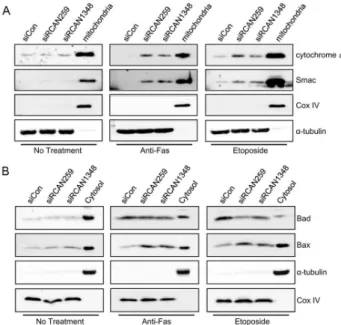
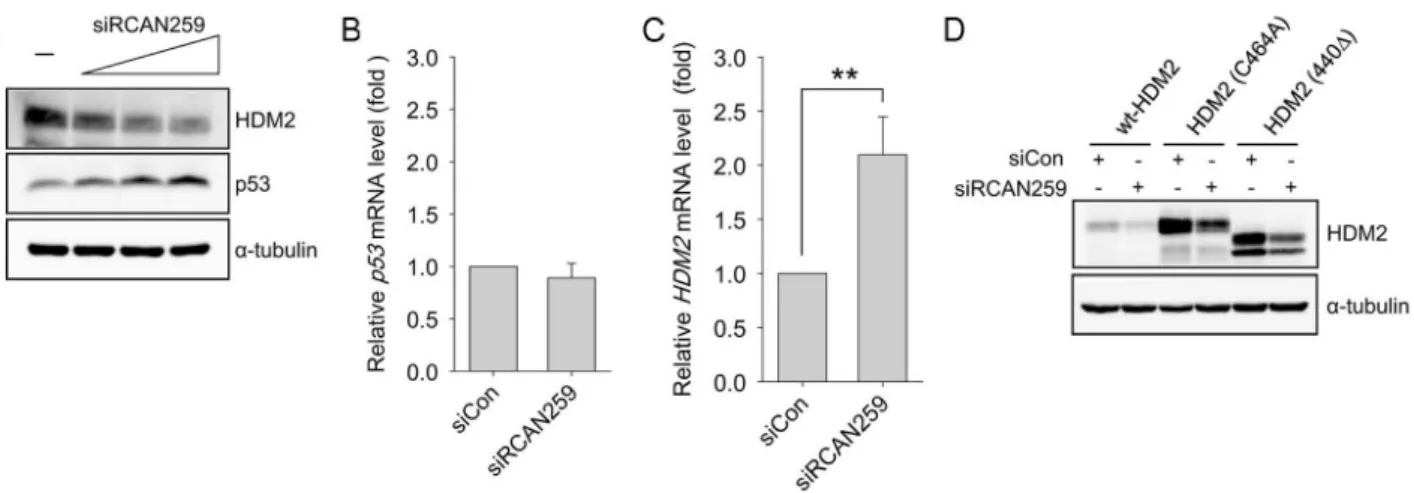
Knockdown of RCAN1.4 Increases Susceptibility to FAS-mediated and DNA-damage-induced Apoptosis by Upregulation of p53 Expression
7
0
0
전체 글
(2)
(3)
(4)
(5)
(6)
(7)
수치

관련 문서
co-treatment with hispidulin and TGF-β up-regulated the protein of expression E-cadherin and occludin against TGF-β-induced in MCF-7 and HCC38 cells.. The
Here, we found that TAMR-MCF-7 cells had undergone EMT, evidenced by mesenchymal-like cell shape, down-regulation of the basal E-cadherin expression
Activated H-Ras expression in human fibroblast cell lines increases the activity of Ku80 to bind injuried DNA, reduces γ-H2AX expression by UV irradiation,
protein levels in control or PIG3 depleted HCT116 and HeLa cells were tested. by
Recombinant RapA fused with the C-terminus of RapC completely recovered the phenotypes of rapC null cells, indicating that the functions of RapA were modified to become
Fig 4 : Inhibitory effects of classified methanol extracts of Acalypha australis the COX-2 protein expression of the human oral cavity carcinoma KB cells....
Glutamate excitotoxicity induced by excessive activation of NMDA receptor causes various damage to cells, which leads to cell death.. In previous studies, increased ROS
4 > Effects of Taro on COX-2 expression and iNOS expression(hot water) in human thyroid cancer cells. The cells were pretreated for 48hours with either
