Ch. 27.
Biosynthesis
of the Nutritionally Nonessential Amino Acids
Ch. 28. Catabolism of Proteins & of Amino Acid
Nitrogen
Ch. 29. Catabolism of the Carbon Skeletons of Amino Acids
Ch. 30. Conversion of Amino Acids to Specialized Products
AMINO ACIDS METABOLISM
Car-bons
NH
32019 년 1 학기
Text : Harper’s Biochemistry Refs: Devlin’s, Lippincott’s
AMINO ACIDS METABOLISM
Car-bons
NH
3Chapter 29.
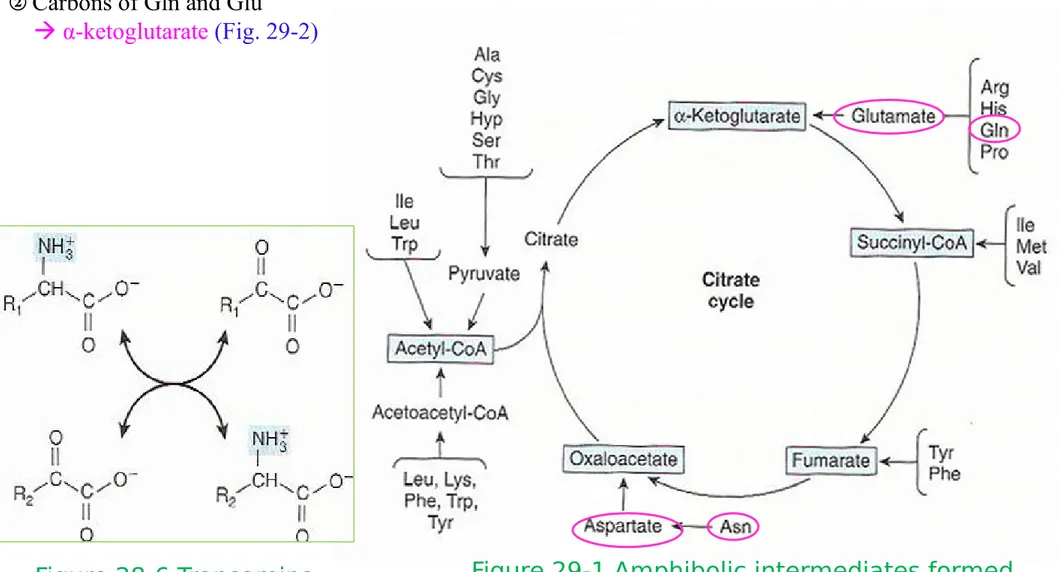
* Transamination typically initiates amino acid catabolism
① Removal of α-amino nitrogen by transamination (Fig. 27-3, Fig. 28-6) is first catabolic reaction
except for Pro, Hyp, Thr, Lys
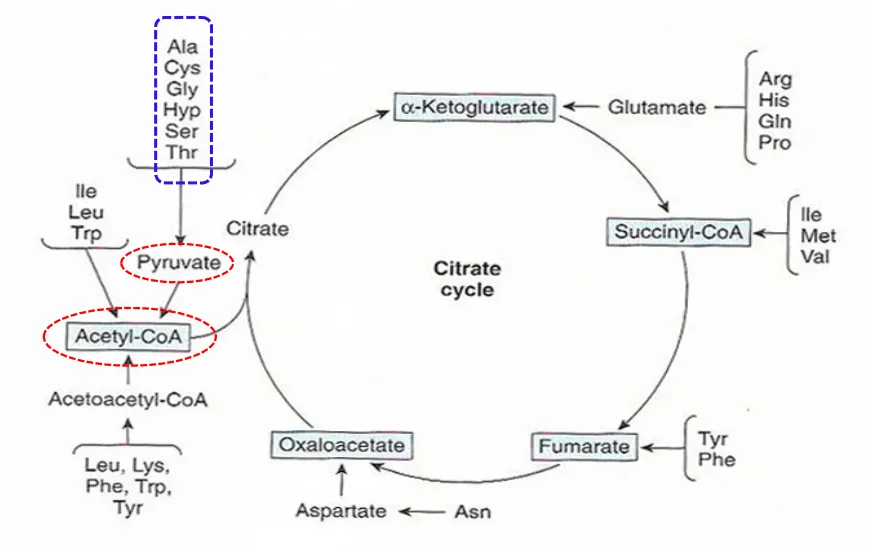
② Residual hydrocarbon skeleton amphibolic intermediates (Fig. 29-1)
Figure 28-6 Transamination
Figure 27-3 Formation of alanine by transamination of pyruvate
Figure 29-1 Amphibolic intermediates formed from the carbon skeletons of amino acids
Alanine (Aspar-tate)
* Transamination typically initiates amino acid catabolism
① Removal of α-amino nitrogen by transamination (Fig. 27-3, Fig. 28-6) is first catabolic reaction
except for Pro, Hyp, Thr, Lys
② Residual hydrocarbon skeleton amphibolic intermediates (Fig. 29-1) Glucogenic (글루코스 생성 )
amino acids
Ketogenic (캐톤 생성 ) amino acids
Both glucogenic and ketogenic amino acids
Figure 29-1 Amphibolic intermediates formed from the carbon skeletons of amino acids
*
글루코스생성 아미노산 (Glucogenic AAs)
• 정의 : 이화되어서 파이루브산을 생성하거나 TCA 회로의 중간산물 중 하나를 생성하는 아미노 산 간에서 글루코스와 글리코겐의 생성을 증가시키고 또한 근육에서의 글리코겐 합성 α-ketoglutarate: 글루타민 , 글루탐산 , 프롤린 , 아지닌 , 히스티딘 , Pyruvate 형성 아미노산 : 알라닌 , 세린 , 글라이신 , 시스테인 , 트레오닌 Fumarate 형성 아미노산 : 페닐알라닌 , 타이로신 , Succinyl-CoA 형성 아미노산 : 메티오닌 , 발린 , 아이소류신 , 트레오닌 , Oxaloacetate 형성 아미노산 : 아스파트산 , 아스파라진 .•
