Riboflavin synthase binds two molecules of 6,7-dimethyl-8- ribityl lumazine as substrate to produce one molecule of riboflavin and pyrimidine derivative. The peculiar structural characteristic of riboflavin synthase is the internal amino acid homology between N-terminal domain half of riboflavin synthase (N-RS) and C-terminal domain half of riboflavin synthase (C-RS). To generate the variant proteins of riboflavin synthase, the recombinant plasmid of pQE vector inserted the DNA coding for N-RS and C-RS from the bioluminescent bacteria of Photobacterium leiognathi were constructed by polymerase chain reaction. The variant riboflavin synthase genes in pQE vector were expressed in Escherichia coli, and the purified whole riboflavin synthase (W-RS), N-terminal domain half of riboflavin synthase (N-RS), and C-terminal domain half of riboflavin synthase (C-RS) were assayed and determined the K
mvalue and V
maxvalues. The K
mvalues of W-RS, N-RS, and C-RS was 2.7 µM, 3.2 µM, and 1.9 µM, respectively. The V
maxvalue of W-RS was 2.5 times higher than that of N-RS and 4 times higher than that of C-RS.
Keywords: Photobacterium, bioluminescence, lux, riboflavin synthase
To make light emission of 495 nm catalyzed by bacterial luciferase in bioluminescent bacteria, the luciferase requires 1 molecule of long chain fatty aldehyde, O
2and reduced flavin
mononucleotide (FMNH
2) as substrates (Meighen, 1988, 1991).
For continuity of light emission in bioluminescence bacteria, the long chain fatty acid and the flavin mononucleotide (FMN) produced by bacterial luciferase are continuously reduced by fatty acid reductase complex (LuxCDE) and flavin reductase (LuxG), respectively (Haddock et al., 2010; Lee et al., 2019).
The genes coding for the proteins and enzymes involved in bioluminescence reaction exist in the lux operon (Meighen, 1988, 1991). The luxAB genes at the center of operon codes for luciferase αβ subunits and the genes of luxCDE codes for fatty acid reductase complex that are related with supplying for long chain fatty aldehyde substrate for luciferase composed of acyl- CoA reductase (LuxC), acyl transferase (LuxD), and acyl-protein synthetase (LuxE) (Meighen, 1991, 1993). The luxG gene codes for flavin reductase for the regeneration of reduced flavin mononucleotide substrate (FMNH
2) for bacterial luciferase (Meighen, 1993; Lee et al., 2016). In the downstream of luxG, ribEBHA genes involved in the flavin synthesis are present (Lee and Meighen, 1992). These genes code for riboflavin synthase (RibE), dihydroxy butane-4-phosphate synthase (RibE), lumazine synthase (RibH), and GTP cyclohydrolase II (RibA), respectively (Lee et al., 1994).
Flavin mononucleotide (FMN) is a key element not only in light emission but also in homeostasis including electron
Generation, purification, and enzyme activity of riboflavin synthase protein variants from Photobacterium leiognathi
Eugeney Oh, Sun-Joo Lim, Sanggi Lee, HyounJi Yun, and Chan Yong Lee*
Department of Biochemistry, Chungnam National University, Daejeon 34134, Republic of Korea
Photobacterium leiognathi 리보플라빈 생성효소 단백질 변이체의 제조, 정제 및 효소 활성도
오동현 ・ 임선주 ・ 이상기 ・ 윤현지 ・ 이찬용*
충남대학교 생화학과
(Received August 10, 2021; Revised September 10, 2021; Accepted September 10, 2021)
*For correspondence. E-mail: [email protected];
Tel.: +82-42-821-5482; Fax: +82-42-822-7548
transportation system in bioluminescent bacteria (Fischer and Bacher, 2005, 2008). Riboflavin (Vitamin B
2) is the direct precursor of flavin mononucleotide (riboflavin 5’-phosphate), and both chemicals have high similarity of molecular structure (Marcus and Coulston, 1990; Fischer and Bacher, 2005). The only one difference between them is the phosphorylation at ribityl end of flavin mononucleotide. The riboflavin synthase is a potential target for the chemotherapy of infections by Gram- negative bacteria which are unable to absorb riboflavin from the environment due to the absence of absorbing system and are dependent on endogenous synthesis (Markus and Coulston, 1990). Therefore, the knowledge of a detailed understanding
on the structure and mechanism of the riboflavin synthase, could serve as a starting point for the rational design of enzyme inhibitor with antibacterial activity.
