analysis of variance (ANOVA), and the stastical significance between groups was determined by Duncan's multiple range test. Statistical significance was accepted with a p<0.05.
. RESULTS
1. The Influence of Stress on H. pylori-associated Gastropathy; Macroscopic and Microscopic Observation
1) The macroscopic changes
H. pylori infection alone only provoked the gastric inflammations in rats.
There were neither ulcerative lesions nor tumorous lesions in the stomach upto 24 weeks after H. pylori infection. WIRS, on the other hand, formed multiple erosions with hemorrhagic changes. By the way, stress significantly increased gastric lesions of H. pylori infection than without H. pylori infection. WIRS of 30 min duration alone did not develop any GML, but when stress was imposed in H. pylori-infected rat, multiple scattered hemorrhagic erosions were noted even after 30 min of WIRS, severe than rats without H. pylori infection. The gastric lesions including erosions and linear ulcerations were appeared after 120 min of WIRS and multifocal hemorrhagic lesion at 480 min. Hemorrhagic, erosive lesions shown at 30 min after WIRS increased number and size of hemorrhagic lesion at 120 and 480 min in H. pylori-infected group and linear ulcerations were noted thereafter. Bleeding index of WIRS alone increased the score by 0.33, 0.75, 0.88 and 0.99 at 30, 120, 480 and 720 min (Fig. 1) and bleeding index of WIRS with H. pylori infection increased the score by 0.38, 0.93, 1.35 and 1.97 at 30, 120, 480 and 720 min, of which index was increased in a time-dependent manner. Compared to WIRS alone, bleeding index of WIRS with
difference (p<0.05). Bleeding rates of WIRS alone increased the percentage by 0, 59.6, 69.7 and 80.4 % at 30, 120, 480 and 720 min in time-dependent manner (Fig. 2). Compared to WIRS alone, bleeding rates of WIRS with H. pylori infection significantly increased the percentage by 41.6, 79.9, 90.8 and 100 at 30, 120, 480 and 720 min respectively (p<0.05).
2) The microscopic changes
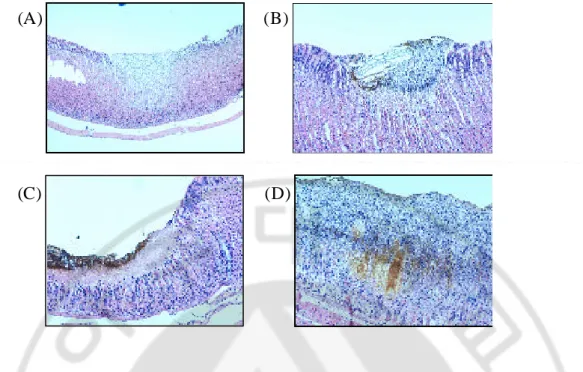
The WIRS caused serious pathological changes in the stomach, of which severity was significantly increased in accordance with the times of restraint stress. Pathologically, overt gastric ulcer with necrotic bases and exudative changes and inflammatory cell infiltrations, mostly neutrophils were observed. Only 30 min duration of WIRS caused some erosive changes with inflammatory cells. However, in group affected with both stress and H. pylori infection, the gastric lesions were more brisk and aggravated. In a same time point, the overall pathological changes including hemorrhages and erosions were apparently severe in rats affected with both stress and H. pylori infection than groups of rat only done with WIRS. Microscopic examination of the mucosa after 480 min WIRS-induced GMD revealed widespread damage of the surface epithelium with many cells sloughed off into the gastric lumen and focal area of deep hemorrhagic necrosis. Severe hemorrhagic GMD are observed WIRS-induced GMD with H. pylori infection animals (Fig. 3). Inflammatory cell infiltration and hemorrhage were noted in the lamina proporia as well as submucosa. The degree of mucosal inflammation became more prominent and a mild to moderate decreased thickness layers was also seen during this period.
