ORIGINAL CONTRIBUTION
Proximal Dominant Hereditary Motor and Sensory
Neuropathy With Proximal Dominance Association
With Mutation in the TRK-Fused Gene
Sang-Soo Lee, MD; Hye Jin Lee, MSc; Jin-Mo Park, MD; Young Bin Hong, PhD; Kee-Duk Park, MD; Jeong Hyun Yoo, MD; Heasoo Koo, MD; Sung-Chul Jung, MD; Hyung Soon Park, MD; Ji Hyun Lee, PhD; Min Goo Lee, MD; Young Se Hyun, MSc; Khriezhanou Nakhro, MSc; Ki Wha Chung, PhD; Byung-Ok Choi, MD
Importance:Hereditary motor and sensory neuropa-thy with proximal dominance (HMSN-P) has been re-ported as a rare type of autosomal dominant adult-onset Charcot-Marie-Tooth disease. HMSN-P has been de-scribed only in Japanese descendants since 1997, and the causative gene has not been found.
Objectives:To identify the genetic cause of HMSN-P in a Korean family and determine the pathogenic mecha-nism.
Design:Genetic and observational analysis.
Setting:Translational research center for rare neuro-logic disease.
Participants:Twenty-eight individuals (12 men and 16 women) from a Korean family with HMSN-P.
Main Outcome Measures: Whole-exome
sequenc-ing, linkage analysis, and magnetic resonance imaging .
Results: Through whole-exome sequencing, we
re-vealed that HMSN-P is caused by a mutation in the
TRK-fused gene (TFG). Clinical heterogeneities were re-vealed in HMSN-P between Korean and Japanese patients. The patients in the present report showed faster progres-sion of the disease compared with the Japanese patients, and sensory nerve action potentials of the sural nerve were lost in the early stages of the disease. Moreover, tremor and hyperlipidemia were frequently found. Magnetic reso-nance imaging of the lower extremity revealed a distinct proximal dominant and sequential pattern of muscular involvement with a clearly different pattern than pa-tients with Charcot-Marie-Tooth disease type 1A. Par-ticularly, endoneural blood vessels revealed marked nar-rowing of the lumen with swollen vesicular endothelial cells.
Conclusions and Relevance: The underlying cause
of HMSN-P proves to be a mutation in TFG that lies on chromosome 3q13.2. This disease is not limited to Japa-nese descendants, and marked narrowing of endoneu-ral blood vessels was noted in the present study. We be-lieve that TFG can affect the peripheral nerve tissue. JAMA Neurol. 2013;70(5):607-615. Published online March 18, 2013. doi:10.1001/jamaneurol.2013.1250
C
HARCOT-MARIE-TOOTHdisease (CMT), which is also called hereditary mo-tor and sensory neuropa-thy (HMSN), is a clini-cally and geneticlini-cally heterogeneous disorder of the peripheral nervous system.1To date, more than 50 causative genes or loci have been reported to be associated with CMT at the Inherited Peripheral Neuropathies Mutation Database (http://www.molgen.ua .ac.be/CMTMutations/Mutations/) or in other reports.2-5However, for many pa-tients, the genetic causes of their disease are unknown.
Hereditary motor and sensory neuropa-thy with proximal dominance (HMSN-P), which is characterized by predominant proximal muscle weakness and distal sen-sory disturbances, is a rare autosomal
dominant, adult-onset, peripheral neuro-degenerative disorder.6 Until now, HMSN-P has been reported only in Japan or in Japanese descendants.7,8Takashima et al6mapped the locus on chromosome 3q14.1-q13. Later, they suggested a nar-rowed locus on 3q13.1 within 3.1 cM.9 Maeda et al7also reported a candidate lo-cus that resides within 7.3 Mb from chro-mosome 3q11-q13.1.
Whole-exome sequencing is an effi-cient tool for identifying underlying ge-netic causes. It has been proven to provide potentially important contributions to re-veal the genetic causes of rare human
dis-Video available online at
www. jamaneuro.com
Author Aff Departmen Chungbuk School of M (Dr S.-S. Le Biological S National Un (Mss H. J. L Mr Hyun, a Departmen (Drs J.-M. P K.-D. Park, Radiology ( (Dr Koo), a (Dr Jung), University Seoul; and Pharmacolo Project for Yonsei Univ Medicine, S J. H. Lee, an Korea. Author Affiliations: Department of Neurology, Chungbuk National University School of Medicine, Chungbuk (Dr S.-S. Lee); Department of Biological Science, Kongju National University, Gongju (Mss H. J. Lee and Nakhro, Mr Hyun, and Dr Chung); Departments of Neurology (Drs J.-M. Park, Hong, K.-D. Park, and Choi), Radiology (Dr Yoo), Pathology (Dr Koo), and Biochemistry (Dr Jung), Ewha Womans University School of Medicine, Seoul; and Department of Pharmacology, Brain Korea 21 Project for Medical Science, Yonsei University College of Medicine, Seoul (Drs H. S. Park, J. H. Lee, and M. G. Lee), Korea.eases.10,11Whole-exome sequencing has recently been in-troduced in studies of CMT and has exhibited great accuracy in identifying rare genetic causes.4,5,12
The TRK-fused gene (TFG) (MIM 602498), which lies on chromosome 3q13.2, was first identified in human pap-illary thyroid carcinoma as a fusion partner of the NTRK1 gene (MIM 191315).13Despite the fact that TFG is ubiq-uitously expressed across several cancerous and normal tissues, its function remains unclear.14,15Recent studies have suggested that TFG protein is implicated in regu-lating cargo export at the endoplasmic reticulum16and in putative metastatic melanoma tumor suppression17; however, its biological function was still largely un-known. In this study, we report what we believe to be the first non-Japanese family with clinical manifesta-tions of HMSN-P and suggest a heterozygous mutation in TFG as the cause of the disease.