케톤생성 아미노산 (Ketogenic AAs)
이화되어 아세토아세테이트를 형성하거나 그것의 전구체인 아세틸 CoA 를 형성하는 아 미노산 류신과 ( 라이신 ) 은 오로지 케톤생성으로만 작용하는 아미노산*
Both glucogenic and ketogenic AAs
Figure 29-1 Amphibolic intermediates formed from the carbon skeletons of amino acids
•Transamination typically initiates amino acid catabolism
1) Asparagine, aspartate, glutamine, and glutamate
① Carbons of Asn and Asp
oxaloacetate
② Carbons of Gln and Glu
α-ketoglutarate(Fig. 29-2)
Figure 28-6 Transamina-tion
Figure 29-2 Catabolism of L-asparagine (top) and of L-glutamine (bottom) to amphibolic intermediates
1) Asparagine, aspartate, glutamine, and glutamate
Carbons of Asn and Asp
oxaloacetate
Carbons of Gln and Glu
α-ketoglutarate
1) Asparagine, aspartate, glutamine, and glutamate
Carbons of Asn and Asp
oxaloacetate
Carbons of Gln and Glu
α-ketoglutarate
Figure 29-3 Catabolism of proline (mitochondria)
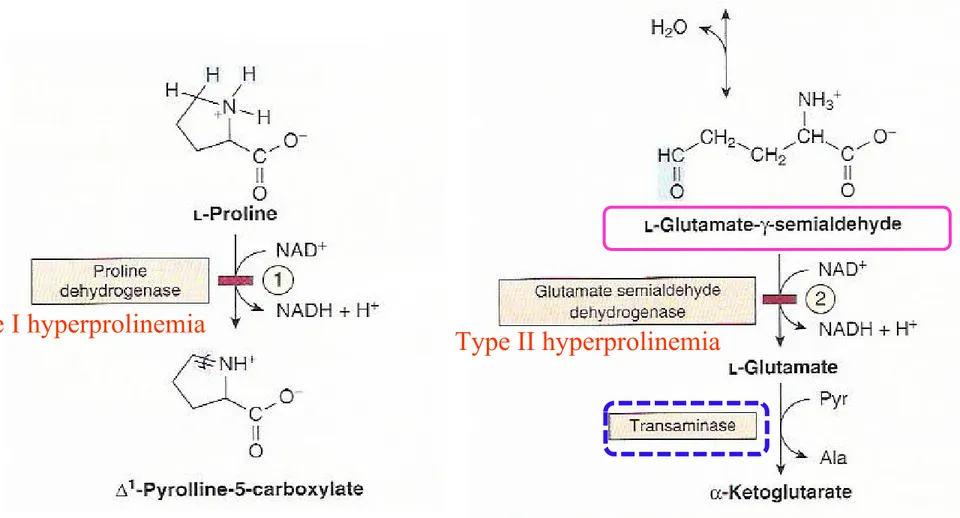
2) Proline
① Pro dehydroproline glutamate-γ-semialdehyde glutamate α-ketoglutarate (Fig. 29-3)
② Metabolic block in type I hyperprolinemia : Pro dehydrogenase
③ Type II hyperprolinemia : glutamate- γ-semialdehyde dehydrogenase
type I hyperprolinemia
Figure 27-8 Biosynthesis of proline from glutamate
Proline
2) Proline
① Pro dehydroproline glutamate-γ-semialdehyde glutamate
α-ketoglutarate
(Fig. 29-3)
Figure 29-4 Catabolism of arginine
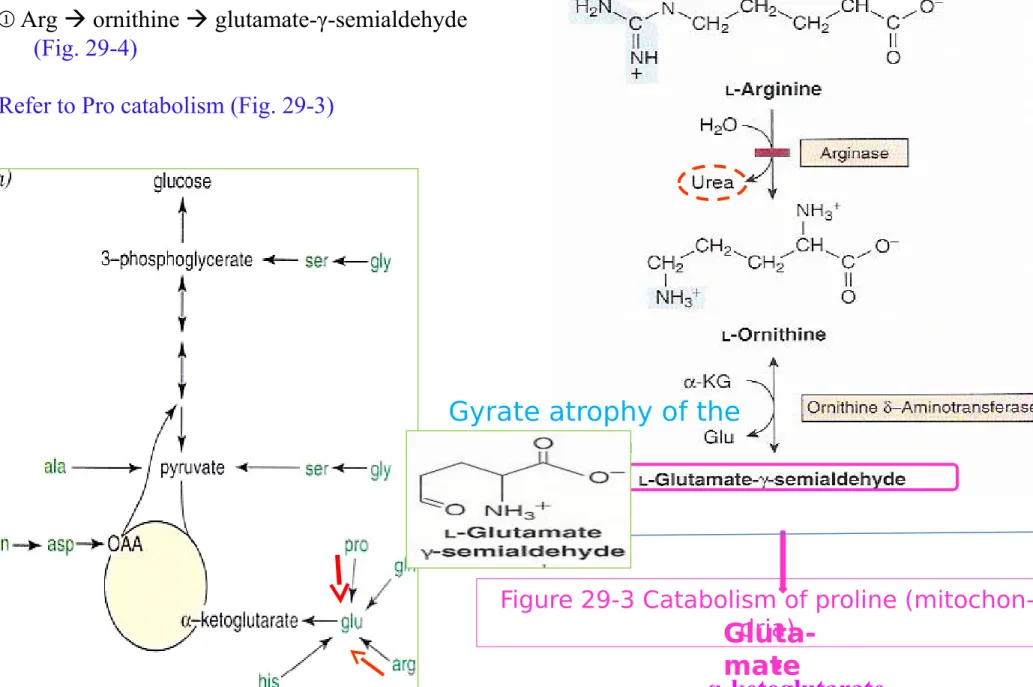
3) Arginine and ornithine
① Arg ornithine glutamate-γ-semialdehyde (Fig. 29-4)
Refer to Pro catabolism (Fig. 29-3)
Gluta-mate
α-ketoglutarate
Figure 29-3 Catabolism of proline (mitochon-dria)
Gyrate atrophy of the
retina
Figure 29-4 Catabolism of argi-nine
