Biomedical Science Letters 2014, 20(2): 85~95 eISSN : 2288-7415
The GSK-3β/Cyclin D1 Pathway is Involved in the Resistance of Oral Cancer Cells to the EGFR Tyrosine Kinase Inhibitor ZD1839
Nam Kyeong Jeon1, Jin Kim2,† and Eun Ju Lee3,†
1Seegene Institute of Life Sciences, Seoul, 138-050, Korea;
2Department of Oral Pathology, Oral Cancer Research Institute, BK21 Project for Medical Science, Yonsei University College of Dentistry, Seoul 120-750, Korea;
3Department of Clinical Laboratory Science, Daejeon Health Science College, Daejeon 300-711, Korea
Activation of the epidermal growth factor receptor (EGFR) and downstream signaling pathways have been implicated in causing resistance to EGFR-targeted therapy in solid tumors, including the head and neck tumors. To investigate the mechanism of antiproliferation to EGFR inhibition in oral cancer, we compared EGFR tyrosine kinase inhibitor (Gefitinib, Iressa, ZD1839) with respect to its inhibitory effects on three kinases situated downstream of EGFR: MAPK, Akt, and glycogen synthase kinase-3β (GSK-3β). We have demonstrated that ZD1839 induces growth arrest and apotosis in oral cancer cell lines by independent of EGFR-mediated signaling. An exposure of oral cancer cells to ZD1839 resulted in a dose dependent up-regulation of the cyclin-dependent kinase inhibitor p21 and p27, down regulation of cyclin D1, inactivation of GSK-3β and of active MAPK. In resistant cells, GSK-3β is constitutively active and its activity is negatively regulated primarily through Ser 9 phosphorylation and further enhanced by Tyr216 phosphorylation. These results showed that the resistance to the antiproliferative effects of ZD1839, in vitro was associated with uncoupling between EGFR and MAPK inhibition, and that GSK-3β activation and degradation of its target cyclin D1 were indicators of high cell sensitivity to ZD1839. In conclusion, our data show that the uncoupling of EGFR with mitogenic pathways can cause resistance to EGFR inhibition in oral cancer.
Key Words: Cyclin D1; Glycogen synthase kinase-3β; Human oral carcinoma cells; p21CIP1/WAF1; p27KIP1; ZD1839
INTRODUCTION
Oral cancer is the sixth most common cancer in the world, and its incidence varies in different ecogeographic
regions (Mishra, 2010; Cheong et al., 2009; Mishra et al., 2010). Oral malignancy, commonly found as squamous cell carcinoma (SCC), constitutes a major worldwide health problem (Jemal et al., 2003). The clinical observation that patients with oral squamous cell carcinoma (OSCC) in comparable stages may run different clinical courses and yet respond differently to similar treatments is not adequately understood, but several prognostic markers have been proposed (Molinolo et al., 2009). One such factor, the EGFR, is a transmembrane phosphoglycoprotein whose over expression has been shown to correlate with decreased disease free survival and increased metastasis in tumors including OSCC (Chang et al., 2004). Activation of the receptor by the binding of any of its six known ligands leads to receptor dimerization and trans autophosphorylation on
Original Article
*Received: June 18, 2014 / Revised: June 30, 2014 Accepted: June 30, 2014
†Corresponding author: Eun Ju Lee. Department of Clinical Laboratory Science, Daejeon Health Science College, 77-3 gayang 2-dong, Dong-Gu, Daejeon 300-711, Korea.
Tel: +82-42-670-9163, Fax: +82-42-670-9160 e-mail: [email protected]
†Corresponding author: Jin Kim. Department of Oral Pathology, Oral Cancer Research Institute, BK21 project for Medical Science, Yonsei University College of Dentistry, Seoul 120-750, Korea.
Tel: +82-2-2228-3031, Fax: +82-2-361-2639 e-mail: [email protected]
○CThe Korean Society for Biomedical Laboratory Sciences. All rights reserved.
tyrosine residues. Phosphorylated tyrosine residues on the receptor then serve as binding sites to recruit and activate downstream signaling pathways that control cellular pro- liferation and the progression of invasion and metastasis (Shintani et al., 2004; Gossage and Eisen, 2010). Many epithelial cancers, especially the SCC, display EGFR over- expression with or without the EGFR gene amplification often associated with an increased production of EGFR ligands (Laskin and Sandler, 2004; Cataldo et al., 2011;
Van Erp et al., 2009). Recently, a new EGFR inhibitor, ZD1839 (Iressa), has been developed and used in clinical trials for cancer patients. ZD1839 inhibits EGFR-associated tyrosine kinase (EGFR-TK) and shows marked antipro- liferative activity towards cancer cells and other host- dependent processes promoting cancer growth (Shrader et al., 2007; Ciardiello et al., 2000).