METHODS PARTICIPANTS
This study included 28 members of a Korean family with au-tosomal dominant CMT (family ID: FC457; 12 males and 16 females) with proximal dominant involvement (Figure 1and eTable 1). The study also included 750 healthy controls who had no clinical features or family history of CMT. Informed con-sent was obtained from all participants and from the parents of participants younger than 18 years according to the
proto-col approved by the institutional review board for Ewha Wom-ans University, Mokdong Hospital, Seoul, Korea.
CLINICAL AND ELECTROPHYSIOLOGIC ASSESSMENTS
The clinical evaluation was performed by 2 independent neu-rologists (S.-S. Lee and B.-O. Choi). Information on deceased family members was obtained from the available relatives. Strength of the flexor and extensor muscles was assessed manu-ally using the standard Medical Research Council scale. To de-termine the extent of physical disability, we used a functional disability scale.18Sensory impairments were assessed in terms
of the level and severity of pain, temperature, vibration, and position.
Neurophysiologic studies were carried out on 5 affected in-dividuals (4 males and 1 female). Motor nerve conduction ve-locities and sensory nerve conduction veve-locities were deter-mined. Amplitudes of compound muscle action potentials were measured from positive to negative peak values. Amplitudes of sensory nerve action potentials (SNAPs) were measured from positive peaks to negative peaks. Electromyography was per-formed in bilateral proximal and distal upper and lower ex-tremity muscles.
HIP, THIGH, AND LEG
MAGNETIC RESONANCE IMAGING STUDIES
Four individuals (III-23, III-27, III-29, and IV-17) with TFG mutation were studied with magnetic resonance imaging (MRI)
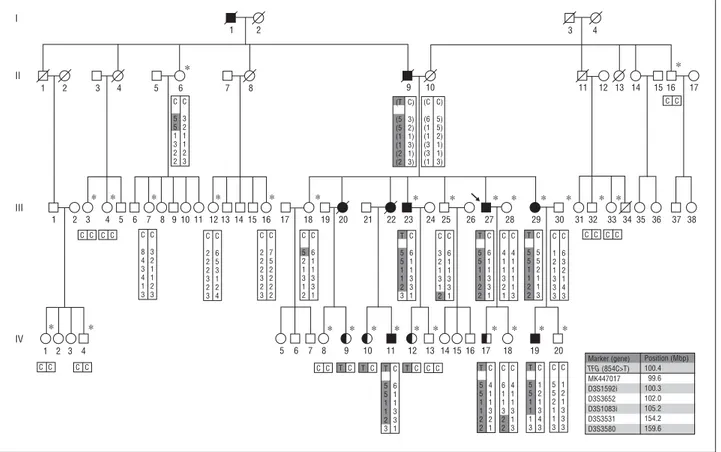
T 5 5 1 1 2 3 C 6 1 1 3 3 1 C C T C T C T C C C T 5 5 1 1 2 2 C 4 1 1 3 2 1 C 6 1 1 3 2 2 C 4 1 1 3 1 3 T 5 5 1 1 1 3 C 1 2 1 3 4 3 C 5 5 2 1 1 3 C 1 2 1 3 3 3 Marker (gene) TFG (854C>T) MK447017 D3S1592i D3S3652 D3S1083i D3S3531 D3S3580 Position (Mbp) 100.4 99.6 100.3 102.0 105.2 154.2 159.6 C 5 5 1 3 2 2 C 3 2 1 1 2 3 C 8 4 3 4 1 3 C 3 2 1 1 2 3 C 2 2 2 3 2 3 C 6 5 3 1 2 4 C 2 2 2 3 2 3 C 7 5 2 2 2 2 C 5 2 1 3 1 2 C 6 1 1 3 3 1 T 5 5 1 1 2 3 C 6 1 1 3 3 1 C 3 2 1 3 1 2 C 6 1 1 3 3 1 T 5 5 1 1 2 2 C 6 1 1 3 3 1 C 4 1 1 3 2 1 C 4 1 1 1 1 3 T 5 5 1 1 2 2 C 5 5 2 1 1 3 C 1 2 1 3 3 3 C 6 3 2 1 4 3 (T (5 (5 (1 (1 (2 (2 C) 3) 2) 1) 3) 1) 3) (C (6 (1 (1 (3 (3 (1 C) 5) 5) 2) 1) 1) 3) C C C C C C C C C C C C C C 1 2 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 3 4 1 2 3 4 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ I II III IV
Figure 1. Pedigree of the FC457 family. *Indicates an individual whose DNA was used for this study. Alleles of TFG (c.854C⬎T) as well as 6 microsatellites on 3q12-q25 are indicated in the rectangles beneath the identifiers of examined individuals. Alleles within parentheses were inferred. Although the II-9 affected man is the elder brother of the II-8 unaffected woman, II-9 is indicated on the right side of II-8 for the convenience of pedigree drawing. Mbp indicates megabase pair; square, male; circle, female; black-filled, affected; half-filled, unaffected but with TFG mutation; no fill, unaffected; arrow, proband; diagonal bar across symbol, deceased.
of the hip, thigh, and leg using a 1.5-T system (Siemens Vi-sion) equipped with a phase-array multicoil. Leg imaging was carried out in axial (field of view, 24-32 cm; slice thickness, 10 mm; and slice gap, 0.5-1.0 mm) and coronal planes (field of view, 38-40 cm; slice thickness, 4-5 mm; and slice gap, 0.5-1.0 mm). The following protocol was used in all patients: T1-weighted spin echo (repetition time/echo time [TR/TE], 570-650/14-20; 512 matrixes), T2-weighted spin echo (TR/TE, 2800-4000/96-99; 512 matrixes), and fat-suppressed T2-weighted spin echo (TR/TE, 3090-4900/85-99; 512 matrixes).19
HISTOPATHOLOGIC STUDIES
Histopathologic analyses, including an immunohistochemis-try study with anti-TFG antibody (Abcam) of the distal sural nerves, were performed in 2 patients: 1 patient (III-27) at age 50 years and the other patient (III-29) at 48 years. The density of myelinated fibers (MFs), axonal diameter, and myelin thick-ness were determined directly from the semithin transverse sec-tions using a computer-assisted image analyzer (AnalySIS; Soft Imaging System). Additional analysis also was performed at lower magnification of an electron microscope (⫻3000) for inci-dence of abnormal myelin, onion bulb formation, regenera-tive axonal clusters, and other findings.