WIRS alone WIRS + H. pylori
WIRS (min)
Bleeding index
0.5 1.0 1.5 2.0 2.5 0 30 120 480 720*
*
WIRS alone WIRS + H. pyloriWIRS (min)
Bleeding index
0.5 1.0 1.5 2.0 2.5 0 30 120 480 720*
*
Fig. 1. Bleeding index of WIRS-induced gastric mucosa in rats with or
without H. pylori infection. WIRS was applied for 30, 120, 480 and
720 min at 40 weeks after H. pylori infection. Bleeding index was expressed as Table 1 and all values are expressed as mean ± S.E.M. for 6 8 rats. Asterisk indicates stastical difference from WIRS alone (* :
WIRS alone WIRS + H. pylori
WIRS (min)
Bleeding rates (%)
20 40 60 80 100 0 30 120 480 720 * * * * WIRS alone WIRS + H. pylori WIRS alone WIRS + H. pyloriWIRS (min)
Bleeding rates (%)
20 40 60 80 100 0 30 120 480 720 * * * *Fig. 2. Bleeding rates (%) of WIRS-induced gastric mucosa in rats with or
without H. pylori infection. WIRS was applied for 30, 120, 480 and
720 min at 40 weeks after H. pylori infection. Bleeding rates was expressed as ratio of hemorrhage recurrence and all values are expressed as Mean ± S.E.M. for 6 8 rats. Asterisk indicates stastical difference from WIRS alone (* : p<0.05)
(A) (B)
(C) (D)
Fig. 3. Microscopic observation of WIRS-induced gastric mucosa in rats with
or without H. pylori infection after stress loaded 480 min. (A) and
(B) were observed inflammation in epithelium on WIRS alone, (C) and (D) were checked severe gastritis and hemorrhage in epithelium on WIRS with H. pylori infection. (A, C ×40, B, D ×100)
2. The Influence of Stress on H. pylori-associated Gastropathy; Changes of Several Mediators Involved in H. pylori-induced Gastric Mucosal Damages
1) Oxidative mediators
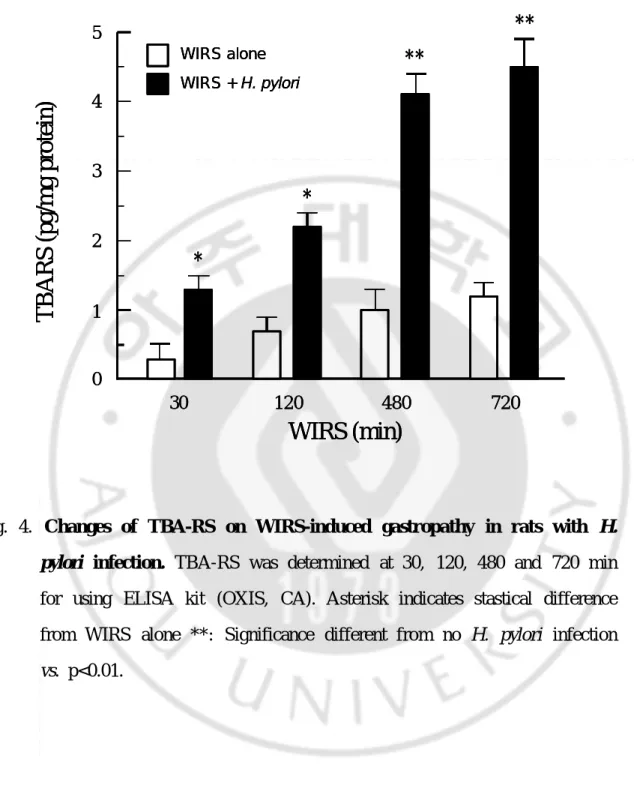
The contents of thiobarbituric acid reactive substances (TBA-RS, a marker of lipid peroxidation) were significantly increased in WIRS with H. pylori infection compared to WIRS group without H. pylori infection at 30, 120, 480 and 720 min (*p<0.05, **p<0.01) (Fig. 4). After 720 min of WIRS, TBARS was seen maximum level (4.4 pg/mg of protein).
2) Cytokines
Production of INF- was significantly increased in WIRS with H. pylori infection than without H. pylori infection on time-dependent manner (Fig. 5A). After 480 min of WIRS, IFN- was seen maximum level (79.0 pg/mg protein), but IFN- was decreased after 720 min. The group of without H. pylori infection didn't change level of IFN- .
The content of TNF- and IL-10 were increased in WIRS with H. pylori infection and no H. pylori infection on time-dependent manner (Fig. 5B). TNF-was not significantly differently among groups.
3) Proteome markers
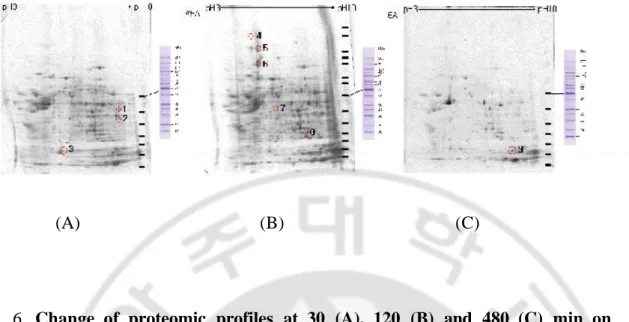
Changes of proteomic profiles were differentially expressed on WIRS-induced GMD in H. pylori-infected rats (Fig. 6). Identifications of changes spot by SELDI-TOF analysis were seen with the check of genebank identification number. After 30 min in WIRS, Fig. 6A identified three spots into (1) PART1 (putative novel phosphatidylinositol 3 kinase, AAB32956, gi:806955), (2) BMI-1 (murine leukemia viral oncogene homolog, XP_031361, gi:14747007) and (3) hypothetical protein (XP_035122, gi:14783675). After 120 min in WIRS, Fig. 6B identified five spots into (4) nestin (P48681, gi:1346682), (5) mitotic spindle associated protein (AAK91712, gi:15193198), (6) hypothetical protein (XP_044197, gi:14744107), (7) caspase-9 (N92123, gi:1264432) and (8) solute carrier family 26, number 1, isoform b (AAH15517, gi:15930164). And, after 480 min in WIRS, Fig. 6C identified one spot (9) ZA (AAB36122, gi:1478361).