In preclinical studies, ZD1839 showed antitumor activity in a variety of human cancer cell lines expressing EGFR and it was active in a range of Xenograft models (Wakeling et al., 2002; Sirotnak, 2003). Other studies have shown that tumor cells may acquire resistance to anti-EGFR therapies without altering EGFR expression but rather through up- regulation and activation of other proliferate and/or anti- apoptotic activities (Cunnick et al., 1998; Chakravarti et al., 2002; Roudabush et al., 2000; Saito et al., 2001). Although targeted therapies yield better outcomes than non-targeted therapies, frequent treatment failure suggests the need for new treatments or targets for this disease. In oral cancer, active transcription of various genes leads to rapid cell division, faster invasion and reduction of cell death. Although it has been largely overlooked, there is a potential link between key players in oral cancer, including transcription factors, cell cycle regulators, invasion/metastasis-promoting factors, and cell survival regulators, and their regulation under the control of glycogen synthase kinase 3β (GSK-3β) (Kassouf et al., 2005).
Glycogen synthease kinase-3β (GSK-3β) is a serine/
threonine kinase that plays a crucial role in mammalian development by regulating the wnt signaling pathway (Nicolle et al., 2006). GSK-3β is a critical component among several receptor coupled signaling pathways, including those emanating from growth factor-stimulated receptors that
activate the intermediary protein kinase AKT or ribosomal S6 kinase (RSK), which in turn phosphorylates and inhibits GSK-3β and other signaling pathway (Farr et al., 2000;
Kassouf et al., 2005). Mitogens, such as EGF, inactivate GSK-3β via a pathway involving Ras/phosphatadylinositol -3 kinase (PI3K)/protein kinase B/AKT or Ras/MAPK/RSK.
Furthermore, alternations in the subcellular distribution of cyclin D1 during the cell cycle may also regulate cyclin D1/
cdk4 function. Thus, cyclin D1 accumulates in the nucleus throughout G1 phase, but it relocalizes to the cytoplasm during the reminder of interphase (Farr et al., 2000; Kassouf et al., 2005). Cyclin D1 redistribution and its degradation are correlated with its phosphorylation on Thr286 by GSK- 3β (Kassouf et al., 2005). We hypothesized that specific signal coupling between the EGFR and downstream kinases would dictate the biological response obtained when cells are treated with an EGFR inhibitor. In this study, we provide evidence that ZD1839 inhibition of the MAPK pathway is associated with GSK-3β activation, cyclin D1 degradation, and cell cycle distribution in G1 phase. The results of these studies indicate that EGFR-dependent GSK-3β regulation may predict the cytostatic effect of ZD1839 on OSCC cells
MATERIALS AND METHODS Tumor cell lines and culture medium
Oral epithelial cancer cell lines YD-8, YD-9, YD-10B, YD-15, YD-15M, YD-17, YD-17M, and YD-38 from Yonsei University College of Dentistry was used for this study (Lee et al., 2005). The cancer cells were fed with a mixture of Dulbecco's modified Eagle's medium (DMEM;
Gibco BRL, USA) and Ham's nutrient mixture F12 (Gibco BRL, USA) at a 3:1 ratio supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT), 1 × 10-10 M cholera toxin, 0.4 mg/ml hydrocortisone, 5 μg/ml insulin, 5 μg/ml transferrin, and 2 × 10-11 M triiodotyronine (all purchased from Sigma, St. Louis, USA). All cell lines were cultured at 37℃ in a humidified atmosphere Containing 5%
CO2 and maintained as a monolayer. The culture medium was changed every 2 or 3 days (Lee et al., 2005).
Reagents and antibodies
AstraZeneca generously provided gefitinib (Iressa, ZD- 1839). It was dissolved in DMSO to generate a concentrated stock solution, and this stock was diluted in medium just before use so that the concentration of DMSO never ex- ceeded 0.1%. The following antibodies were used: p27KIP1, p21CIP1/WAF1 and EGFR were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Phospho-EGFR (Tyr1068), HER2/Erb2, HER3/Erb3, HER4/Erb4, p38, phospho-p38 (Thr180/Tyr182), p44/42 MAPK, phospho- p44/42 MAPK (Thr202/Tyr204), Akt, phospho-Akt, phospho- GSK-3β, Rb, phospho-Rb, P16LNK4A, cyclin D1 and β-tublin were purchased from cell signaling Technology, Inc.
(Beverly, MA). GSK-3β, phospho-GSK-3β (Y216) and lamin A/C were purchase from BD (San Diego, CA).