CHROMOSOMAL MAPPING OF 3q1 REGION AND DETERMINATION OF 17p12 DUPLICATION
For the FC457 family, linkage analysis was performed on the reported HMSN-P locus (3q1-q2) and the surrounding region by genotyping of 19 fluorescent-labeled microsatellites (eTable 2).7,9The 1.4⫻ 106–base pair duplication/deletion of 17p12,
which is the most frequent genetic cause of CMT, was deter-mined by genotyping of 6 microsatellites.20Polymerase chain
reaction products were resolved on the automated genetic ana-lyzer ABI3130xl, and data were analyzed using the GENESCAN program (Applied Biosystems).
WHOLE-EXOME SEQUENCING
Whole-exome sequencing was performed for 4 individuals (af-fected, III-23, III-27, and III-29; unaf(af-fected, III-25). A stan-dard shotgun exome sequencing library was prepared using the Human SeqCap EZ Capture Array, version 2.0 (23 and III-27) or version 3.0 (III-25 and III-29) (Roche-NimbleGen), and sequencing was performed using the HiSeq2000 Genome Ana-lyzer (Illumina). Paired-end sequences were first mapped to the human reference genome UCSC assembly hg19 (http://genome .ucsc.edu), and reads mapping was achieved by the BWA pro-gram (http://bio-bwa.sourceforge.net/). Variants (single-nucleotide polymorphisms [SNPs] and insertions or deletions [indels]) were detected by the SAMTOOLS program (http: //samtools.sourceforge.net/) and compared with common vari-ants registered in dbSNP135 (http://ncbi.nlm.nih.gov/) and the 1000 Genomes Project database (http://www.1000genomes .org/). Functionally significant cosegregating variants were se-lected from 4-sample exome sequencing data. The comple-mentary DNA numbering was achieved with⫹1 corresponding to the A of the ATG initiation codon.
TRANSCRIPTOME SEQUENCING
The RNA-sequencing data were generated from a patient (III-29, a 48-year-old woman [F/48]) and 2 individuals serving as healthy controls (F/37 and F/38). The complementary DNA li-brary was prepared using the TruSeq RNA lili-brary kit (Illu-mina). The sequencing protocol consisted of total RNA
extrac-tion from the biopsy of distal sural nerve and polyA RNA extraction, RNA fragmentation, random hexamer primed re-verse transcription, and 100nt paired-end sequencing by HiSeq2000. To estimate expression levels and identify alterna-tive spliced transcripts, the RNA-Seq reads were mapped to the human genome using TopHat (version 1.3.3; http://www.tophat .cbcb.umd.edu). The transcript levels were calculated and the relative transcript abundances were measured in FPKM (frag-ments per kilobase of exon per million frag(frag-ments mapped) using Cufflinks. To discover gene fusion from RNA-seq data, the de-Fuse program (version 0.4.3; http://sourceforge.net/apps /mediawiki/defuse/) was used.
CAPILLARY DNA SEQUENCING AND IN SILICO ANALYSIS
The candidate variants considered to be causative mutations were confirmed by the capillary sequencing method using the automatic genetic analyzer ABI3130xl with the BigDye termi-nator cycle sequencing kit (Applied Biosystems). In silico pre-dictions were performed using SIFT (http://sift.jcvi.org/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/).
LOCALIZATION AND CYTOTOXICITY OF TFG
Mammalian-expressible TFG plasmids were constructed using pcDNA 3.1 vector. For mutant TFG, Pro-285 residue (CCT) was substituted with Leu (CTT) by oligonucleotide-directed mutagenesis. Immunoblot assays were performed with rou-tine methods using anti-TFG antibody (Abcam) after expres-sion of TFG in HEK 293T cells. The cytotoxic effects of TFG mutant on the NSC-34 cells were determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay kit (Invitrogen) in the NSC-34 cells, following the manufac-turer’s instructions. To determine the localization of TFG, wild-type and mutant-wild-type pEGFP-N1-TFG were transfected into the NSC-34 cell using Lipofectamine 2000 (Invitrogen). Location of TFG protein was visualized by N-terminal–fused green rescent protein in the differentiated NSC-34 cells under fluo-rescent microscope.
RESULTS
CLINICAL MANIFESTATIONS
The clinical manifestations are summarized inTable 1. The ages at onset ranged from 27 to 48 years for 5 af-fected individuals. In 3 patients (23, 27, and III-29), cramping as well as fasciculation were noted, and all 3 of them experienced hand tremor and rapid disease progression compared with patients in previous re-ports.6,7,21In the fourth generation, patient IV-11 re-ported proximal lower limb weakness at 29 years, and patient IV-19 felt frequent fasciculations at 27 years. How-ever, no symptoms were noticed in participants IV-9 (F/ 32), IV-10 (F/33), IV-12 (F/28), and IV-17 (M/23).