3) Arginine and ornithine
② Hyperornithinemia-hyperammonia syndromes: A defective mito. ornithine-citrulline antiporter
(Fig. 28-12) : impaired transport of ornithine into mitochondria for use in urea synthesis
Gluta-mate
Figure 29-5 Catabolism of L-histidine to a-ketoglu-tarate
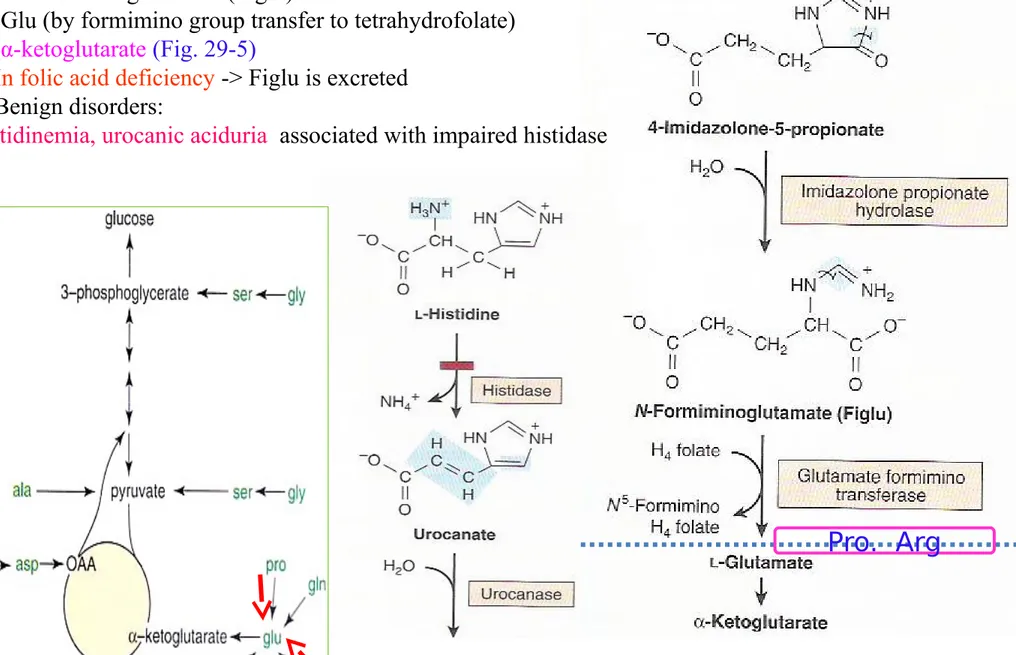
4) Histidine
① His urocanate 4-imidazolone-5-propionate N-forminoglutamate (Figlu)
Glu (by formimino group transfer to tetrahydrofolate) α-ketoglutarate(Fig. 29-5)
② In folic acid deficiency -> Figlu is excreted ③ Benign disorders:
histidinemia, urocanic aciduria associated with impaired histidase
*Catabolism of Glycine, Serine, Alanine, Cysteine, Threonine and 4-Hydroxyproline
① All of carbons of Gly, Ser, Ala, Cys, Thr, 4-Hyp form pyruvate acetyl-CoA
Figure 29-1 Amphibolic intermediates formed from the carbon skeletons of amino acids
1) Glycine
① Gly cleavage complex of liver mito: splits Gly to CO2 and NH4+, and forms N
5,N10-methylene THF (Fig. 29-6)
② Glycinuria : defect in renal tubular reabsorption
③ Defect in primary hyperoxaluria : failure to catabolize glyoxalate formed by deamination of glycine ④ Oxidation of glyoxalate to oxalate urolithiasis, nephrocalcinosis, and early mortality
Figure 29-6 The glycine cleavage system of liver mitochondria
Glycine dehydrogenase (decarboxylating) Amino-forming aminomethyltrans-ferase Dihydrolipoamide dehydrogenase hyperoxaluria Glycine aminotransferase Glu or Ala ; Amino donor
2) Serine
① Ser to Gly by Ser hydroxymethyltransferase (Fig. 29-7)
② Ser catabolism Gly , pyruvate
3) Alanine
① Transamination of Ala pyruvate
② No known metabolic defect
Figure 29-7 Interconversion of serine and glycine by serine