WIRS alone WIRS + H. pylori
WIRS (min)
TBARS (pg/mg protein)
1 2 3 4 5 0 30 120 480 720**
**
*
*
WIRS alone WIRS + H. pylori WIRS alone WIRS + H. pyloriWIRS (min)
TBARS (pg/mg protein)
1 2 3 4 5 0 30 120 480 720**
**
*
*
Fig. 4. Changes of TBA-RS on WIRS-induced gastropathy in rats with H.
pylori infection. TBA-RS was determined at 30, 120, 480 and 720 min
for using ELISA kit (OXIS, CA). Asterisk indicates stastical difference from WIRS alone **: Significance different from no H. pylori infection
(A) WIRS alone WIRS + H. pylori WIRS (min) Interferon -γ (pg/mg protein) 20 40 60 80 100 0 30 120 480 720 * * * * WIRS alone WIRS + H. pylori WIRS alone WIRS + H. pylori WIRS (min) Interferon -γ (pg/mg protein) 20 40 60 80 100 0 30 120 480 720 ** ** ** ** (B) WIRS alone WIRS + H. pylori WIRS (min) TNF -α (pg/mg protein) 25 50 75 100 125 0 30 120 480 720 WIRS alone WIRS + H. pylori WIRS alone WIRS + H. pylori WIRS (min) TNF -α (pg/mg protein) 25 50 75 100 125 0 30 120 480 720
Fig. 5. (A) Changes of IFN- on WIRS-induced GMD in rats with H. pylori infection. IFN- was determined at 30, 120, 480 and 720 min for using ELISA kit. *: Significance different from no H. pylori infection vs. p<0.05. **: Significance different from no H. pylori infection vs. p<0.01.
(B) Changes of TNF- on WIRS-induced GMD in rats H. pylori infection. TNF- was determined at 30, 120, 480 and 720 min for using ELISA kit. IFN- and TNF- were checked severe gastritis and hemorrhage in epithelium on WIRS with H. pylori infection. *: Significance different from no H. pylori infection vs. p<0.05. **: Significance different from no H. pylori infection vs. p<0.01.
(A) (B) (C)
Fig. 6. Change of proteomic profiles at 30 (A), 120 (B) and 480 (C) min on
WIRS-induced GMD in H. pylori-infected rats. Identification for
differentially expressed spot by 2-dimensional gel electrophoresis analysis from genebank identification numbers as described in the mateials and methods. At 30, 120 and 480 min, we observed detection of three, five and one spot on WIRS-indued GMD in H. pylori-infected rats.
3. The Influence of Stress on H. pylori-associated Gastropathy; Changes of Heat Shock Proteins (HSPs)
1) Western blot analysis
H. pylori infection alone decreased expression of HSP70 and HSP27, but
heat-induced stress increased expression of HSP90, HSP70 and HSP27 in AGS cells (Fig. 7). In H. pylori infected AGS cell, heat shock increased HSP70, but decreased HSP27. The changes of other HSPs including HSP90 and HSC70 were not significant.
2) 2-D gel electrophoresis with blotting
Fig. 8 showed the overall change of proteome spots according to the times after heat shock in AGS cell. There was no significant change of HSP90 in spite time passages. However, when we transferred the whole proteome spots separated by 2D-gel electrophoresis in to NC paper and applied with each specific antibodies of various HSPs, the change of HSP expression were significantly different according to time points. As times passed, significant phosphorylation of HSP90 and HSP27 wre noted and also heat shock or H.
pylori infection triggered the significant phosphorylation of these proteins (Fig.
9). When the cells were treated with both H. pylori and heat shock, these phosphorylations of HSP90 were significantly increased, reflecting that H. pylori or stress and both provoked the activation of HSP90 rather than the changes of amounts.
-
+ -
+
-
-
+ +
Hsp 90
70
60
27
H. pylori
Heat shock
α−
tubulin
-
+ -
+
-
-
+ +
Hsp 90
70
60
27
H . p y l o r i H e a t s h o c k α −t u b u l i nFig. 7. Detection of various HSPs expression in cultured AGS cells by
Western blot analysis. Either heat shock and/or H. pylori infection were
induced in AGS cells. After extracting proteins from each group, proteins were separated and transferred onto NC paper, blotted with HSP 27, 60, 70, and 90 antibodies, respectively. Proteins amounts were balanced with
Control + Heat shock + Heat shock
presence of H. pylori
Fig. 8. 2 D-gel electrophoresis. Isoelectric focusing with PAGE separation was done in each group, control, heat shock, and H. pylori infection. Each spots were analyzed with Image Analyser.