The ribE gene from the lux operon of the bioluminescent bacteria of Photobacterium leiognathi ATCC25521 used in this study codes for riboflavin synthase (Lee and Meighen, 1992). The peculiar feature of riboflavin synthase is the internal amino acid homology in protein. Comparing the amino acid sequences between N-terminal domain half of riboflavin synthase (N-RS) and C-terminal domain half of riboflavin synthase (C-RS), it shows that 16 identical amino acids and 17 similar amino acids exist (Fig. 1). This high similarity of internal amino acid (A)
(B)
Fig. 1. (A) Internal amino acid sequence homology between N-terminal domain half of riboflavin synthase (N-RS) and C-terminal domain half of riboflavin
synthase (C-RS) from P. leiognathi. High similarity exists between N-terminal domain half and C-terminal domain half of riboflavin synthase. Sixteen amino
acids are identical (black box) and 17 amino acids are similar (grey box) Putative amino acids involved in active sites in N-terminal half and C-terminal half
of riboflavin synthase are shown in asterisks. (B) Diagram for cloning strategy of the gene coding for the whole riboflavin synthase (W-RS), N-terminal
domain half of riboflavin synthase (N-RS), and C-terminal domain half of riboflavin synthase (C-RS). The PCR product DNA using the primers in Table 1
were inserted into pQE vector. The Original amino acids from Met 1 to Ser 218 in W-RS, from Met 1 to Glu 111 in N-RS, and from Leu 98 to Ser 218 in C-RS
are present, respectively.
sequence in protein can be explained for the fact that two substrate binding sites in riboflavin synthase (Illarionov et al., 2001, 2005; Fischer and Bacher, 2008). Based on the structure of riboflavin synthase from Escherichia coli (Illarionov et al., 2001; Kim et al., 2010), the putative amino acids located in active sites in N-terminal half and C-terminal half of riboflavin synthase are shown in asterisks.
As shown in Fig. 2, riboflavin synthase produces 1 molecule of riboflavin and of 5-amino-6-ribitylamino-2,4-pyrimidinedione from the two molecules from 6,7-dimethyl 8-ribityllumazine (it is hereafter referred as lumazine) (Plaut et al., 1970; Volker and Bacher, 1988). The 4 carbon units from lumazine that located in the donor site are transferred to another lumazine in the acceptor site (Illarionov et al., 2001, 2005). Research revealed that this reaction is catalyzed through forming pentacyclic intermediate (Illarionov et al., 2001; Kim et al., 2010).
Up to now, there is no clear evidence that which part of riboflavin synthase’s binding site play role as acceptor site or donor site. The fact that both recombinant domains have independent ligand binding sites (Schott et al., 1990) provides direct evidence for the hypothesis that the catalytic sites of the
riboflavin synthase are located at interfaces of N-terminal half and C-terminal half. This model implicates that the individual binding sites of the N-terminal and C-terminal domains are closely adjacent in the subunit interface region, therefore enabling the enzyme to bind two substrate molecules in close proximity as a prerequisite for the 4-carbon transfer reaction (Illarionov et al., 2001; Kim et al., 2010).
To find the enzyme characteristics of riboflavin synthase from Photobacterium leiognathi, the ribE genes coding for the whole riboflavin synthase (W-RS) as well as for the variants of riboflavin synthase including N-terminal domain half of (N-RS) and C-terminal domain half of riboflavin synthase (C-RS) were inserted in pQE 30 vector which is suitable for purification of protein owing to 6X His-tag. The genes coding for whole riboflavin synthase (W-RS), N-terminal domain half of riboflavin synthase (N-RS) and C-terminal domain half of riboflavin synthase (C-RS) genes were expressed and purified by multiple steps of purification. Specially, the C-RS was purified for the first time from whole organisms, and the purified proteins were analyzed by enzymatic studies using the lumazine as substrate. Despite considerable efforts for the purification of C-RS, it was failed to purify it due to its unstable character or forming inclusion body from many different organism. Therefore, success of the protein purification of N-RS and C-RS altogether is a crucial step of further enzymatic and binding studies of the variants of riboflavin synthase, which is leading to identify the role of N-terminal domain half and of C-terminal domain half of riboflavin synthase as well as to search for the compound with the inhibition of enzyme activity.