hydroxymethyltrans-ferase
Figure 27-3 Formation of ala-nine
4) Cysteine
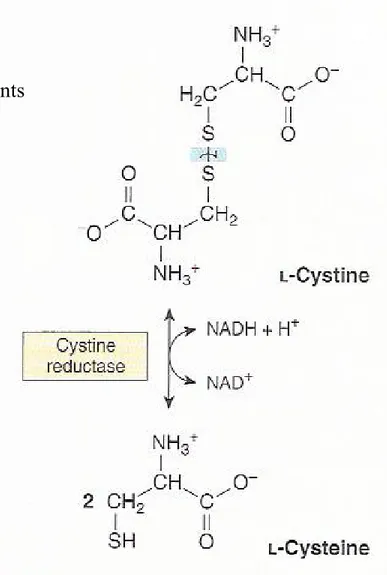
① Cystine to cysteine by cystine reductase (Fig. 29-8)
② Two different pathways : cysteine to pyruvate (Fig. 29-9)
Figure 29-8
The cystine reductase reaction
Figure 29-9 Catabolism of L-cysteine via the cysteine sulfinate pathway and by the 3-mercaptopyruvate pathway
Figure 29-8 The cystine reductase reac-tion
4) Cysteine
① Cystine to cysteine by cystine reductase (Fig. 29-8)
② Two different pathways : cysteine to pyruvate (Fig. 29-9)
③ Numerous abnormalities of Cys metabolism: Cystine-lysinuria (cystinuria; benign);
* defects in renal absorption of AAs (Cys, Lys. Arg . Ornithine) * 신장의 근위세관 (proximal tubule) 에서
시스틴 , 라이신 , 아지닌 , 오니틴을 재흡수하는 체계가 결핍되어 일어나는 질환 .
* 시스틴을 재흡수하지 못하여 신장결석이 생성
mixed disulfide of cysteine and homocysteine excreted by cystinuria patients is more soluble than cystine and reduces formation of cystine calculi
(시스틴 결석 ) (29-10)
Figure 29-10 Mixed disulfide of cysteine and homocysteine
4) Cysteine
① Cystine to cysteine by cystine reductase (Fig. 29-8)
② Two different pathways : cysteine to pyruvate (Fig. 29-9)
③ Numerous abnormalities of Cys metabolism:
Cystine-lysinuria (cystinuria; 시스틴뇨증 ) (benign); mixed disulfide of cysteine and homocysteine (29-10)
Homocystinurias:
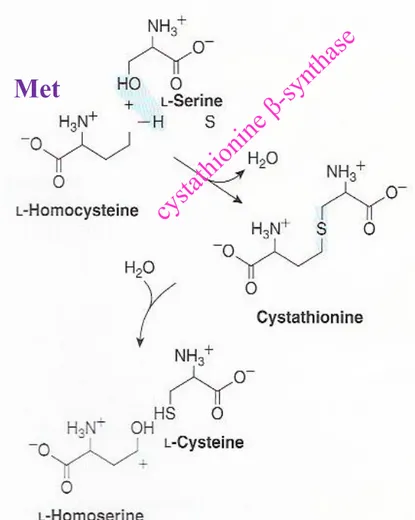
* deficiency in the reaction catalyzed by cystathionine β-synthase * osteoporosis ( 골다공증 ), mental retardation ( 정신지체 )
Figure 27-9 Conversion of homocysteine and serine
to homoserine and cysteine
cy
sta
thi
on
ine
β-sy
nth
as
e
Met
4) Cysteine
① Cystine to cysteine by cystine reductase (Fig. 29-8)