HSP90 HSP70 HSP60 HSP27 Control H. pylori Heat shock H. pylori + Heat shock HSP90 HSP70 HSP60 HSP90 HSP70 HSP60 HSP27 Control H. pylori Heat shock H. pylori + Heat shock
Fig. 9. Activation of HSP90, HSP70, HSP60 and HSP27 in cultured AGS
cells. After 2D-PAGE separatiom, the gels were transferred into NC
paper and blotted with each antibody. H. pylori infection alone or heat shock provoked significant phosphorylation of HSP90 and 27. Both H.
pylori and heat shock together induced more apparent phosphorylation of
HSP90, which might be the principal mechanism of gastric mucosal damages.
4. Prevention of Stress-induced Augmented Gastric Lesions with Antioxidants
1) Macroscopic and Microscopic evidences
As shown in Fig. 10, treatment of DA-9601 (6, 20 mg/kg, po) or -tocopherol (40 mg/kg, po) significantly attenuated the severity of WIRS-induced GMD in H. pylori-infected rats. Rats were sacrificed and fixed with 10% neutral formalin buffer at 30, 120 or 480 min after WIRS. At 30 min, control was observed several GMD in stomach, DA-9601 (6 mg/kg) and -tocopherol were equal effect (1-2 scatter GMD). But, DA-9601 (20 mg/kg) was not at all seen GMD in stomach. At 120 min, control was seen a lot of linear and hemorrhagic GMD and DA-9601 were decreased GMD in dose-dependently manner. -Tocopherol seem to GMD in administration of DA-9601 6 mg/kg. Finally, at 480 min after WIRS induced GMD, control was seen high severe, a lot of thick-linear and hemorrhagic GMD, even observed a few gastric ulcer in any animals. Administration of DA-9601 (6 and 20 mg/kg) were not change gross observations and number of GMD, and not seen linear and hemorrhagic GMD at all. -Tocopherol was observed more severe than DA-9601 6 mg/kg.
In the control group, pathological changes were noted with hemorrhagic GMD at 120 and 480 min after WIRS in H. pylori-infected rats (Fig. 11A). At 120 min after WIRS, control was mainly observed hemorrhagic site and inflammation-related factors in epithelium of fundic area, and severe hemorrhagic gastritis at 480 min after WIRS. DA-9601 (20 mg/kg) was not seen GMD at 120 min after WIRS, and observed weakly GMD without hemorrhage at 480 min after WIRS (Fig. 11B).
2) Molecular evidences; Western blot of HSP changes
The changes of various HSPs was determined by Western blotting with antibody against HSP90, HSP70 and HSP27 (Fig. 12). DA-9601 significantly increased HSP70 and HSP27 expressions on WIRS-induced GML of H.
pylori-infected rats.
3) Molecular evidences; RNase protection assay
The changes of mRNA of inflammation-related cytokines and apoptosis-related factors were measured by RPA using the RNA obtained from rat gastric mucosa of each group. The TNF- , TGF- 1, MIF and IFN- mRNA transcript with two housekeeping gene, L32 and GAPDH were detected in the rat gastric mucosa using rck-3 multi probe template set (Fig. 13). Antioxidant treatment significantly decreased these inflammatory cytokines mRNA expression levels at 0.5, 2 or 8 h after WIRS inducetion in H. pylori-infected rats.
Similarily Bcl-xL, caspase-1, caspage-2, caspase-3 mRNA transcript with two housekeeping gene, L32 and GAPDH were detected in the rat gastric mucosa using rAPO multi probe template set, although the level of mRNA expression weak (Fig. 14). Antioxidant increased these apoptosis-related mRNA expression levels at 30, 120 or 480 min after WIRS inducetion in H.
pylori-infected rats, suggesting the preventive induction of apoptosis related gene
4) Molecular evidences; NF- B DNA bindings
Fig. 15 shows the NF- B complex of nuclear proteins extracted from each group. Pretreatment with DA-9601 significantly decreased NF- B DNA binding in a dose dependent manner, suggesting the antioxidant regulated the transcriptional binding for either cytokines or other inflammation related genes.
(A)
(B)
(C)
(D)
(A)
(B)
(C)
(D)
Fig. 10. Representative macroscopic findings of the gastric mucosa of rats. At 480 min of WIRS-induced stress, we observed gastric mucosa in H.
pylori infection rats. These groups are (A) control, (B) DA-9601 6
(A )
(B )
(C )
(D )
Fig. 11. Microscopic observation of WIRS-induced GMD in H. pylori-
infected rats. (A) and (B) denoted the representive gastric pathology in
rats affected with both H. pylori infection and WIRS 120 and 480 minutes, respectively. (A) and (B) denoted the gastric pathology in group treated with DA-9601 at 120 and 480 min, respectively. Significant protection was noted in rats pretreated with antioxidant.