Physical examination revealed no pes cavus in any af-fected individuals. In addition, dysphagia, dysarthria, and bulbar symptoms were not observed. Hip abduction (Medical Research Council, G0/5 [no power] to G2/5 [mild power]) was markedly weaker than hip adduc-tion (G1/5 [low power] to G5/5 [full power]) and ex-tension (G1/5 to G5/5). Vibration and position senses were more severely disturbed than were pain and
tempera-ture senses. Areflexia was noted in the very early stages of the disease, but pathologic reflexes were not found. The frequency of hyperlipidemia was high (80% [4 tients]) but that of hyperglycemia was low (20% [1 tient]) compared with the frequency in Japanese pa-tients.6,21Elevated serum creatine kinase levels were noted in patients III-27 and III-29.
Sensory nerve action potentials of the sural nerves were lost in the early stage of the disease. Although com-pound muscle action potentials in the tibial motor nerves of patient III-29 were within the normal range, SNAPs in sural nerves were not elicited. Needle electromyogra-phy showed abundant fasciculation potentials and neu-rogenic motor unit action potentials.
Family members reported that the proband’s de-ceased grandfather (I-1), father (II-9), and 2 sisters (III-20 and III-22) had proximal muscle weakness. The initial symptoms started after their fourth decade, and they be-came bedridden 6 to 8 years later.
HIP, THIGH, AND LOWER LIMB MRI FINDINGS Results of an MRI were normal in an asymptomatic in-dividual (IV-19), but hyperintense signal abnormalities were observed in the lower limb muscles of 3 sympto-matic individuals (III-23, III-27, and III-29). The T1-weighted images demonstrated marked signal changes and atrophies in the hip muscles rather than the thigh or leg muscles; therefore, MRI findings of fatty infiltra-tions were more affected proximally (Figure 2A, E, I, and M). It is noteworthy that gluteus minimus and me-dius muscles were initially involved and revealed the most severe fatty hyperintense signal changes, but tibialis an-terior muscles were spared in the later stages.
We observed a sequential pattern of muscle involve-ment associated with disease duration and severity. In the early disease stage, diffuse fatty infiltration was ob-served in the gluteus minimus and to a lesser degree in the gluteus medius; in later stages, the infiltration was Table 1. Clinical Manifestations of 7 Individuals With P285L Mutation in the TFG Gene
Characteristic
Patients
III-23 III-27 III-29 IV-11 IV-12 IV-17 IV-19
Sex Male Male Female Male Female Male Male
Age at examination, y 56 51 48 31 28 23 27
Age at disease onset, y 45 48 46 29 . . . 27
Symptom at disease onset Proximal leg weakness Fasciculation Proximal leg weakness Proximal leg weakness . . . Fasciculation Muscle weakness Proximal limb P P P P A A A Distal limb P P P A A A A Muscle atrophy Proximal limb P P P A A A A Distal limb P P P A A A A
Sensory loss Yes Yes Yes Yes No No Yes
Areflexia Yes Yes Yes Yes No No Yes
Fasciculation Yes Yes Yes No No No Yes
Cramping Yes Yes Yes Yes No No No
Hand tremor Yes Yes Yes Yes No No Yes
Plantar response No No No No No No No Pes cavus No No No No No No No FDSa 8 3 3 2 0 0 0 Laboratory test CK, IU/Lb 72 1614 494 NE NE 102 128 Hyperglycemia Yes No No NE NE No No
Hyperlipidemia No Yes Yes NE NE Yes Yes
Tibial nerve, right/leftc
CMAP, mV NP/NP 3.9/2.3 10.9/10.1 NE NE 13.2/16.6 21.4/18.7
MNCV, m/s NP/NP 37.4/35.7 45.8/46.2 NE NE 48.1/49.5 48.3/48.3
Sural nerve, right/leftc
SNAP, µV NP/NP NP/NP NP/NP NE NE 14.0/13.0 6.0/6.9 SNCV, m/s NP/NP NP/NP NP/NP NE NE 43.5/49.2 40.7/44.3 Electromyography Neurogenic MUAPs Neurogenic MUAPs Neurogenic MUAPs NE NE Normal Frequent fasciculations
Nerve biopsy NE Axonal neuropathy Axonal neuropathy NE NE NE NE
Abbreviations: A, absent; CK, creatine kinase; CMAP, compound muscle action potential; ellipsis, disease onset had not occurred at the time of examination; FDS, functional disability scale; MNCV, motor nerve conduction velocity; MUAPs, motor unit action potentials; NE, not examined; NP, no potential; P, present; SNAP, sensory nerve action potential; SNCV, sensory nerve conduction velocity.
SI conversion factor: To convert CK to microkatals per liter, multiply by 0.0167.
aOn the FDS, 0 indicates normal; 1, normal but with cramps and fatigability; 2, inability to run; 3, walking with difficulty but without assistance; 4, walking with a
cane; 5, walking with crutches; 6, walking with a walker; 7, wheelchair bound; and 8, bedridden.18
bReference range,⬍185 IU/L.
cNormal NCV values: tibial nerve, 41.1 m/s or greater; sural nerve, 32.1 m/s or greater. Normal amplitude values: tibial nerve, 6 mV or greater; sural nerve,
present in the gluteus maximus (Figure 2B, F, J, and N). At the thigh level, there was selective severe involve-ment of the semitendinosus muscles, but the adductor muscles, vastus medialis, and intermedius muscles were relatively spared (Figure 2C, G, K, and O). In the legs, gastrocnemius and peronei muscles showed initially equal involvement; however, tibialis anterior muscles were not involved until the later stages (Figure 2D, H, L, and P).