② Two different pathways : cysteine to pyruvate (Fig. 29-9)
③ Numerous abnormalities of Cys metabolism:
Cystine-lysinuria (cystinuria; 시스틴뇨증 ) (benign);
Cystine calculi ; mixed disulfide of cysteine and homocysteine (29-10)
Homocystinurias:
* deficiency in the reaction catalyzed by cystathionine β-synthase
* osteoporosis, mental retardation Cysinosis (cysteine storage disease):
* defective carrier-mediated transport of cystine * deposition of cystine crystals in tissues
* early mortality from acute renal failure ( 급성신부전 )
Figure 29-8 The cystine reductase reac-tion
Cysteine
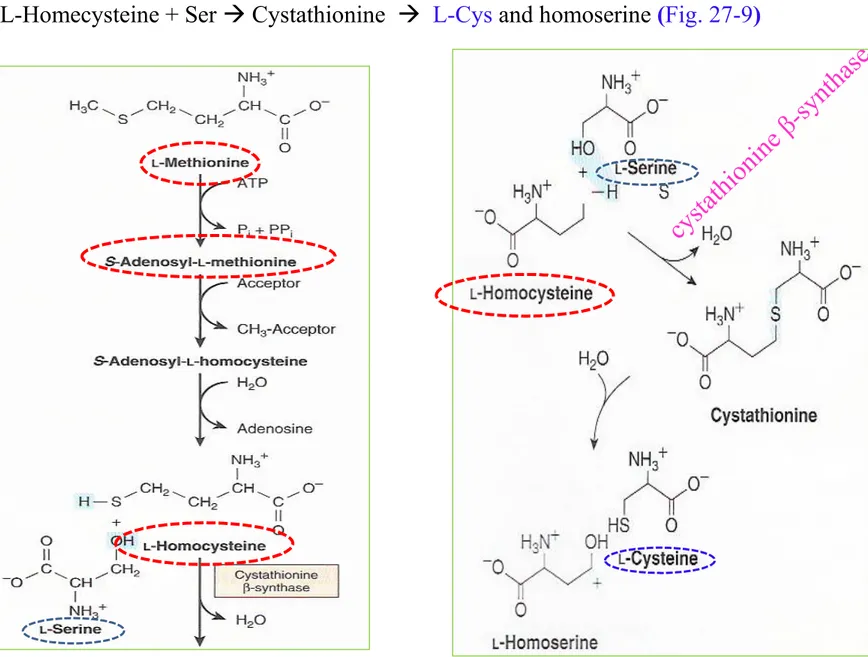
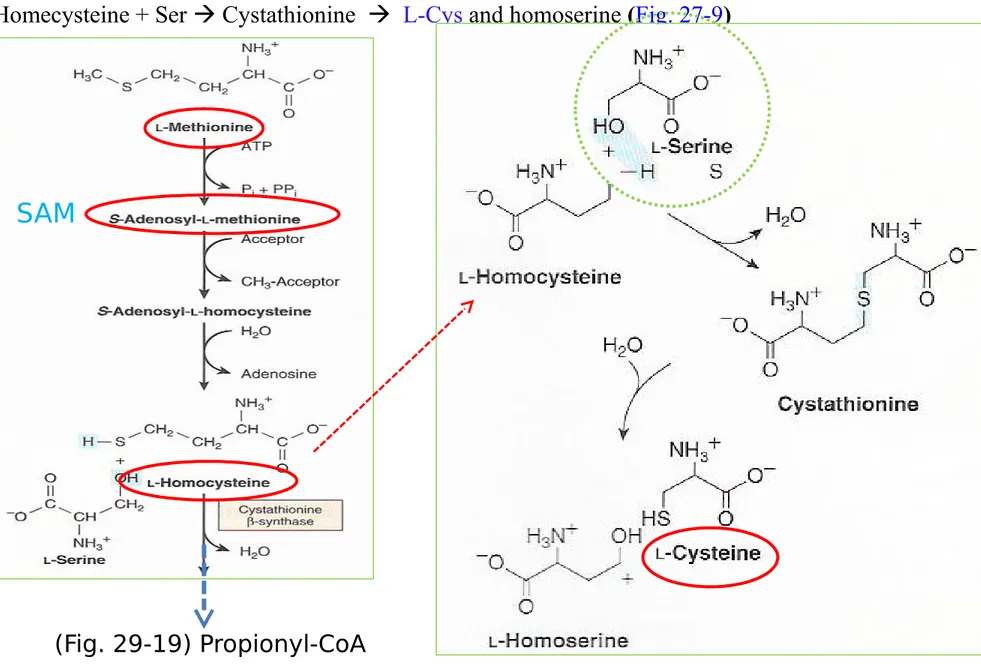
Cys from Met (nutritionally essential AA): Met SAM homocysteine (Fig. 29-19)
L-Homecysteine + Ser Cystathionine L-Cys and homoserine (Fig. 27-9)
Figure 27-9 Conversion of homocysteine and serine to homoserine and cys-teine
cy
sta
thi
on
ine
β-sy
nth
as
e
Cysteine
Cys (nutritionally nonessential AA) from Met (nutritionally essential AA) : Met SAM homocys-teine (Fig. 29-19)
L-Homecysteine + Ser Cystathionine L-Cys and homoserine (Fig. 27-9)
Figure 27-9 Conversion of homocysteine and serine to homoserine and cys-teine
SAM
5) Threonine
① Thr acetaldehyde + Gly and pyruvate
② Oxidation of acetaldehyde to acetate formation of acetyl-CoA (Fig. 29-11)
Figure 29-11
Conversion of threonine to glycine and acetyl-CoA
pyruvate
Ser
Figure 29-7 Interconversion of
serine and glycine by serine hydroxymethyl-transferase
Figure 29-12 Intermediates in L-hydroxyproline catabolism
6) 4-Hydroxyproline
① Catabolism of Hyp α-keto- γ-hydroxyglutarate
② An aldol-type cleavage glyoxylate + pyruvate (Fig. 29-12)