Treatment with ZD1839
Cells were treated with 0.01, 0.1, 1, 10, and 100 μM ZD1839 (AstraZenca, London, UK), 24 hours after seeding.
The cells were treated with culture for 2 days. This ensured that the cells were in an exponential growth phase at the time of treatment. ZD1839 was dissolved in DMSO (Sigma, St. Louis MO, USA). Each treatment condition contained the same amount of DMSO. Because DMSO modulates keratinocyte differentiation and proliferation, blank controls and solvent controls were also included to confirm that DMSO had not extended such effects at the concentrations used 0.1 % (v/v) for experiments with doses up to 100 μM, and 0.005~0.01% (v/v) for low dose experiments (1~10 μM) (Lee et al., 2007).
Cell proliferation assay
The percentage of growth inhibition was determined by using an MTT (Sigma, St. Louis MO) assay for the measure- ment of viable cells. A total of 3.0 × 103 cells/well were seeded onto a 96-well plate for 24 hours, treated with various concentrations of ZD1839, and incubated for an additional 2 days at 37℃ (Lee et al., 2007). Subsequently, 10 μl of MTT at a concentration of 5 mg/ml was added to each well, and the cells were incubated for an additional 4~6 hours.
The supernatant was aspirated, and 100 μl of DMSO was
added to the wells in order to dissolve any precipitate present.
The absorbance was then measured at a wavelength of 570 nm using an ELX800 reader (Bio-Tek Instruments, Inc., Winooski, VT).
Cell cycle analysis
A total of 1.5 × 105 cells/well were plated onto 6-well plates, and incubated for 24 hours at 37℃. Various concen- trations of ZD1839 were added to the wells, and then incubated for an additional 2 days. Cells were then washed, pelleted, fixed with cold 100% ethanol for at least 30 minutes, and incubated with 100 μg/ml RNase A and 50 μg/ml propidiumiodide in PBS for 30 minutes at 37℃. Samples were immediately analyzed by flow cytometry (Becton Dickinson, San Jose, CA). Cell cycle phase distribution was determined using the Modfit software (Verity Software House, Topsham, ME).
Immunoprecipitation and western blot analysis
EGF-stimulated and non stimulated cells were treated with ZD1839, and the proteins were extracted from the cells using a buffer which contained 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1% SDS, 1% NP40, and 1x protease inhibitors (Roche Applied Science, Indianapolis, IN). 15 μg of protein were fractionated by electrophoresis through a 10% SDS polyacrylamide gel, and the proteins were then transferred onto a nitrocellulose membrane. The membrane was blocked for 1 hour in a blocking buffer which contained 5% dry milk, 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 0.1% Tween 20, and then incubated with a polyclonal antibody. The membrane was then incubated with an antirabbit antibody conjugated with horseradish peroxidase (Amersham, Arlington Heights, IL, USA), and the protein was detected using the chemiluminescence method, followed by autoradiography. The same membrane was then used to detect the β-actin protein, using a monoclonal anti-β-actin antibody (1:2,000 dilution; Sigma, Indianapolis, IN) as described above.
Apoptosis assay
Cells were grown in appropriate medium in the presence or absence of ZD1839 for 24 and 48hours. We harvested 5
× 105 cells by treatment with trypsin and detected apoptosis using annexin V Detection kit (Santa Crutz Biotechnology, Santa Cruz, CA) according to the instructions of the manufacturer. Sample was analyzed using flow cytometer (Becton Dickinson, Bedford, MA).
Immunofluorescence and confocal analysis
Cellular localization of EGFR phosphorylation was determined using indirect immunofluorscence. Cells grown on glass coverslips were fixed in 3.7% paraformaldehyde at room temperature for 10 minutes and then extracted with ice-cold acetone. Cells were treated with or without anti EGFR rabbit polyclonal antibody and then treated with Alexa-488-labeled goat anti-rabbit antibody (Molecular Probes, Inc., Eugene, OR). Confocal analysis was carried out using a Zeiss lase-scanning confocal microscope and established methods, involving processing of the same section for each detector (two excitations corresponding to 546 and 488) and comparing image pixel by pixel.
Statistical analysis
All results represent the average of at least three separate
experiments and expressed as mean ± SD unless otherwise indicated. Student's t-tests were performed in order to compare cell growth and the invasion/migration index, as well, as densitometric values of Western blot bands among different concentrations of ZD1839. P values of <0.05 were considered to be statistically significant.
RESULTS
Antiproliferative effects of ZD1839 in Human OSCC
We initially examined the human OSCC cell lines for EGFR expression by Western and found that all the cell lines tested expressed various levels of EGFR protein (Fig.