3 0 1 2 0 4 8 0 3 0 1 2 0 4 8 0 WIRS alone WIRS pretreated With DA -9601 HSP27 HSP70 HSP90 3 0 1 2 0 4 8 0 3 0 1 2 0 4 8 0 WIRS alone WIRS pretreated With DA -9601 HSP27 HSP70 HSP90
Fig. 12. Effect of antioxidant on HSPs expressions according to group. DA-9601 treatment induced significant levels of HSP70 and HSP27, which might be involved in the protective action against WIRS in rats.
1 2 3 4 5 6 7 8 9 1 0 I F N -b T N F -b G M - C S F T G F-b1 T G F-b2 L T b T N F -a M I F I F N -g L 3 2 G A P D H T G F -b3 1 2 3 4 5 6 7 8 9 1 0 I F N -b T N F -b G M - C S F T G F-b1 T G F-b2 L T b T N F -a M I F I F N -g L 3 2 G A P D H T G F -b3
Fig. 13. Results of RNase protection assay (RPA) for cytokine (rCK-3). The RNA extracted from each group according to the treatment of DA-9601 were hybridized with radiolabelled multi-probes hybridizing with inflammation associated cytokines, which included IFN- , TNF- , GM-CSF, TGF- 1, TGF- 3, TGF- 2, LT-b, TNF- , MIF and IFN- with two housekeeping gene products, L32 and GAPDH and digested with RNase enzyme. Each lane signify the levels offf transcript of each gene. Lane denotes; 1: probe, 2: control RNA, 3: yeast RNA, 4: normal, 5: WIRS alone for 30 min, 6: WIRS for 30 min pretreated with DA-9601, 7: WIRS alone for 120 min, 8: WIRS for 120 min pretreated with DA-9601, 9: WIRS alone for 480 min and 10: WIRS for 480 min pretreated with DA-9601.
F a s A g ( 4 3 5 ) B c l-x L ( L ) ( 3 9 3 ) B c l-x L ( S ) ( 3 4 3 ) F a s L ( 3 1 5 ) C a s p a s e -1 ( 2 8 2 ) C a s p a s e - 3 ( 2 5 5 ) C a s p a s e - 2 ( 2 3 1 ) B a x ( 2 1 0 ) B c l- 2 ( 1 8 9 ) L 3 2 ( 1 4 1 ) G A P D H ( 1 2 5 ) 1 2 3 4 5 6 7 8 9 1 0 F a s A g ( 4 3 5 ) B c l-x L ( L ) ( 3 9 3 ) B c l-x L ( S ) ( 3 4 3 ) F a s L ( 3 1 5 ) C a s p a s e -1 ( 2 8 2 ) C a s p a s e - 3 ( 2 5 5 ) C a s p a s e - 2 ( 2 3 1 ) B a x ( 2 1 0 ) B c l- 2 ( 1 8 9 ) L 3 2 ( 1 4 1 ) G A P D H ( 1 2 5 ) 1 2 3 4 5 6 7 8 9 1 0
Fig. 14. Results of RNase protection assay (RPA) for apoptosis (rAPO). Genes of Fas Ag, Bcl-xL (L), Bcl-xL (S), FasL, Caspase-1, Caspase-2, Caspase-3, Bax and Bcl-2 were examined by RNase protection assay with two housekeeping gene products, L32 and GAPDH according to the drug treatment. Each lane denotes. 1: probe, 2: control RNA, 3: yeast RNA, 4: normal, 5: WIRS alone for 30 min, 6: WIRS for 30 min pretreated with DA-9601, 7: WIRS alone for 120 min, 8: WIRS for 120 min pretreated with DA-9601, 9: WIRS alone for 480 min and 10: WIRS for 480 min pretreated with DA-9601.
N F -κB
1 2 3 4 5 6 7 8
N F -κB
1 2 3 4 5 6 7 8
Fig. 15. Effect of NF-κ B by DA-9601 on WIRS-induced GMD in rats with
H. pylori infection. NF-κB DNA binding activities were measured by
electrophoretic mobility shift assay using 32P-labeled oligonucleotide coding NF-κB binding sites Significant increased in NF-κB binding activities were noted after H. pylori infection or H. pylori infection and heat shock and these radiactivities were markedly decreased with antioxidant treatment Lane 1: no probe 2: control cells (30 min) 3: H.
pylori infection 4: H. pylori infection + heat shock 5: H. pylori + heat
shock pretreated with DA-9601 6: H. pylori infection 7: H. pylori infection + heat shock 8: H. pylori + heat shock pretreated with DA-9601.
. DISCUSSION
The present study showed for the first time that the gastric lesion by H.
pylori could be augmented by both stress and oxidative stress and the
dysregulation of heat shock protein might be responsible for augmented gastric lesion. These findings can explain why the incidence of gastric cancer is so high in Korea, because we can infer that the high prevalence of H. pylori infection and complex social structure might lead to notoriously high incidence of gastric cancer in Korea.