③ hyperhydroxyprolinemia (benign) : A defect in hydroxyPro dehydrogenase
Type II hyperprolinemia
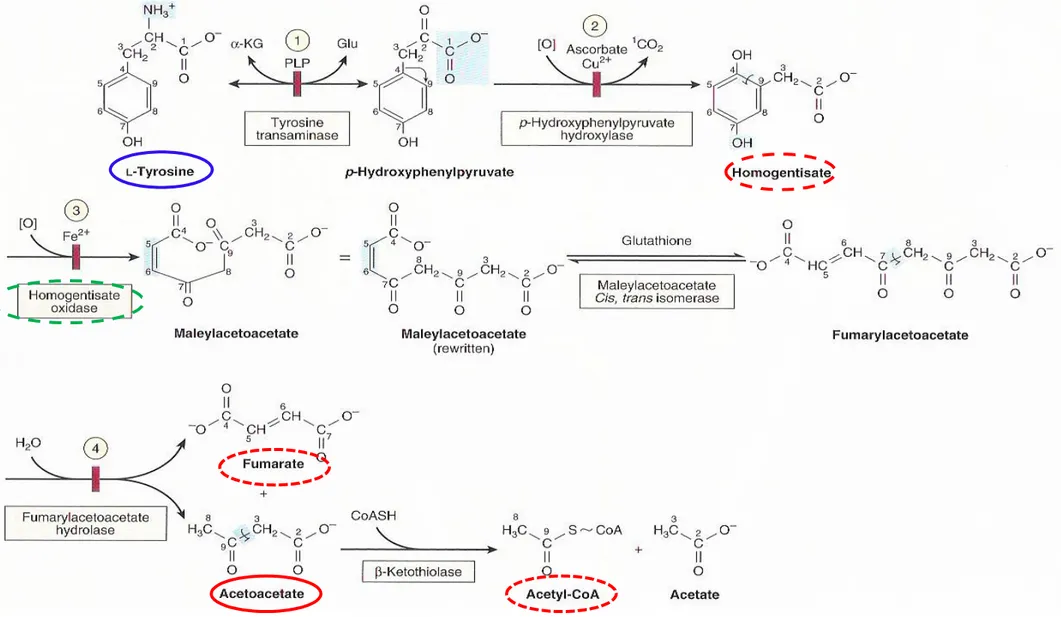
Figure 29-13 Intermediates in tyrosine catabolism
Additional AAs that form acetyl-CoA
1) Tyrosine
① Tyr p-hydroxyphenylpyruvate homogentisate maleylacetoacetate fumarylacetoacetate
Figure 29-1 Amphibolic intermediates formed from the carbon skeletons of amino acids
Figure 29-13 Intermediates in tyrosine catabolism
Additional AAs that form acetyl-CoA
1) Tyrosine
Type I tyrosinemia (tyrosi-nosis) (reaction 4)
Type II tyrosinemia (Richner-Hanhart syn-drome) (reaction 1)
Neonatal tyrosinemia : lower p-hydroxyphenylpyruvate hydroxylase activity (reaction 2)
Additional AAs that form acetyl-CoA
1) Tyrosine
Alkaptonuria (reaction 3)
Alkaptonuria (1859, Garrod’s classic ideas of heritable metabolic disorder) : lack of homogentisate oxidase
: urine darkens due to oxidation of homogentisate
2) Phenylalanine
① Phe Tyr (Fig 27-10) subsequent reactions of tyrosine (Fig. 29-13)
② Hyperphenylalaninemias : defects in phenylalanine hydroxylase
(Type I, classic phenylketonuria or PKU), or in dihydrobiopterin re-ductase (Types II and III), or in dihydrobiopterin biosynthesis
(types IV and V)
③ Alternative catabolites (Fig. 29-14)
Figure 27-10 The phenylalanine hydroxylase re-action
phenylalanine hydroxylase
Figure 29-14 Alternative pathways of phenylalanine catabolism in
*
효소의 결핍 , 진단 , 페닐케톤뇨증의 치료
•
페닐케톤뇨증 (PKU):
-
페닐알라닌 하이드록실레이스의 결핍으로 인해 발병하는 질환 . 고페닐
알라닌혈증은 마찬가지로 하이드록실레이스의 보효소인 4 중 수소바이오
테린을 합성하거나 분해하는 효소의 결핍으로 인하여 생기는 질환 .
-
치료하지 않은 PKU 환자의 경우 정신지체 , 걸음 혹은 대화장애 , 간
질 , 과운동성 , 떨림 , 성장장애가 발생 .
-
신생아가 단백질을 섭취하기 시작한 후 48 시간에 채취한 혈액을 이용하
여 조기에 진단 .
-
치료는
페닐알라닌의 섭취 제한
.