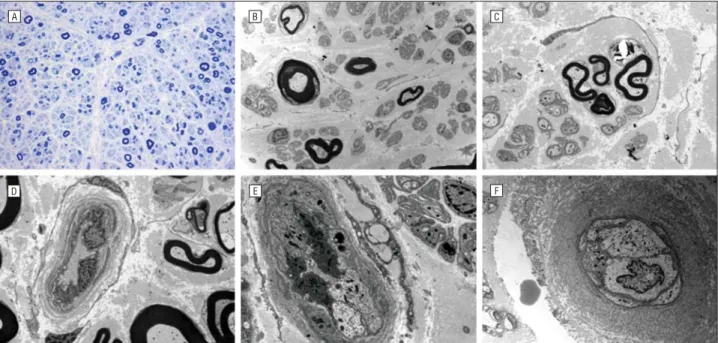
HISTOPATHOLOGIC FINDINGS
The sural nerve biopsy of III-29 revealed the absence of large MFs, with remaining medium and small MFs (4653/ mm2), and showed the unimodal distribution pattern with loss of large MFs (Figure 3A). Ultrastructural micro-graphs showed MFs with an irregular thickness of my-elin, thick MFs, and occasional regenerating axonal clus-ters (Figure 3B and C). The histopathologic features of patient III-27 were similar to those of patient III-29 but became more severe in its degree of changes. Immuno-histochemical analysis with anti-TFG antibody showed similar TFG protein distribution between 2 control cases (37- and 38-year-old women) and 2 patients (III-27 and III-29) (data not shown).
ENDONEURAL BLOOD VESSEL WITH SWOLLEN VESICULAR ENDOTHELIAL CELLS AND
NARROWED LUMEN
One patient’s (III-29) endoneural blood vessels dis-played marked narrowing of the lumen with swollen ve-sicular endothelial cells (Figure 3E) compared with the healthy control (Figure 3D, F/38). In particular, patient III-27 revealed extensive edematous changes of endo-thelial cells with a nearly obstructed lumen (Figure 3F). Concentric multilamella collections of basal lamina were prominent in both patients.
IDENTIFICATION OF LINKAGE DISEQUILIBRIUM IN THE 3q12-q25 REGION
Chromosomal mapping of the 3q region revealed a link-age disequilibrium region in the 3q12-q25 from D3S3652 to D3S3531, which spans 52.2 megabase pairs (Mbp) (Figure 1 and eFigure 1). Although the linkage region could not be narrowed into a short distance, it overlaps with the Japanese HMSN-P locus6,7between D3S1592i and D3S1083i (4.8-Mbp interval) but slightly deviates from the narrow locus (eFigure 1).9
B D A C H G F E L K I P O N M J
Figure 2. Hip, thigh, and leg magnetic resonance images (MRIs) of a clinically unaffected individual (IV-17, A-D) and 3 symptomatic individuals (III-29, E-H; III-27, I-L; and III-23, M-P) with TFG mutation. A 23-year-old man (IV-17) did not experience any symptoms, and MRIs of the hip (A and B), thigh (C), and leg (D) revealed normal findings. A 48-year-old woman (III-29) with 2 years’ disease duration showed atrophy and fatty changes in gluteus minimus and medius muscles (arrow, E) but relatively preserved gluteus maximus muscle (arrowhead, F). At the thigh level (G), the semitendinosus muscle (open arrowhead) was severely damaged, and vastus lateralis and sartorius muscles (arrowhead) were involved. On the leg (H), we found mild fatty changes in the gastrocnemius muscle (arrow); however, the tibialis anterior muscle (arrowhead) was not involved. A 51-year-old man (III-27; I-L) with 3 years’ disease duration had more progression of fatty infiltrations than his younger sister (III-29). In a 56-year-old man (III-23; M-P) with 11 years’ disease duration, we observed diffuse severe atrophy and fatty change of his whole lower limb muscles, including the hip (N). Very minimal muscle fiber remained in the adductor longus on his thigh (O), and the muscle fiber was relatively intact in the anterior compartment, especially the tibialis anterior muscles on the leg (P).
FILTERING OF NONSYNONYMOUS VARIANTS FROM WHOLE-EXOME DATA
The exome sequencing data of 4 samples are summa-rized inTable 2. The mean total sequencing yield was approximately 8.79 giga base pairs /sample, and the cov-erage rate of targeted exon regions (ⱖ10⫻) was 92.6%. The average number of observed variants per sample was 70 645 SNPs and 8427 indels, respectively. Of these, func-tionally significant variants (eg, missense, frameshift, stop-gain, stoploss, stop site, and coding indel) were 9856.8/ sample. When the nonsynonymous variants were eliminated by the dbSNP135 and 1000 Genomes Proj-ect database, each sample exhibited 165 to 348 variants.
Finally, we filtered cosegregating variants from the exome data of 4 individuals (eg, present in 3 affected individu-als [III-11, III-14, and III-16] but absent in an unaf-fected individual [III-6]), and then only 22 functionally significant variants remained (eTable 3). Except for a non-sense mutation (c.753G⬎A, W251X) in the PAICS gene, all other variants were missense mutations.
IDENTIFICATION OF A P285L MUTATION IN TFG When the 22 nonsynonymous variants were examined in all family members, a c.854C⬎T (P285L) mutation in TFG variants was cosegregated only with the affected members (Figure 1). The TFG is located within the link-B
D
A C
F E
Figure 3. Distal sural nerve biopsies and narrowed endoneural blood vessels. A, Transverse semithin sections of a patient (III-29) with toluidine blue stain revealed the absence of large myelinated fibers, with remaining medium and small fibers and occasional regenerating axonal clusters. B and C, Ultrastructural micrographs by electron microscope showed myelinated fibers with irregular thickness of myelin, thick myelinated fibers, and regenerating axonal clusters. D, Healthy control’s (woman, age 38 years) endoneural blood vessels with intact vascular lumen. E, The endoneural blood vessels in the sural nerve of patient III-29, however, showed marked narrowing of the lumen with swollen vesicular endothelial cells. F, Patient III-27 displayed extensive edematous change of endothelial cells with an almost obstructed lumen. Concentric multilamella collections of basal lamina were prominent. Original magnifications: ×40 (A), ×3000 (B), ×7000 (C), ×6000 (D), ×10 000 (E), and ×8000 (F).