As major etiologic factors leading to peptic ulcer disease, and hypersecretion, smoking, alcohol, NSAIDs, bile acid, and stress are considered. The German blitz in London, the Kobe Hanshin great earthquake, economic crisis in Sophia, and sovereignty negotiations in Hong Kong have all been followed by an abrupt increase in peptic ulcers of both the stomach and the duodenum.31-33 By the way the evidence that psychological stress is one of those factors is not invalidated by the discovery of H. pylori. Among potential mediators, several known behavioral risk factors for ulcers - smoking, alcohol abuse, and lack of sleep - have clear associations with real-life stress and are known to impair wound healing through their effects on immune function.34 On the physiological side, stress is known to modify gastric blood flow, which plays an important role in the gastric mucosal barrier, and to affect possible mediators such as thyrotropin-releasing hormone, cytokines, and corticotropin-releasing hormone. Besides, stress seems to have variable effects on gastric motility:
delayed gastric emptying could increase the risk of gastric ulcer, while accelerated emptying could increase the net acid load delivered to the duodenum at any given level of gastric secretion, enhancing the risk of duodenal ulcer; skipped meals and poor sleep might increase duodenal acid load still further.35 Psychological stress may also promote the growth of H. pylori in the duodenum since the H. pylori-inhibitory effects of bile seem to be reversed by acid.36
The WIRS model is one of the most widely used methods for studying the pathogenesis of peptic ulcer.15,16,37-41 It has been established that exposure to WIRS rapidly induces gastric mucosal lesions in rats, but the interaction between
H. pylori infection and WIRS was not clearly defined because that the results
have been rather conflicting.42 Using BALB/c mice, Matsushima et al. reported that H. felis infection itself did not cause gastric mucosal erosions, but it did augment the stress-induced erosions.16 And, Yamamoto reported that H. pylori
infected C57BL/6 mice increased the severity of mucosal injury at 30 min of stress exposure, but at 720 min, there was no difference in mucosal lesions between mice with or without H. pylori infection.15 In the present study, long-term WIRS induced gastric mucosal damages no respected of H. pylori infection, but mean severity was significantly higher in group infected with H.
pylori compared to no H. pylori infection. We observed that the bleeding index
and bleeding rate in WIRS-induced rats with H. pylori infection were significantly higher than without H. pylori infection. In histological observation and inflammation-mediated factors, stress augmented inflammation of gastric mucosa, and increased TBA-RS, IFN- , and TNF- in gastric mucosa layer with H. pylori infection.
The changes in oxygen-free radicals and pro-inflammatory cytokines are induced by both stress and H. pylori, but their roles in the development of gastric ulcers are not fully understood.15,16,43 Recent reports suggest that the effect of altered cytokines may be different in stress-induced and H. pylori-associated ulcers. For example, Matsushima et al. reported that both Helicobater felis infection and water-immersion stress induced a similar IL-1 response.40 And, we found that the Th1 lymphocytic responses were augmented in group affected with both stress and H. pylori infection, suggesting the intense involvement of gastric inflammation after H. pylori infection was increased. The production of IFN- and TNF- was significantly higher in the infected than in the uninfected group of rats, which supports the findings.44-46 The changes in IFN- and
TNF-induced by H. pylori infection and stress treatment indicate that these cytokines may be candidates for the cause of acute mucosal injury of the stomach. On the other hand, the time lag between the occurrence of mucosal injuries and the alterations in cytokine production, and the absence of inflammatory cell reaction suggest that additional mechanisms may be involved in the formation of gastric mucosal injuries in the early phase of stress treatment.
In H. pylori-infected gastric mucosa, significant generations of oxidative radicals was involved. NH3 derived from H. pylori reacts with HOCl to yield
monochloramine (NH2Cl) and these liphophilic compounds freely penetrates
biological membranes to oxidize intracellular components.47 Therefore, a new therapeutic approach using agents that inhibit reactive oxygen species (ROS) production from activated neutrophils or scavenging ROS has been proposed for
H. pylori associated gastric mucosal inflammation. We and other groups have
demonstrated that some kinds of gastroprotective drugs, such as rebamipide, polaprezine, and ecabet sodium, have these properties documented in vitro and in
vivo.48,49 In the stress and H. pylori infected condition, ROS from neutrophils could play an important role in the pathogenesis of gastric mucosal injury.50-52 Under WIRS, gastric mucosal oxidative stress as well as lesion formation were attenuated by treatment with the xanthine oxidase inhibitor, allopurinol, suggesting the importance of xanthine/xanthine oxidase system for the development of ischemic-reperfusion injury in the stomach.53 The most important physiological function of antioxidant enzymes like peroxidase, catalase, and glutathione peroxidase is to detoxify cellular H2O2 to prevent oxidative damage
to the cell.54 SOD dismutase O2- into H2O2, which is then scavenged by
peroxidase and catalase so that the highly reactive OH or HOCl formed by MPO-catalysed Cl- oxidation is not generated.55 H2O2 is diffusible and may go to
any cellular compartment where it locally produces highly reactive OH in presence of O2- through a metal-catalysed Haber-Weiss reaction.56 Neutrophil
accumulation in stomach clogged the microvasculature, thus causing tissue ischemia.57 Accumulation of H2O2, which not only causes oxidative damage of
the gastric mucosa, but also leads to apoptotic cell death during ulceration.58,59 Our results confirmed that H. pylori increased contents of TBA-RS in WIRS-induced GMD.