2) Phenylalanine
② Hyperphenylalaninemias : defects in phenylalanine hydroxylase (Type I, classic phenylketonuria or PKU) , or in dihydrobiopterin reductase (Types II and III), or in dihydrobiopterin biosynthesis (types IV and V)
④ Diagnoses :
* Prenatal DNA probes (Phe hydroxylase or dihydrobiopterin reductase), * Neonatal FeCl3 (detect urinary phenylpyruvate)
⑤ A diet low in Phe
Figure 29-15 Reactions and intermediates in the catabolism of L-lysine
3) Lysine
① Lys crotonyl-CoA (Fig. 29-15)
acetyl-CoA + CO2 by the reactions of fatty acid catabolism
(Fig. 22-3)
② Hyperlysinemia:
elevated lysine (ornithine) inhibits liver arginase hyperammonemia
Mitochon-dria
Figure 29-16 Catabolism of L-tryptophan
4) Tryptophan
① Trp via kynurenine-anthranilate pathway (Fig. 29-16) N- L-formylkynurenine (by Trp oxygenase + O2) L-kynurenine
② Kynureninase require PLP and excretion of xanthurenate (Fig. 29-17) diagnosis of vitamin B6 deficiency
③ Hartnup disease impaired intestinal & renal transport of Trp ④ Pellagra-like signals and symptoms : Trp deficiency
Figure 29-18 Formation of S-adenosylmethionine
5) Methionine
Figure 29-19 Conversion of methionine to propionyl-CoA
5) Methionine
① Met + ATP S-adenosylmethionine (SAM), “active Met” (Fig. 29-18)
② Met SAM S-adenosyl-L-homocysteine-> L-homocysteine -> Cystathione -> L-cysteine + α-ketoglutarate-> propionyl-CoA (Fig 29-19) and ultimately succinyl-CoA (Fig. 20-2)
methyl-
Malonyl-CoA
B12 coenzymeSuccinyl-CoA
Intermediates ofCitric acid cycle
Cysteine
Cys (nutritionally nonessential AA) from Met (nutritionally essential AA) : Met SAM homocys-teine (Fig. 29-19)
L-Homecysteine + Ser Cystathionine L-Cys and homoserine (Fig. 27-9)
Figure 27-9 Conversion of homocysteine and serine to homoserine and cys-teine
SAM
The Initial Reactions are Common to All Three Branched-chain Amino Acids Transamination , Oxidative decarboxylation (decarboxylase, a transacylase, and
a dihydrolipoly dehyrogenase resembles pyruvate dehydrogenase), Dehydrogenation
Maple syrup urine
dis-ease
(branched-chain
ke-tonuria)
Figure 29-20 The analogous first three reactions in the catabolism of leucine, valine, and isoleucine
6. Metabolic Disorders of Branched-chain Amino Acid Catabolism
① Maple syrup urine disease (branched-chain ketonuria) <- defect in α-keto acid dehydrogenase complex
(reaction 2, Fig. 29-20) -> elevated plasma & urine levels of Leu, Ile, Val, α-keto acids, and α-hydroxy acids
@
효소 결핍 , 진단 , 단풍시럽뇨증의 치료
단풍시럽뇨증 (MSUD, Maple syrup urine disease):
- 류신 , 아이소류신 , 발린을 탈카복시화 하는 효소인 가지사슬 - 케토산 디하이드로저네이스가 부분적이거나 완전하게 결핍되어 발병하는 질환 . – 이러한 아미노산과 그에 따른 α- 케토산은 혈중에 축적되어 뇌의 기능을 저해 . 증상 은 섭식 장애 , 구토 , 탈수 , 심각한 대사성 산증 , 뇨에서의 특징적인 냄새 . – 만약 치료하지 않는다면 환자는 심각한 정신지체 , 물리적 운동장애를 겪게 되고 심지 어는 죽음에까지 이름 . – 이 질환은 생후 24 시간 이내의 혈액 샘플을 채취하여 진단 . – MSUD 환자는 류신 , 아이소류신 , 발린의 함량을 제한한 식이로 치료 .
The Initial Reactions are Common to All Three Branched-chain Amino Acids
* Subsequent catabolism of each AAs (Fig. 29-21, 22, 23)
Acetoacetate + Acetyl- CoA
(Fig. 29-21)Propionyl-CoA + Acetyl-CoA
(Fig. 29-22)
Succunyl-CoA
Chapter 30.
Conversion of Amino Acids to Specialized Products
Biomedical importance
① Modified amino acid in protein for a specific function
② Amino acid derivatives: heme, purines, pyrimidines, hormones,
neurotransmitters, and biological active peptides
③ Small peptides or peptide like molecules: histidine
④ Neurotransmitters and many drugs
α-amino acids
Alanine:
- Carrier of ammonia and of the carbons of pyruvate from skeletal muscle to liver via cori cycle (Fig. 20-4)
Figure 28-3 The glucose-alanine cycle Fig 20-4 Cori cycle
Figure 28-4 Summary of amino acid exchange between organs immediately after feeding
glycogen glycogen
Figure 30-1 Arginine, ornithine, and proline metabolsm
α-amino acids
Arginine:
- formamidine donor for creatine synthesis (Fig. 30-12)
Figure 30-12 Biosynthesis of creatine
Figure 30-1 Arginine, ornithine, and proline metabolsm
α-amino acids
Arginine:
- formamidine donor for creatine synthesis (Fig. 30-12)
Figure 30-8 Intermediates and enzymes that participate in the biosynthesis of spermidine and spermine
Figure 30-8 Intermediates and enzymes that participate in the biosynthesis of spermidine and spermine
Methion-ine SAM
NH3
N
H
3
N
H
3
Ornithine Spermi-dine SpermineFigure 30-9 Catabolism of polyamines
Polyamin
e
oxidase
Polyamin
e
oxidase
Figure 30-1 Arginine, ornithine, and proline metabolsm
α-amino acids
Arginine:
- formamidine donor for creatine synthesis
- Ornithine Polyamine (putrescine spermine and spermidine )(Fig. 30-8)
Figure 30-2 The reaction catalyzed by phosphopantothenate-cysteine ligase
α-amino acids
Cysteine:
- biosynthesis of coenzyme A (Fig. 44-18) by forming 4-phosphopanthothenoyl-cysteine
(Fig. 30-2)
- taurine conjugates with bile acids (Fig. 26-7; Biosynthesis and degradation of bile acids)
Figure 30-4 Biosynthesis of hippurate
α-amino acids
Glycine:
① Metabolites and pharmaceuticals excreted as water-soluble glycine conjugates:
glycocholic acid (Chap 26) and hippuric acid (Fig. 30-4) formed from the food additive bezoate
α-amino acids
Glycine:
① Metabolites and pharmaceuticals excreted as water-soluble glycine conjugates: glycocholic acid (Chap 26), hippuric acid (Fig. 30-4)
② Gly incorporated into creatine(Fig. 30-12)
Figure 30-12 Biosynthesis of creatine and creati-nine Arg
SA
M
Glycine
Creatine
α-amino acids
Glycine:
① Metabolites and pharmaceuticals excreted as water-soluble glycine conjugates glycocholic acid (Chap 26), hippuric acid (Fig. 30-4)
② Gly incorporated into creatine (Fig. 30-12)
③ Heme (Chap. 31)
- Nitrogen and a-Carbon are incorporated into pyrrole rings and methylene bridge carbons of heme ④ Purine biosynthesis (Fig. 33-1)
- Entire glycine molecule becomes atoms of 4,5, and 7 of purines
Figure 30-5 The reaction catalyzed by histidine decarboxylase
α-amino acids
Histidine
① Decarboxylation of His to histamine by broad specificity of aromatic L-amino acid decarboxylase (Dopa, 5-hydroxytryptophan, Phe, Tyr, and Trp) (Fig. 30-5) : 알레르기 반응 , 위산분비 , 모든 조직 에 존재 .