1A). We tested the antiproliferative effect (Fig. 2) of ZD1839 in the seven oral cancer cell lines. Sensitivity to ZD1839 was defined as a 50% inhibition of cell growth at a various concentration. Of the cell lines tested, YD-38 was most sensitive to the antiproliferative effect of ZD1839, with an IC50 <0.01 μM; whereas other cell lines were relatively resistant to ZD1839. Because YD-9, YD-15, and YD-38 express similar amount of EGFRs as tested by western blot but displayed differences in ZD1839 sensitivity, Fig. 1. (A) Expression of three EGFR family members, EGFR, Her2 and Her 4 in YD-8, YD-9, YD-10B, YD-15, YD-15M, YD-17, YD-17M, and YD-38 oral cancer cell lines. (B) Confocal analysis of YD-10B and YD-38 oral cancer cell lines for phosphorylated EGFR.
we conclude that expression of EGFR does not predict sensitivity to the antiproliferative effects of ZD1839. Thus we further characterized the achievable levels of EGFR autophosphorylation upon ligand addition.
We then compared the effects of ZD1839 on the baseline EGFR phosphorylation observed in serum-starved cells.
The two cell lines expressed high level of constitutively phosphorylated EGFR, and this phosphorylation was in- hibited in a concentration dependent fashion by ZD1839 (Fig. 4A). Following treatment with ZD1839 and 10 ng/ml EGF for 2 days, EGF-stimulated growth was not inhibited by 0.01 μM ZD1839, but was inhibited at 0.1, 1 and 10 μM ZD1839 in the YD-10B cell line. At these higher concen- trations, similar growth curves were obtained with ZD1839 alone or in the presence of EGF. Similar results were obtained using 50 ng/ml EGF, with 0.1 μM ZD1839 inhibiting the EGF stimulatory effect (data not shown).
Because recent reports have indicated that internalization of EGFR following treatment with an inhibitor predicts for the sensitivity of NSCLC to ZD1839 (Cheng et al., 1999), we determined the subcellular localization of the activated
EGFR in YD-10B and YD-38 cells by immunofluorescence microscopy following ZD1839 treatment. Confocal analysis of tumor cells double-stained for auto phosphorylated EGFR (green) and nucleus (red) confirmed the existence of baseline and EGF-inducible EGFR auto phosphorylation (Fig. 1B).
We observed complete EGFR phosphorylation in YD-38 cells following treatment with ZD1839. A high baseline activation of the EGFR did not seem to be a predictor of ZD1839 sensitivity, because YD-10B which displayed the highest activation has also different response patterns.
EGFR signaling pathways and effect of ZD1839
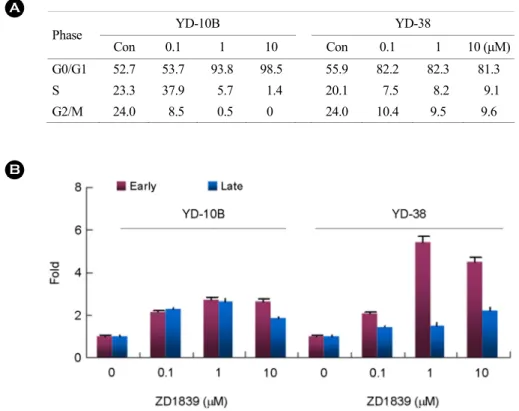
To determine whether ZD1839-induced suppression of YD cell lines proliferation were associated with cell cycle arrest, YD-10B and 38 cells were incubated in the presence of ZD1839 for 24 and 48 hours. As shown in Fig. 3, 1~10 μM ZD1839 markedly affected cell-cycle distribution (Fig.
3A). We initially focused on the effects on the G1/S transition in the more sensitive YD-38 cells. These cells contain wild-type Rb, a 24h exposure to 0.1 to 10 umol/L ZD1839 resulted in a dose-dependent inhibition of both EGFR phosphorylation and constitutive MAPK activity, hypophos- phorylation of Rb, down-regulation of cyclin D1 protein, an increase in p27 protein levels, and recruitment of YD-38 cells into G1 phase of the cell cycle, but without any effect on EGFR protein level (Fig. 4A and Fig. 4B).