Table 2. Whole-Exome Sequencing Analysis for 4 Individuals
Sample
Affected
Unaffected III-25
III-23 III-27 III-29
Total yield, Gbp 5.48 6.88 14.08 8.71
Target coverage⬎10 times, % 91.4 93.0 93.7 92.3
Rate of mappable reads per total reads, % 88.5 94.1 98.2 98.4
Mean read depth of target region 57.7 91.7 83.0 53.9
Total SNPs, No. 48 208 56 343 89 299 88 730
Total indels, No. 6298 9640 8662 8569
Filtering, No.a
Coding SNPs 19 371 19 221 20 316 20 545
Nonsynonymous variantsb 10 030 9780 9724 9893
Excluded from dbSNP135 and 1000 Genomes Project databasesc 165 348 343 376
Abbreviations: Gbp, giga base pairs; indels, insertions and deletions; SNPs, single-nucleotide polymorphisms.
aTwenty-two cosegregating, nonsynonymous variants were present in the 3 affected individuals but not in the unaffected individual. bNonsynonymous variants include splicing site, frameshift, stopgain, stoploss, and coding indels.
age disequilibrium region between D3S1592i and D3S3652 (eFigure 1). It is located on the HMSN-P link-age maps by Maeda et al7and Takashima et al6; how-ever, it is deviated from the 1-cM–narrowed map.9
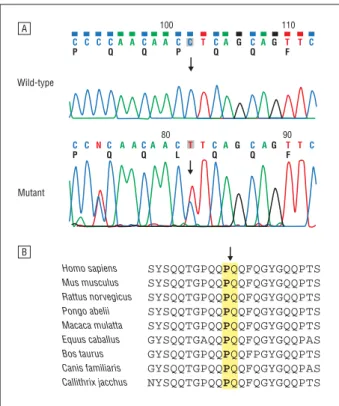
Specifically, the P285L mutation in TFG was con-firmed by the Sanger sequencing method (Figure 4A). It was completely cosegregated with affected individu-als of second and third generations in the FC457 family (eTable 1 and Figure 1). Although the mutation was also identified in 4 unaffected individuals of the fourth gen-eration (IV-9, IV-10, IV-12, and IV-17), they are younger than the age at onset (23-33 years), whereas the affected individuals’ mean age at onset was 39 years.
The TFG mutation was not found in 750 healthy con-trols, and the mutation site was well conserved between different animal species (Figure 4B). In silico analysis also predicted that the mutation may affect protein function, with scores of 0.01 (SIFT) and 2.25 (PolyPhen-2).
In addition to the TFG P285L mutation, a Y317H mu-tation in GPR128 was identified within the linkage dis-equilibrium region (eFigure 1). The GPR128 is located on chromosome 3q12.12 near the TFG with 1 Mbp of distance. Both genes were located in the linkage disequi-librium region between D3S1592i and D3S3652; how-ever, the Y317H mutation was found in an elderly unaf-fected woman (II-6, 89 years old) who revealed no symptoms of HMSN in careful clinical examinations. The GPR128 mutation site was not conserved in different spe-cies, and in silico analysis also predicted toleration of the mutation. Therefore, the GPR128 mutation was not con-sidered the genetic underlying cause, although it was not found in 500 controls. All functionally significant vari-ants in the CMT-relevant genes could be excluded from the causative mutation. Exome data from the proband (III-14) revealed 15 nonsynonymous variants in 10 CMT genes; however, no variant was cosegregated with af-fected individuals (eTable 4). The 17p12 duplication that is the underlying cause of CMT type 1A disease was also excluded by pretesting before the exome sequencing.
RNA-seq data revealed TFG expression in the periph-eral nerves but with no significant difference in the af-fected individuals compared with the healthy controls (eFigure 2). Although the TFG was first reported to be a fusion partner of the NTRK1 gene,13the RNA-seq data revealed no TFG fusion with other genes, including NTRK.
COMMENT
In the literature,6-8HMSN-P has been described only in Japanese descendants; however, we could not find any record of a blood relationship between the present Ko-rean family and the Japanese. Therefore, to our knowl-edge, this is the first report of a family with HMSN-P out-side of Japan who are not Japanese descendants. Through whole-exome sequencing of 4 individuals, we identified a novel heterozygous P285L mutation in TFG as the un-derlying cause of HMSN-P. The TFG locus is in accor-dance with previous reports.1,2On completion of the pre-sent study, Ishiura et al21independently reported a TFG mutation as a cause for HMSN-P in a Japanese family. Interestingly, the mutation is the same as in our cases.
Clinical heterogeneities were revealed between Ko-rean and Japanese patients. In our group of patients, hand tremors were present in the early stages of the disease, but this had not been recorded in the Japanese pa-tients.6,7,21The disease duration from symptom onset un-til a bedridden state was relatively short in the Koreans (6-8 years) compared with the Japanese (10-25 years).6,21 Furthermore, Ishiura et al21suggested that SNAPs of the sural nerve were lost in the later stage of this disease, but the present study revealed it in the early stage. Even though patient III-29 had normal compound muscle ac-tion potentials in the tibial motor nerve, she showed no SNAPs in the sural nerve. Furthermore, SNAPs in pa-tient IV-19 showed a low limit of the normal range in the very early stage of the disease. The frequency of hy-perlipidemia was high (80%) in the Korean patients com-pared with that of the Japanese individuals (38%).21 More-over, hyperlipidemia was observed in the clinically asymptomatic individual (IV-17); thus, it may be useful as a biomarker to evaluate presymptomatic evidence of HMSN-P.