For construction of recombinant plasmids inserting the genes for the wild type and variants of riboflavin synthase, the recombinant plasmid of PlXba pT7-3 (Lee et al., 1991) containing the ribE gene and all the lux genes from P. leiognathi was used to as a template to perform PCR. The amplifications of the DNA containing the ribE genes coding for W-RS, N-RS, and C-RS were carried out with primers manufactured from Bioneer Co. (Table 1). PCR are composed of 5 steps of pre-denaturation at 95°C for 5 min, denaturation at 95°C for 30 sec, annealing at 50°C for 30 sec, extension at 72°C for 1 min and post-extension at 72°C for 5 min. Denaturation, annealing and extension steps were repeated 30 cycles. PCR products were loaded in 1%
Fig. 2. Plausible mechanism of riboflavin synthase. Riboflavin synthase
binds 2 molecules of 6,7-dimethyl-8-ribityl lumazine as substrate at the
donor and acceptor site, producing 1 molecule of riboflavin and 1
molecule of 5-amino-6-ribitylamino-2,4-pyrimidinedione (Plaut et al.,
1970; Volker and Bacher, 1988).
agarose gel, and purified Monarch DNA gel extraction kit from NEB. The pQE 30 vector from Qiagen was used to insert the DNA containing the genes coding for W-RS, N-RS, and C-RS.
The insert DNA and pQE 30 vector DNA were digested with restriction enzyme BamHI, SalI, and HindIII. Cloning of gene experimental works were carried out with the strain of E. coli XL-1 blue which have lacI
qmutation (Stüber et al., 1990).
After ligation process, the mixture was transformed to E. coli XL-1 blue. Colony PCR was done and genes were isolated from 1% agarose gel by mini-prep.
To verify the generating recombinant plasmid, the DNA containing the genes were isolated by using Hybrid-q Plasmid rapid-prep and requested sequence analysis to Bioneer Co. The DNA sequences were confirmed by using NCBI blast and shown to 100% identical to the original DNA sequences. The DNA containing the genes coding for W-RS were 674 bp, 359 bp for N-RS, and 383 bp for C-RS, respectively. The con- structed recombinant plasmids (Fig. 1B) derived from pQE 30 vector were transformed to M15[pREP4]. As the pQE 30 vector is composed of T5 promoter system, pREP4 plasmid was used for expression of lac repressor proteins to repress the protein synthesis of recombinant plasmid (Stüber et al., 1990).
Transformed bacteria were incubated in 3 L of Luria Bertani
(LB) media with ampicillin (100 µg/ml) and kanamycin (25 µg/ml). 0.8 mM of Isopropyl-β-thiogalactopyranoside (IPTG) was added when the optical density at 600 nm reached to 0.6, incubation was continued additionally for 2 h at 37°C. Culture of bacteria was harvested by centrifugation under conditions of 3,000 rpm, 4°C, 15 min and stored at -20°C.
Expressed proteins were analyzed by 15% SPS-PAGE. Band of target protein was shown on the gel (Fig. 3). Cell lysates were added 60 ml of lysis buffer (Table 2) and sonicated using BRANSON Sonifier 450. Sonications were repeated for 12 cycles with 30 sec of sonication and 30 sec of pause time.