FITC-conjugated Annexin V was used to detect cells in apoptosis by flow cytometry. Apoptosis was measured by Sub-G1 fraction on flow histograms and annexin V- and propidium iodide-stained fraction on flow histogram. Apop- totic cells possess brightly stained pyknotic nuclei and/or cell surface staining of phosphatidylserine by Annexin V;
non-apoptotic cells possess large weakly stained nuclei and no Annexin V staining. Cells were counter-stained with calceiun-AM (calcein) to obtain the number of live cells (stain green with calcein-AM, 5 l M for 15 min) or pro- pidium iodide (PI) to obtain the number of dead cells (staining red with PI) displaying permeabilised plasma membranes indicative of end-state apoptosis or necrotic cells. Thus, annexin V positive and PI negative cells are early apoptosis and annexin V positive and PI positive cells are late apoptosis or necrosis. Annexin-V negative and PI Fig. 2. A growth-inhibitory effect of ZD1839 on YD-8, YD-9,
YD-10B, YD-15, YD-15M, YD-17, YD-17M, and YD-38 oral cancer cell lines exposed to various concentrations of drug for 48 hours, as assesses by MTT.
positive cells are necrosis and Annexin-V negative and PI negative cells are live cells. The results showed that 1 and 10 μM ZD1839 induced significantly greater apoptotic cell death in the YD38 cell line than in the YD-10B cell lines (Fig. 3B).
The YD-38 and YD-10B cell lines had the highest pro- portion of phosphorylated receptors and displayed a decrease in phosphorylation following ZD1839 treatment for 48 hours.
This decrease in receptor phosphorylation was associated with a strong decrease in phosphorylation of the MAPKp44/
42 (T202, Y204) and AKT (S473) downstream effectors.
In other cell lines, 1umol/L ZD1839 had little or no effect on EGFR phosphorylation and downstream signaling pathways.
We next investigated the pattern of EGF-induced activation of PI3K/Akt and of Ras/MAPK pathways in both cell lines following ZD1839 treatment. Interestingly, the addition of 0.1 μM ZD1839 effectively blocked MAPK p38 and posphorylation p38 in YD-38 but not in the YD- 10B cells and was associated with the inhibition of Akt phosphorylation (Fig. 4A). We quantified by western blot
analysis the level of its inactive pool in YD-38 and YD-10B cells in the presence or absence of ZD1839 (Fig. 4A and Fig. 4B). We observed a significant reduction in the inactive form of GSK-3β in YD-38 cells following treatment with 0.1 μM ZD1839, whereas fails to reduce the size of inactive pool of GSK-3β in the YD-10B cells.
Significance reduction of cyclin D1 could be detected after 0.1 μM ZD1839 treatments in YD-38 cells (Fig. 4B), whereas no change in its level was observed in YD-10B cells. To explore whether ZD1839 treatment might regulate Cyclin D1 localization through modifyimg GSK-3β activity, we examined the phosphorylation status of GSK-3β using an antiphospho-ser9-specific antibody in OSCC cells. The total level of GSK-3β in OSCC cells remained constant under all ZD1839 treatment compared with that in the untreated control cells. Treatment with ZD1839 suppressed the GSK-3β-Ser9 phosphorylation was at or below the level of detection in the YD 38 cells. In contrast, YD10B cells showed no change after ZD1839 treatment. In addition to this inhibitory Ser9 phosphorylation, GSK-3β is also sub-
Phase YD-10B YD-38
Con 0.1 1 10 Con 0.1 1 10 (μM)
G0/G1 52.7 53.7 93.8 98.5 55.9 82.2 82.3 81.3
S 23.3 37.9 5.7 1.4 20.1 7.5 8.2 9.1
G2/M 24.0 8.5 0.5 0 24.0 10.4 9.5 9.6
Fig. 3. (A) Effect of ZD1839 on cell cycle distribution in YD-10B and YD-38 cells. (B) apoptosis assays. Apoptosis was measured using Annexin V kit and flow cytometer.
jected to the phosphorylation at Tyr216 that stimulates its activity. Using an anti-GSK-3β-Tyr216-specific antibody, we determined whether ZD1839 regulated GSK-3β activity through this Tyr216 phosphorylation (Fig. 4A). ZD1839 treatment did not change the phosphorylation level of GSK- 3β at Tyr216.
Phosphorylation of pRB can be effected by several CDKs acting with their coordinate cyclin partners. Thus, to confirm that cyclin D1 contributed to this pRB phosphorylation, we used a phosphospecific antibody that specifically recognized pRB proteins when phosphorylated at ser807/811, the site whose phosphorylation is catalyzed by cyclin D1-CDK4/6 kinases. The association among GSK-3β activation, loss of cyclin D1, and the cytostatic effects of ZD1839 suggests that loss of phosphorylated GSK-3β after the first predict the antiproliferative effects of the inhibitor in OSCC.