In the current experiment, antioxidant treatment including -tocopherol or DA-9601 reported to possessing the antioxidative actions in the stomach, ameliorated the gastric lesions provoked by both stress and H. pylori infection.
-Tocopherol is known to antioxidant, and DA-9601 is ethanol extract of
Artemisia asiatica possesses antioxidative and antiinflammatory activities. DA-9601 mainly contribute to its protective effects against experimentally induced gastric damage (gastritis, gastric ulcer and duodenal ulcer) in animal models.60-62 The mechanisms of DA-9601 are known as antioxidantive effect, anti-inflammatory effect, reepithelization of gastric mucosa and cytoprotective effects, potentially.63-65 Recently, DA-9601 evaluated chemopreventive effect on phorbol ester-induced ornithine decarboxylas activity, papilloma foemation, COX2 expression, iNOS expression, NF- B activation in mous skin.66
Similarily the antioxidant administration markedly decreased the mRNA of several inflammatory cytokines and even transcription factor like NF- B, known to be inflammation-associated transcription factor. H. pylori activated NF- B in gastric mucosa in vivo and cultured epithelial cells in vitro.67 Maeda et al. identified upstream mediators that regulate H. pylori-induced NF- B-dependent IL-8 production.68 H. pylori-induced apoptosis in gastric epithelial cells is suppressed by inhibition of NF- B activation using catalase, pyrrolidine dithiocarbamate, an antisense oligonucleotide for NF- B subunit p50, or peroxisome proliferator-activated receptor ligands. Kanai et al. who noted that TGF- plays an anti-apoptotic role in gastric mucosal cells by enhancing the expression of Bcl-2 family proteins via an NF- B-dependent pathway.69 Antioxidant, -tocopherol and DA-9601, weakened inflammation via inhibition of ROS production and DA-9601 especially decreased inflammation and apoptosis-mediators in RPA and reduced NF- B activation in EMSA.
were the changes of molecular chaperones in our study. One of these key moleculaes involved in the response against stress might be heat shock protein (HSP), which are necessary for survivals of cells under stress conditions.70,71 The heat shock response was first discovered in 1962 by Ritossa, who observed a pattern of Drosophila salivary gland chromosome puffs that were induced in response to transient exposures to elevated temperature.72 They have been classified into six major families according to their molecular size (HSP100, HSP90, HSP70, HSP60, HSP40 and small HSP27), of which HSP70 is one of the most extensively studied in mammalian cell.73,74 HSP family is induced in cultured gastric mucosal cells by heat stress, and this protein has a cytoprotective function in vivo.75 ROS-mediated rabbit gastric cell injury was also reportedly to be partially protected through the induction of HSP70 by antioxidative drug application.76
Although the relationship between H. pylori infection and host HSPs response has not been fully clarified, a recent study has shown that H. pylori infection reduces and cancelled the inductions of HSP70 in gastric mucosa.77 All organisms respond to exposure to sublethal temperatures and to a variety of other stresses such as heavy metal ions, ethanol, anoxia, and oxidants, through the rapid induction and synthesis of HSPs.78 Their induction and synthesis has been correlated with the acquisition of thermotolerence, a property of cells and organisms to show transient protection from the adverse effects of subsequent heat or chemical stresses.79 Evidence for the protective effects of individual heat shock proteins has been reported for human HSP27, yeast HSP104, human HSP70, mammalian HSP90, and Drosophila HSP27.80-84 The level of HSP90 is
generally similar, ubiquitously existed and expressed, in most tissues, with the highest level observed in the intestine and the germ tissue. HSP25 was particularly abundant in stomach, colon, and bladder, with reduced levels in ovary, lung, and skin.85 Restraint and water-immersion stress caused rapid expression of HSP90, HSC70, and HSP70 mRNAs in the hypothalamus, and these expression were followed by inductions of the respective HSP proteins.86 The stress protein HSP70 is an inducible protein synthesized in response to a stressor. Nakamura et al. suggested that synthesis of HSP by sublethal heat played an important role in the intracellular mechanism of gastric protection against ethanol.87 HSP70 induction after a conditioned emotional stress would be cytoprotective for gastric mucosal oxidative injury.88 Koh et al. study showed a significant increase in MPO activity in the conditions without HSP70 induction and a significant reduction in MPO activity with HSP induction.89
In the current our experiment, very important finding was grawn from the 2D-gel electrophoresis experiment showing the active phosphorylation of HSP90 was noted after either H. pylori infection or heat shock. These phosphorylation was further active after the challenge of both stress and H. pylori infection. Similar finding was also observed in HSP27 (Fig. 9). Recently, HSP90 is regarded as significant oncoprotein, based on which, multiple targets for HSP90 were tried in clinical field as promising anticancer agents. HSP90 is constitutively expressed at 2 10 fold higher levels in tumor cells compared with their normal counterparts, suggesting that it could be crucially important for the growth and/or survival of tumors. These can be explained by the fact that as client proteins chaperoned by HSP90, mutated p53, Akt kinase, Raf-1 kinase,
Bcr-Abl kinase, ErbB2 transmembrane kinase, CDK4, Wee1, and certain basic helix-loop-helix transcription factors including HIF-1 were revealed. Therefore, active phosphorylation of HSP90 shown in our experiment result might participate in the propagation or perpetuation of stress-associated gastropathy and extentively in possible carcinogenesis.