Detailed MRI analysis revealed a distinct pattern of muscular involvement in HMSN-P. Marked hyperin-tense signal changes in the hip muscles compared with those in the thigh or the leg muscles were well related to the proximal dominance; however, those signal changes were clearly different from the ones in patients with CMT having length-dependent neuropathy.19,22Because the ear-liest and most severe changes were seen in the gluteus minimus and medius muscles, the present group of pa-tients showed a waddling gait (video). Furthermore, leg
A B Wild-type Mutant 100 110 80 90 C P C C C Q A A C Q A A C P C T C Q A G C Q A G T F T C C P C N C Q A AC Q A A C L T T C Q A G C Q AG T F T C Homo sapiens Mus musculus Rattus norvegicus Pongo abelii Macaca mulatta Equus caballus Bos taurus Canis familiaris Callithrix jacchus SYSQQTGPQQPQQFQGYGQQPTS SYSQQTGPQQPQQFQGYGQQPTS SYSQQTGPQQPQQFQGYGQQPTS SYSQQTGPQQPQQFQGYGQQPTS SYSQQTGPQQPQQFQGYGQQPTS GYSQQTGAQQPQQFQGYGQQPAS GYSQQTGPQQPQQFPGYGQQPTS GYSQQTGPQQPQQFQGYGQQPAS NYSQQTGPQQPQQFQGYGQQPTS
Figure 4. Sequencing chromatograms and conservation analysis of c.854C⬎T (Pro285Leu) in TFG. A, Confirmation of the mutation by capillary sequencing. The heterozygous c.854C⬎T mutation was cosegregated in the affected individuals but was not found in the controls. B, Conservation analysis. The mutation site (arrow) and surrounding amino acid sequences are well conserved between the different species.
MRI showed that tibialis anterior muscles were not in-volved until the later stage of the disease. Those were very different from the MRI features of CMT type 1A19,22and might explain why all reported patients with HMSN-P did not have pes cavus, which usually is seen in CMT.
The endoneural blood vessels in the sural nerve of the patients clearly revealed moderate to marked nar-rowing of the lumen and enlarged endothelial cells with swollen vesicular appearance (Figure 3D-F). Although the significance of the blood vessel change is uncertain and further studies are needed, the narrowing of the endoneural blood vessels may explain the proximal dominant weakness.
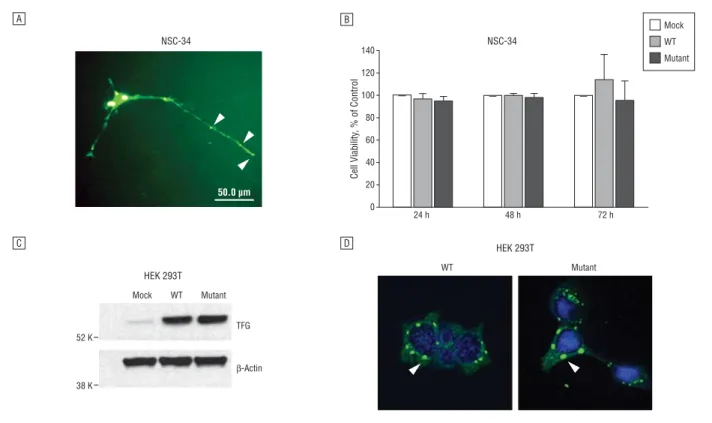
The in vitro expression of wild-type TFG revealed that TFG proteins can be expressed in the neurites of the NSC-34 motor neuron cells (Figure 5A). However, the P285L mutation did not affect the cellular localization of the TFG protein (not shown) and viability of the trans-fected cells (Figure 5B). In addition, the length of the neu-rites and size of soma in cells with P285L-TFG were un-changed compared with those of the wild type (data not shown). In HEK 293T cells, expression levels were not different between wild-type TFG and P285L-TFG (Figure 5C). Some cells with P285L-TFG transfection showed punctate or inclusion-like structures. However, this was also observed in cells with wild-type TFG (Figure 5D). These data may indicate that simple tran-sient expression of mutant TFG in neuronal or easily trans-fectable mammalian cells is not enough to demonstrate the neurodegeneration in vitro.
The present study suggests that a missense mutation in TFG provides the underlying cause of HMSN-P with dominant inheritance. We also suggest that TFG could play an important role in the peripheral nervous system. Accepted for Publication: October 4, 2012.
Published Online: March 18, 2013. doi:10.1001 /jamaneurol.2013.1250
Correspondence: Byung-Ok Choi, MD, Department of Neurology, Ewha Womans University School of Medi-cine, Mokdong Hospital, 911-1 Mokdong, Yangcheon-ku, Seoul 158-710, Korea (bochoi@ewha.ac.kr); and Ki Wha Chung, PhD, Department of Biological Science, Kongju National University, 182 Sinkwandong, Gongju 314-702, Korea (kwchung@kongju.ac.kr).
Author Contributions: Study concept and design: S.-S. Lee, M. G. Lee, Chung, and Choi. Acquisition of data: S.-S. Lee, H. J. Lee, J.-M. Park, K.-D. Park, H. S. Park, M. G. Lee, Hyun, Nakhro, Chung, and Choi. Analysis and interpre-tation of data: H. J. Lee, J.-M. Park, Hong, K.-D. Park, Yoo, Koo, Jung, J. H. Lee, M. G. Lee, Hyun, Nakhro, and Chung. Drafting of the manuscript: S.-S. Lee, H. J. Lee, Hong, K.-D. Park, Yoo, Koo, H. S. Park, M. G. Lee, Hyun, Nakhro, Chung, and Choi. Critical revision of the manuscript for im-portant intellectual content: S.-S. Lee, J.-M. Park, Jung, J. H. Lee, and M. G. Lee. Statistical analysis: H. J. Lee, Hyun, Nakhro, and Chung. Obtained funding: M. G. Lee, Chung, and Choi. Administrative, technical, and material support: S.-S. Lee, J.-M. Park, K.-D. Park, Jung, and H. S. Park. Study supervision: K.-D. Park, M. G. Lee, Chung, and Choi. 0 24 h 48 h 72 h 120 80 Cell Viability , % of Control 40 100 60 140 20 A B C D Mock WT Mutant NSC-34 HEK 293T NSC-34 50.0 µm WT Mutant HEK 293T WT Mock Mutant TFG β-Actin 52 K 38 K
Figure 5. In vitro expression of TFG in NSC-34 and HEK 293T cells. A, NSC-34 cells were transfected with green fluorescent protein (GFP)–tagged wild-type (WT)
TFG. The green fluorescence image was obtained by fluorescent microscope. The TFG proteins are expressed in neurites (arrowheads). B, Cell viability was
measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Wild-type and mutant TFG vectors were transfected into the NSC-34 cells. Mock transfected cells were used as controls. C, Protein expression of WT and P285L (mutant) TFG in HEK 293T cells. D, Expression of GFP-tagged TFG in HEK 293T cells. In some cells, both WT and P285L TFG induced punctate or inclusion-like structures (arrowheads).