We have shown that ZD1839 had an activity when GSK-3β activation and loss of cyclin D1 was dependent on constitutive EGFR activation. Thus, we studied EGFR phosphorylation, GSK-3β and AKT activation and loss of cyclin D1 in a panel of 7 human OSCC cell lines to make
it possible to compare our finding with the situation in vitro in humans (Fig. 5). We did this analysis by westernblot, loading equal amount of protein on each well. We observed no EGFR phosphorylation in 5 (70%), moderate phosphory- lation in 2 (30%) and GSK-3β inactivation in 3 (45%) and loss of cyclin D1 in 2 (28.6%) cell lines. Thus the most significant reduction in phosphorylated GSK-3β and cyclin D1 was observed in the YD-38 cell line, in which ZD1839 exerted its most potent cytostatic effect.
DISCUSSION
We sought to examine the cellular and biochemical mechanisms of growth arrest that follow interruption of EGFR signaling in receptor-overexpressing OSCC human tumor cells. In this study, we characterized sensitivity of seven OSCC cell lines to an EGFR TKI with respect to baseline EGFR expression or activation and regulation of downstream pathways. We found that although the target was efficiently blocked in all cell lines, the cell growth inhibitory effects were dependent on regulation of specific Fig. 4. (A) EGFR signaling pathways in YD-38 and YD-10B cell lines. β-Actin was used as a loading control. P.C was positive control for each protein detection. (B) Changes in nuclear proteins with ZD1839 tratment in YD-10B and YD-38 cells. (C) Effect of various concentration of ZD1839 on p27 and p21 levels in YD-38 and YD-10B cell lines.
downstream kinases. Thus we found that cell cycle inhibitory effects of ZD1839 were associated with GSK-3β activation and cyclin D1 degradation. Using two oral squamous cell carcinoma cell lines, the sensitivity YD-38 cell line and the resistant YD-10B cell line, we investigated the mechanism of ZD1839 resistance.
In the initial study, we evaluated the effects of EGF supplementation on the growth and proliferation of these cell lines, but we were unable to detect any significant effects on cell proliferation by EGF supplementation for a concentration up to 50 ng/ml. Consequently, the simul- taneous treatment of cells with EGF (up to 50 ng/ml) did not significantly affect the cytotoxic effect of ZD1839 at various time points. These results are consistent with previous observations and suggest that EGF receptors may already be completely saturated by the relatively high levels of cell- produced EGF present in the cell culture medium (Nicolle et al., 2006). The importance of autocrine factors in OSCC growth is demonstrated by the ability of the YD cell lines to survive and proliferate under serum-free conditions. We
demonstrated that ZD1839 dose-dependently inhibited oral cancer cell growth with different IC50 concentrations. The antiproliferative effect of ZD1839 was mainly cytostatic and associated with a block in the G0/G1 phase of the cell- cycle, which was evident after 24 h of treatment and led to a significant increase in the expression level of the CDK inhibitor p27KIP1 and p21CIP1/WAF1. These findings are consistent with the previous evidence (Lee et al., 2007;
Nicolle et al., 2006), demonstrating that EGF-induced stimu- lation of growth in head and neck cancer cells is associated with down-regulation of p27KIP1. The p21CIP1/WAF1 protein was originally identified as a gene, which was directly regulated by the tumor suppressor protein p53 (Lee et al., 2005; Sgambato et al., 2004). P21CIP1/WAF1 expression is stimulated by a variety of external stimuli, including various growth factors, cytokines, tumor promoters, and DNA damaging agents (Sgambato et al., 2004; Zhang et al., 2004). However, not all stimulatory pathways involve the p53 protein, as some of the agents are also able to induce p21CIP1/WAF1 expression in p53-negative cells (Nicolle et Fig. 5. EGFR signaling pathways in YD-8, YD-9, YD-10B, YD-15, YD-15M, YD-17, YD-17M, and YD-38 oral cancer cell lines.
al., 2006). In this study, YD-10B cells exhibited a nonsense mutation at codon 236 of exon 7, in which the normal TAC sequence was changed to TAA (Lee et al., 2005). Accordingly, a p53-independent pathway might mediate the up-regulation of p21CIP1/WAF1 in the ZD1839-treated YD-10B cells.
Activated or overexpressed GSK-3β has been shown to promote apoptotic signaling in a number of conditions through a p53-dependent mechanism (Sgambato et al., 2004; Zhang et al., 2004), and several apoptotic stimuli were recently found to cause the accumulation of GSK-3β in the nucleus, colocalizing it with p53 (Sgambato et al., 2004; Zhang et al., 2004) and contributing to p53-mediated p21CIP1/WAF1 induction and caspase-3 activation (Nicolle et al., 2006). During G0/G1 phase, the D-type cyclins accumulate and assemble with either cdk4 or cdk6 in response to mitogenic growth factors. The active cyclin D1 holoenzym promotes G1 progression by activating the growth-suppressive properties of the retinoblastoma protein (Rb) through site specific phosphorylation and by virtue of its ability to titrate cdk inhibitors, such as p27KIP1 and p21CIP1/WAF1.