② Histidine compounds present in the human body ergothioneine, carnosine, and anserine (Fig. 30-6)
- Unknown functions,
- major constituents of excitable tissues, brain, and skeletal muscle ③ Wilson’s disease low urinary levels of 3-methylHis
Figure 30-6 Derivatives of histidine
His-Figure 30-7 Biosynthesis of S-adenosylmethionine, cat-alyzed by methionine adenosyltransferase (MAT)
Methionine
①
S-Adenosylmethione :
methyl donor (Fig. 30-7)
- major nonprotein fate
- principal source of methyl group in the body
α-amino acids
SA
M
(Figure 29-19)
Methion-ine
Methionine
① S-Adenosylmethione : methyl donor (Fig. 30-7)
- major nonprotein fate
- principal source of methyl group in the body
②
Biosynthesis of polyamine: spermine and spermidine (
Fig. 31-4
)
α-amino acids
Spermidine and spemine : growth factors
(Fig. 30-8)
Catabolism of polyamines
(Fig. 30-9)
Figure 30-8 Intermediates and enzymes that participate in the biosynthesis of spermidine and spermine
Methion-ine SAM
NH3
N
H
3
N
H
3
Ornithine Spermi-dine SpermineFigure 30-9 Catabolism of polyamines
Polyamin
e
oxidase
Polyamin
e
oxidase
Serine
• Biosynthesis of
sphingosine (ceramide)
(Chap. 24)
• Biosynthesis of
purine , pyrimidines (Chap. 33)
- carbons 2 and 8 of purines
- methyl group of thymine
• Conversion of serine and homocysteine : cystathione β-synthase :
Cysteine
cy
sta
thi
on
ine
β-sy
nth
as
e
Met
Figure 27-9 Conversion of homocysteine and serine
to homoserine and cysteine
SAM
α-amino acids
Ser-ine
Cys-teine
Figure 30-10 Biosynthesis and metabolism of serotonin and melatonin
Tryptophan (Trp)
Trp 5-hydroxyTrp by Trp hydroxylase
serotonin (5-hydroxytryptamine)
by decarboxylation
(Fig. 30-10)
* serotonin : a potent vasoconstrictor, stimulator of smooth muscle contraction
α-amino acids
Serotonin
Tryptophan
5-OH
Trypto-phan
Trypto-phan
hydroxy-lase
Figure 30-10 Biosynthesis and metabolism of serotonin and melatonin
MAO
Melatonin
Serotonin
N-acetylation of serotonin
and O-methylation in pineal body
Tyrosine (Catecholamines)
* In neural cells, Tyr (L-dopa , dopamine) norepinephrine and epinephrine (Fig. 30-11)
* In melanocytes, different enzyme hydroxylase Tyr (tyrosinase melanin) * In adrenal medulla, phenylethanolamine-N-methyltransferase utilizes SAM epinephrine
* Tyr is also precursor of triiodothyronine and thyoxine (Ch. 42)
Figure 30-11 Conversion of tyrosine to epinephrine and norepinephrine in neuronal and adrenal cells
α-amino acids
SA
M
Tyrosine
Dopa
Dopamin
e
Norepineph-rine
Epinephrine
Figure 30-12 Biosynthesis of creatine and creati-nine
Creatinine
① Creatinine in muscle from creatine
(Fig. 30-12)
② 24-hour urinary excretion of creatinine is proportionate to muscle mass
③
Gly, Arg, and Met
creatinine biosynthesis
Arg
SA
M
Methionine
Creatine
Creatinine
Glycine
-
Present in tissues in a free form-alanine, -aminoisobutyrate,
γ-Aminobutyrate (GABA)
Non-α-amino acids
-alanyl dipeptides
: Activate myosin ATP ase
; Chelate copper
Figure 30-13 Metabolism of g-aminobutyrate