Conflict of Interest Disclosures: None reported. Funding/Support: This work was supported by grant A111218-GM07 from the National Project for Personal-ized Genomic Medicine, Ministry for Health & Welfare, Republic of Korea.
Online-Only Material: The eTables, eFigures, and video are available at http://www.jamaneuro.com.
REFERENCES
1. Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 2009;8(7):654-667.
2. Guernsey DL, Jiang H, Bedard K, et al. Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot-Marie-Tooth disease. PLoS Genet. 2010; 6(8):e1001081. doi:10.1371/journal.pgen.1001081.
3. Boyer O, Nevo F, Plaisier E, et al. INF2 mutations in Charcot-Marie-Tooth dis-ease with glomerulopathy. N Engl J Med. 2011;365(25):2377-2388. 4. Weedon MN, Hastings R, Caswell R, et al. Exome sequencing identifies a DYNC1H1
mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease.
Am J Hum Genet. 2011;89(2):308-312.
5. Beetz C, Pieber TR, Hertel N, et al. Exome sequencing identifies a REEP1 muta-tion involved in distal hereditary motor neuropathy type V. Am J Hum Genet. 2012; 91(1):139-145.
6. Takashima H, Nakagawa M, Nakahara K, et al. A new type of hereditary motor and sensory neuropathy linked to chromosome 3. Ann Neurol. 1997;41(6): 771-780.
7. Maeda K, Kaji R, Yasuno K, et al. Refinement of a locus for autosomal dominant hereditary motor and sensory neuropathy with proximal dominancy (HMSN-P) and genetic heterogeneity. J Hum Genet. 2007;52(11):907-914.
8. Patroclo CB, Lino AM, Marchiori PE, Brotto MW, Hirata MT. Autosomal domi-nant HMSN with proximal involvement: new Brazilian cases. Arq Neuropsiquiatr. 2009;67(3B):892-896.
9. Takashima H, Nakagawa M, Suehara M, et al. Gene for hereditary motor and sen-sory neuropathy (proximal dominant form) mapped to 3q13.1. Neuromuscul
Disord. 1999;9(6-7):368-371.
10. Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45): 19096-19101.
11. Hoischen A, van Bon BW, Gilissen C, et al. De novo mutations of
SETBP1 cause Schinzel-Giedion syndrome. Nat Genet.
2010;42(6):483-485.
12. Choi BO, Koo SK, Park MH, et al. Exome sequencing is an efficient tool for ge-netic screening of Charcot-Marie-Tooth disease. Hum Mutat. 2012;33(11): 1610-1615.
13. Greco A, Fusetti L, Miranda C, et al. Role of the TFG N-terminus and coiled-coil domain in the transforming activity of the thyroid TRK-T3 oncogene. Oncogene. 1998;16(6):809-816.
14. Mencinger M, Aman P. Characterization of TFG in Mus musculus and
Caenorhab-ditis elegans. Biochem Biophys Res Commun. 1999;257(1):67-73.
15. Roccato E, Pagliardini S, Cleris L, et al. Role of TFG sequences outside the coiled-coil domain in TRK-T3 oncogenic activation. Oncogene. 2003;22(6):807-818.
16. Witte K, Schuh AL, Hegermann J, et al. TFG-1 function in protein secretion and oncogenesis. Nat Cell Biol. 2011;13(5):550-558.
17. Dutton-Regester K, Aoude LG, Nancarrow DJ, et al. Identification of TFG (TRK-fused gene) as a putative metastatic melanoma tumor suppressor gene. Genes
Chromosomes Cancer. 2012;51(5):452-461.
18. Birouk N, LeGuern E, Maisonobe T, et al. X-linked Charcot-Marie-Tooth disease with connexin 32 mutations: clinical and electrophysiologic study. Neurology. 1998;50(4):1074-1082.
19. Chung KW, Suh BC, Shy ME, et al. Different clinical and magnetic resonance imaging features between Charcot-Marie-Tooth disease type 1A and 2A. Neuromuscul
Disord. 2008;18(8):610-618.
20. Choi BO, Kim J, Lee KL, Yu JS, Hwang JH, Chung KW. Rapid diagnosis of CMT1A duplications and HNPP deletions by multiplex microsatellite PCR. Mol Cells. 2007; 23(1):39-48.
21. Ishiura H, Sako W, Yoshida M, et al. The TRK-fused gene is mutated in heredi-tary motor and sensory neuropathy with proximal dominant involvement. Am
J Hum Genet. 2012;91(2):320-329.
22. Gallardo E, Garcı´a A, Combarros O, Berciano J. Charcot-Marie-Tooth disease type 1A duplication: spectrum of clinical and magnetic resonance imaging features in leg and foot muscles. Brain. 2006;129(pt 2):426-437.