Table 1. Primers used in PCR
Primer Restriction site Nucleotide sequences
W-RS forward BamHI 5’-CCAATAGATGGATCCATTTATAGGGATATTATG-3’
W-RS reverse HindIII 5’-CCTTATTTATCAAGCTTACATGTGTTATG-3’
N-RS forward BamHI 5’-CCAATAGATGGATCCATTTATAGGGATATTATG-3’
N-RS reverse SalI 5’-CGCCCATTTCGTCGACACTCTATAACTTC-3’
C-RS forward BamHI 5’-ACAACTCGGCTAGGCGGATCCCTTGTTTCA-3’
C-RS reverse HindIII 5’-CCTTATTTATCAAGCTTACATGTGTTATG-3’
(A) (B) (C)
Fig. 3. SDS-PAGE of expressed (A) W-RS, (B) N-RS, and (C) C-RS proteins. Lanes: 1, Marker; 2, W-RS without induction; 3, W-RS 0.8 mM IPTG induction; 4, W-RS sonicated, centrifuged, supernatant; 5, W-RS sonicated, centrifuged, pellet; 6, Marker; 7, N-RS without induction; 8, N-RS 0.8 mM IPTG induction; 9, N-RS sonicated, centrifuged, supernatant; 10, N-RS sonicated, centrifuged, pellet; 11, Marker; 12, C-RS without induction; 13, C-RS 0.8 mM IPTG induction; 14, C-RS sonicated, centrifuged, pellet; 15, C-RS sonicated, centrifuged, supernatant.
Table 2. Buffers used in protein purification
Buffer Composition of buffer
Lysis buffer 50 mM Tris-Cl, 100 mM NaCl, 8 M urea, pH 8.0 Equilibrium buffer 50 mM Tris-Cl, 100 mM NaCl, 8 M urea,
20 mM imidazole, pH 8.0
Wash buffer 50 mM Tris-Cl, 500 mM NaCl, 8 M urea, 50 mM imidazole, pH 8.0
Elution buffer 50 mM Tris-Cl, 100 mM NaCl, 8 M urea, 300 mM imidazole, pH 8.0
Dialysis day 1 buffer 50 mM Tris-Cl, 100 mM NaCl, 4 M urea, pH 8.0
Dialysis day 2 buffer 50 mM Tris-Cl, 100 mM NaCl, 2 M urea, pH 8.0
Dialysis day 3 buffer 50 mM Tris-Cl, 100 mM NaCl, pH 8.0
Sonicated cell lysates were centrifuged at 14,000 rpm, 4°C, 30 min to separate the pellet and supernatant. The sonication step was processed to identify the solubility of W-RS, N-RS, and C-RS. Target proteins were found in pellet, meaning that the proteins possess insoluble characteristics.
As the target proteins showed that they are insoluble, therefore, 8M urea was added to enhance the solubility of W-RS, N-RS, and C-RS. Cell lysate containing expressed proteins were sonicated with lysis buffer (Table 2). To refold the target proteins, 8 M urea and 300 mM of imidazole were removed by using dialysis. Dialysis was done for 3 days in 4°C and the concentration of urea in dialysis buffer (Table 2) was gradually decreased. Dialysis membranes from Spectra/Por with 10 kDa molecular weight cut off (MWCO) were employed.
The supernatants of lysate were filtered with 0.22 µm syringe- driven filter from Merck Millipore.
Supernatant of cell lysates containing target proteins were purified by using Nuvia IMAC (immobilized metal affinity chromatography) resin from Bio-Rad. The pQE 30 vector has 6X His tag in front of multiple cloning sites which can make affinity bond with nickel ion. FPLC of BioLogic LP from Bio-Rad was employed and the resin in column were calibrated in the velocity of 5 ml/min with equilibrium buffer (Table 2), then the protein samples were injected. For the next step the wash buffer (Table 2) with high salt to weaken the strength of binding was injected in 5 ml/min to remove untargeted proteins.
Finally, the elution buffer (Table 2) in 2 ml/min was used to elute the target proteins. In the FPLC analysis, the protein samples were detected by UV absorbance at 280 nm. The fractions after affinity chromatography of FPLC were analyzed by 15% SDS-PAGE.
The purified protein liquid from Nuvia IMAC were pooled in one tube and additionally purified using Sephacryl S-200 High Resolution from GE Healthcare with 40 ml resin in 40 cm height column in velocity of 0.6 ml/min. The step of size exclusion chromatography was carried out to separate refolded proteins. The fraction of size exclusion chromatography (SEC) step was analyzed by using 15% SDS-PAGE gel. After size exclusion chromatography, W-RS, N-RS, and C-RS fractions were gathered in one tube for quantification of proteins (Fig. 4).