Therefore, the remedy for the prevention of stress-augmented H.
pylori-associated gastropathy might be the induction of cytoprotective HSP70 or
27 and conservation of HSP90. Our trial of antioxidant caould be the ideal target for that since pretreatment of antioxidant, -tocopherol or DA-9601 could induced or preserve the HSP70 and intramucosal GSH with the preservation of HSP90.
.
CONCLUSION
This study showed new proof that stress might be the very important environmental facts determining the outcome of H. pylori infection irrespective of virulence or host factors. Since H. pylori is highly associated with either the peptic ulcer disease or gastric cancer, stress also be the critical contributing factors for that significant oxidative stress and immune derangement were attributed to the cause of augmentation of GMD after both H. pylori and stress. Dysregulation of HSP response is also fundamentally involved in these augmentation of gastric damage. Antioxidant can prevent the degree of H. pylori and stress-associated GMD, for that significant inductions of HSP70 or HSP27 were noted.
BIBLIOGRAPHY
1. Holzer P: Gastroduodenal mucosal defense. Curr Opin Gastroenterol 16:469-78, 2000
2. Kitagawa H, Fujiwara M and Osumi Y: Effect of water-immesion stress on gastric acid secretion and mucosal blood flow in rats. gastroenterology 77:298-302, 1979
3. Tepperman BL and Jacobson ED: Circulatory factors in gastric mucosal defense and repair. In: Physiology of the gastrointestinal tract (ed. Johnson LR) New York, Raven Press, 1994, pp 1131-51
4. Wallace JL: Mechanisms of protection and healing: Current knowledge and future research. Am J Med 110(1A):195-235, 2001
5. Takagi KY, Kayuya Y and Watanabe K: Studies on drugs for pepic ulcer. A reliable method for producing stress ulcers in rats. Chem Pharm Bull 12:465-72, 1964
6. Brzozowski T, Konturek SJ, Kwiecien S, Pajdo R, Drozdowicz D and Sliwowski Z et al.: SV-480, a novel synthetic flavonoid derivative of sophoradin, with potent gastroprotective and ulcer healing activity. J Physiol Pharmacol 46:83-98, 1998
7. Brzozowski T, Konturek PC, Konturek SJ, Brzozowska I, Kwiecien S and Hahn EG: Involvement of ornithine decarboxylase and polyamines in epidermal growth factor-induced recovery of gastric mucosa from gastric lesions provoked by stress. Regul Pept 74:73-84, 1998
8. Brzozowski T, Konturek PC, Pajdo R, Kwiecien S, Ptak A and Sliwowski Z et al.: Brain-gut axis in gastroprotection by leptin and cholecystokinin against ischemia-reperfusion induced gastric lesions. J Physiol Pharmacol 52:583-602, 2001
9. Billroth J: Ueber duidenalgeschwure beisepticame. Wein Med Wochenschr 17:705, 1867
10. Bank S, Misra P, Mausner D, Kurtz L, Rehman M and Wise L: The incidence, distribution and evolution of stress ulcers in surgical intensive care patients. Am J Gastroenterol 74:76, 1980
11. Cook DJ, Fuller HD, Guyatt GH, Marshall JC, Leasa D and Hall R et al.: Risk factors for gastrointestinal bleeding in critically ill patients. N Engl J Med 330:67-381, 1994
12. Lucas CE, Sugawa C, Riddle J, Rector F, Rosenberg B and Walt J: Natural history and surgical dilemma of 'stress' gastric bleeding. Arch Surg 102:266-73, 1971
13. Cushing H: Peptic ulcers and the interbrain. Surg Gynecol Obstetrics 55:1-34, 1932
14. Duerksen DR: Stress-related mucosal disease in critically ill patients. Best Prac Res Clin Gastroenterol 17:327-44, 2003
15. Yamamoto N, Sakagami T, Fukuda Y, Koizuka H, Hori K and Sawada Y et al.: Influence of Helicobacter pylori infection on development of stress- induced gastric mucosal injury. J Gastroenterol 35:332-40, 2000
16. Matsushima Y, Aoyama N, Fukuda H, Kinoshita Y, Todo A and Himeno S et al.: Gastric ulcer formation after the Hanshin-Awaji earthquake: a case