The determination of protein concentrations was carried out by Bradford assay. Standard curve was drawn by BSA (Bovine
Serum Albumin) from Bio-Rad with mixing 5 µl of protein sample and 250 µl of Quick Start Bradford Dye Reagent from Bio-Rad, then the absorbance value was measured in 595 nm.
The concentrations of W-RS, N-RS, and C-RS were 12.5 µM, 9.9 µM, and 7.4 µM, respectively.
Riboflavin synthase utilizes the lumazine as a substrate and yields riboflavin for a product. The riboflavin synthase activity was analyzed by UV-visible spectrophotometer from OPTIZEN.
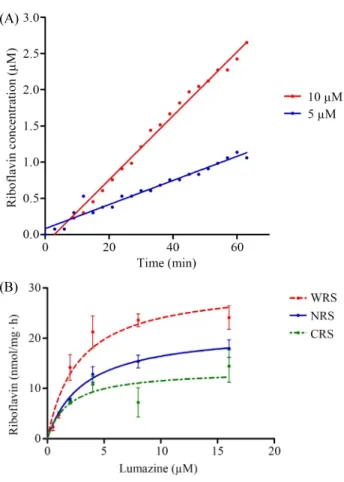
The enzyme activity of riboflavin synthase was measured absorbance at 445 nm for detection of riboflavin product by mixing 20 µM of lumazine and 10 µM, 5 µM, 2 µM of W-RS, N-RS, and C-RS proteins, respectively. Time course assay of W-RS was performed with UV-visible spectrophotometer by measuring absorbance value at 455 nm by increasing incubation time. The proteins and lumazine substrate was incubated in room temperature with blocking of light. The concentration of proteins was 5 µM or 10 µM, respectively, and 20 µM of lumazine substrate was added for each incubation mixtures with the same amount in phosphate buffer solution. In enzymatic reaction, the concentration of produced riboflavin was calculated by Lambert-Beer law with the molar extinction coefficient of riboflavin at 445 nm, 13,201/M·cm. And also, the reaction mixture with 10 µM of whole riboflavin synthase (W-RS) and 20 µM of lumazine as well as with 5 µM of whole riboflavin synthase (W-RS) and 20 µM of lumazine were shown in a linear pattern in terms of the enzyme activity with increase the reaction time (Fig. 5A). As the protein concentration is doubled, the average velocity of the reaction is increased for 2.7 times,
Fig. 4. The purified W-RS, N-RS and C-RS proteins in SDS-PAGE. Lanes:
1, Marker; 2, W-RS; 3, N-RS; 4, C-RS. N-RS was applied to half volume
compare to W-RS and C-RS.
indicating that the W-RS has riboflavin synthase activity.
For the precise measuring of riboflavin synthase activity, the purified W-RS, N-RS, and C-RS proteins were used to check the velocity of producing riboflavin. The same concentration of protein samples was fixed to 2 µM for every point of experiments. The concentrations of substrate of lumazine was varied from 0.5 µM to 16 µM. As the increasing of the substrate concentration, the velocity of riboflavin synthase activity was enhanced and obeyed by Michaelis-Menten’s rectangular hyperbolic curve (Fig. 5B). The enzymatic parameters of K
mand V
maxwere calculated by Lineweaver-Burk plot. A linear Lineweaver-Burk plots were observed with W-RS, N-RS, and C-RS in concentration range from 10
-6to10
-5M. The K
mvalues of W-RS, N-RS, and C-RS was 2.7 µM, 3.2 µM, and 1.9 µM, respectively. The V
maxvalue of W-RS was 2.5 times higher
than that of N-RS and 4 times higher than that of C-RS (Table 3). According to the data shown above, it seems that N-terminal domain half of riboflavin synthase (N-RS) showed enzyme activity. It can be explained the fact that N-terminal domain half of riboflavin synthase in E. coli possesses the strong protein interaction from the previous result (Kim et al., 2011).
The same result was also found in this research experiment after size exclusion chromatography, the N-RS showed 25 kDa band corresponding to dimeric protein of N-RS (data not shown).
By making protein-protein interaction in N-RS to form dimer, it can be speculated that 2 binding sites exist in dimeric N-RS with riboflavin synthase activity. However, the C-RS showed low K
mand V
maxvalues comparing to those of W-RS as well as N-RS, suggesting different protein conformation of C-RS with the low enzymatic activity. The riboflavin synthase activity from the cell extract of E. coli transformed with the recombinant plasmid containing the riboflavin synthase gene coded by ribE from the lux operon of P. leiognathi in this study was 15 fold lower comparing with the specific riboflavin synthase activity from E. coli based on the literature (Illarionov et al., 2001), and it shows similar value to the riboflavin synthase activity from the E. coli transformed with the recombinant plasmid containing the riboflavin synthase gene coded by ribE from the lux operon of P. phosphoreum (Lee et al., 1994). The K
mvalue of P. leiognathi riboflavin synthase for lumazine was 10 fold lower than that of E. coli, indicating that the riboflavin synthase proteins purified from the cell extract harboring recombinant plasmid containing the genes from bioluminescent bacteria of P. leiognathi has high affinity to the lumazine substrate but possesses relatively low enzyme activity.
In summary, the study on the generation and purification of riboflavin synthase variant proteins from the bioluminescent bacteria of P. leiognathi was described. The data reported in
Table 3. The K
mand V
maxvalues of W-RS, N-RS, and C-RS from Photo- bacterium leiognathi
Proteins Enzymatic parameters
K
mV
maxW-RS 2.8 µM 30.6 nmol/mg·h
N-RS 3.2 µM 20.6 nmol/mg·h
C-RS 1.9 µM 12.8 nmol/mg·h
(A)
(B)
Fig. 5. (A) Time course assay of riboflavin synthase with W-RS proteins
with 5 µM and 10 µM of whole riboflavin synthase, respectively. (B)
Riboflavin synthase activities with W-RS, N-RS, and C-RS proteins. All
the proteins concentration were adjusted to 2 µM. The measurement of
riboflavin synthase activity was performed at 50 mM phosphate buffer at
pH 7.0. All the enzyme activities were the average values from the three
times measurements.
this paper demonstrate for the first time that the N-terminal half as well as the C-terminal half of the riboflavin synthase subunit can fold independently and even possesses the enzyme activity.
The N-terminal domain half and C-terminal half of riboflavin synthase showed relatively low riboflavin synthase activity comparing to whole riboflavin synthase. N-terminal domain half of riboflavin showed higher K
mand V
maxvalue than that of C-terminal domain half of riboflavin synthase. The success of the protein purifications of N-RS and C-RS altogether will be valuable for further enzymatic and binding studies of the variants of riboflavin synthase. These results enable us to identify the role N-terminal domain half and C-terminal domain half of riboflavin synthase and to provide a high throughput screening system for searching of the compound with the inhibition of enzyme activity.
적 요
리보플라빈 생성 효소는 두 분자의 6,7-dimethyl-8-ribityl lumazine과 결합하여 리보플라빈과 피리미딘 유도체를 생 성물로 전환시킨다. 리보플라빈 생성 효소는 아미노 말단 절 반 및 카르복시 말단 절반 사이에 자체 아미노산의 상동성을 갖는 특이한 구조적 특징을 갖는다. 본 연구에서는 아미노 말 단 절반 및 카르복시 말단 절반을 갖는 리보플라빈 생성 효소 변이 단백질을 코드하는 유전자를 중합 효소연쇄 반응으로 증폭시켜 pQE30 벡터에 삽입하여 이 재조합 플라스미드를 대장균에 형질 전환하여 이들 단백질들을 발현하여 정제하 였다. 정제된 전체 리보플라빈 생성 효소(W-RS), 아미노 말 단 절반 영역 리보플라빈 생성 효소(N-RS), 카르복시 말단 절 반 영역 리보플라빈 생성 효소(C-RS)들의 효소활성도를 비 교하고 K
m및 V
max값을 결정하였는 바, W-RS, N-RS, C-RS 의 K
m값은 각각 2.7 µM, 3.2 µM 으로 비슷하였으나 V
max는 W-RS가 N-RS에 비교하여 2.5배 C-RS 보다는 4배 높은 값을 가졌다.
Acknowledgments
This work was supported by research scholarship of Chungnam National University.
Conflict of Interest
The authors have no conflict of interest to report.
References