Extracellular mitogens promote cellular proliferation via receptor-mediated signaling. These signals ultimately con- verge on the cell cycle machinery, in which cyclin D may function as a critical sensor of this information. Based on recent observations, increased expression of cyclin D1 is not sufficient for cellular transformation (Cheng et al., 1999).
However, increase expression of GSK-3β and nondegradable cyclin D1 has been shown to permanently reside within the nucleus and to have transforming properties (Kossouf et al., 2005). Our studies showed that GSK-3β activation using the EGFR inhibitor is a necessary event for cell cycle inhibition in vitro and suggest a mechanism that involves cyclin D1 regulation. Initiation of DNA replication at the G1-S phase boundary is a meticulously regulated process that ensures that the cell has assembled the appropriate machinery needed for high-fidelity genome duplication.
Before S-phase entry, pre-replication complexes form at replication origins during G1 phase. Furthermore, it has been shown that cyclin D1/cdk4 kinase is incorporated into chromatin bound protein complexes with the same kinetics as minichromosome maintenance (MCM) proteins and
dissociates Rb-MCM7 complexes, thereby facilitating estab- lishment of the pre-replication complexes composed of cyclinD1/MCM7 (Cheng et al., 1999; LaBaer et al., 2006).
Because cyclin D1 nuclear export has been shown to correlate with GSK-3β regulation, it seems that deregulation of GSK-3β kinase activity through receptor tyrosin kinase inhibitor may be closely associated to DNA replication in tumor cells (Benzeno et al., 1999). Thus, our results link the deregulation of EGFR signals to ZD1839 resistance and cell cycle arrest through modulation of GSK-3β and cyclin D1. In ZD1839 resistant OSCC cell lines, measure- ment of GSK-3β activation together with nuclear cyclin D1 detection may represent potential surrogate prognostic markers for estimating the cytostatic effect of receptor tyrosin kinase inhibition in OSCC. Because the majority of our cell lines (Lee et al., 2007) proliferate in an EGFR independent manner in vitro, by focusing on EGFR alone, we may be overlooking an important therapeutic target in this disease. In conclusion, we proposed that as long as the upstream stimulatory signals act independently of the EGFR kinase activity and keep GSK-3β in a preponderant inactive form with active cyclin D1 inside the nucleus, the cytostatic effect of the EGFR inhibitor will be minor or null. Alter- natively, inhibition of the MAPK pathway and/or GSK-3β activation will translate into cell arrest with cyclin D1 degradation and impaired DNA replication.
Acknowledgements
This study was supported by the Korea Research Foun- dation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund, KRF-2005-005-J05901 and KRF-2006-311-E00461). This paper was supported by Daejeon Health Science College in 2013. We also thank AstraZeneca for the sample of ZD1839 used in this study.
REFERENCES
Benzeno S, Diehl JA. C-terminal sequence direct cyclin D1-CRM1 binding. J Biol Chem. 1999. 279: 56061-56066.
Cataldo VD, Gibbons DL, Perez-Soler R, Quintas-Cardama A.
Treatment of non-small-cell lung cancer with erlotinib or gefitinib. N Eng J Med. 2011. 364: 947-955.
Chakravarti A, Chakladar A, Delaney MA, Latham DE, Loeffler JS. The epidermal growth factor receptor pathway mediates resistance to sequential administration of radiation and chemo- therapy in primary human glioblastoma cells in a RAS- dependent manner. Cancer Res. 2002. 62: 4307-4315.
Chang GC, Hsu SL, Tsai JR, Liang FP, Lin SY, Sheu GT, Chen CY. Molecular mechanism of ZD1839-induced G1-cell cycle arrest and apoptosis in human lung adenocarcinoma A549 cells. Biochem Pharm. 2004. 68: 1453-1464.
Charles JS, James MR. CDK inhibitos: positive and negative regulator of G1-phase progression. Genes Dev. 1999. 13: 1501 -1512.
Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21(cip1) and p27(kip1) 'CDK inhibitor's are essential activator of cyclin D-dependent kinase in murine fibroblast. EMBO J. 1999. 18: 1571-1583.
Cheong SC, Chandramouil GV, Saleh A, Zain RB, Lau SH, Sivakumaren S, Pathmanathane R, Primef SS, Teoa SH, Patelg V, Gutkindg JS. Gene expression in human oral squamous cell carcinoma is influenced by risk factor exposure. Oral Oncol. 2009. 45: 712-719.
Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, Bianco AR, Tortora G. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor- selective tyrosine kinase inhibitor. Clin Cancer Res. 2000. 6:
2053-2063.
Cunnick JM, Dorsey JF, Standley T, Turkson J, Kraker AJ, Fry DW, Jove R, Wu J. Role of tyrosine kinase activity of epidermal growth factor receptor in the lysophosphatidic acid-stmulated mitogen-activited protein kinase pathway. J Biol Chem. 1998. 273: 14468-14475.
Farr GH 3rd, Ferkey DM, Yost C, Pierce SB, Weaver C, Kimelman D. Interaction among GSK-3, GBP, axin, and APC in Xenopus axis specification. J Cell Biol. 2000. 148: 691-702.
Gossage L, Eisen T. Targeting Multiple Kinase Pathways: A change in Paradigm. Clinical Cancer Research. 2010. 16: 1973-1978.
Kassouf W, Dinney CP, Brown G, McConkey DJ, Diehl AJ, Bar-Eli M, Adam L. Uncoupling between Epidermal Growth Factor Receptor and Down-stream Signals Defines Resistance to the Antiproliferative Effect of Gefitinib in Bladder Cancer Cells. Cancer Res. 2005. 65: 10524-10535.
Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ.
Cancer statistics, 2003. CA Cancer J Clin. 2003. 53: 5-26.
LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C,
Chou HS, Fattaey A, Harlow E. New function activaties for the p21 family of CDK inhibitors. Genes Dev. 1997. 11: 847 -862.
Laskin JJ, Sandler AB. Epidermal growth factor receptor: a pro-mising target in solid tumours. Cancer Treat Rev. 2004.
30: 1-17.
Lee EJ, Kim J, Lee SA, Kim EJ, Chun YC, Ryu MH, Yook JI.
Characterization of newly established oral cancer cell lines derived fromsix squamous cell carcinoma and two mucoepi- dermoid carcinoma cells. Exp Mol Med. 2005. 37: 379-390.
Lee EJ, Whang, JH, Jeon, NK, Kim J. The epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 (Iressa) suppresses proliferation and invasion of human oral squamous carcinoma cells via p53 independent and MMP, uPAR dependent mechanism. Annals NYAS. 2007. 1095: 113-128.
Mishra A, Bharti AC, Saluja D, Das BC. Transactivation and expression patterns of Jun and Fos/AP-1 super family protein in human oral cancer. Int J Cancer. 2010. 126: 819-829.
Mishra R. Glycogen synthase kinase 3 beta: can it be a target for oral cancer. Molecular Cancer. 2010. 9: 114.
Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009. 45: 324-334.
Nicolle G, Daher A, Maille P, Vermy M, Loric S, Bakkar A, Bakkar A, Wallerand H, Vordos D, Vacherot F, de Medina SG, Abbou CC, Van der Kwast T, Thiery JP, Radvanyi F, Chopin DK.
Gefitinib inhibits the growth invasion of urothelial carcinoma cell lines in which Akt and MAPK Activation is dependent on constitutive epidermal growth factor receptor activation.
Clin Cancer Res. 2006. 12: 2937-2943.
Roudabush FL, Pierce KL, Maudsley S, Khan KD, Luttrell LM.
Transactivation of the EGF receptor mediates IGF-1-stimulated shc phosphorylation and ERK1/2 activation in COS-7 cells. J Biol Chem. 2000. 275: 22583-22589.
Saito Y, Haendeler J, Hojo Y, Yamamoto K, Berk BC. Receptor heterodimerization: essential mechanism for platelet-derived growth factor-induced epidermal growth factor receptor trans- activation. Mol Cell Biol. 2001. 21: 6387-6394.
Sgambato A, Camerini A, Faraglia B, Ardito R, Bianchino G, Spada D, Boninsegna A, Valentini V, Cittadini A. Targeted inhibition of the epidermal growth factor receptor-tyrosin kinase by ZD1839 ('Iressa') induces cell-cycle arrest and inhibits proliferation in prostate cancer cells. J Cell Physio.
2004. 201: 97-105.
Shintani S, Li C, Mihara M, Yano J, Terakado N, Nakashiro K,
Hamakawa H. Gefitinib ('Iressa', ZD1839), an epidermal growth factor receptor tyrosine kinase inhibitor, up-regulates p27KIP1 and induces G1 arrest in oral squamous cell carcinoma cell lines. Oral Oncology. 2004. 40: 43-51.
Shrader M, Pino MS, Brown G, Black P, Adam L, Bar-Eli M, Dinney CP, McConkey DJ. Molecular correlates of gefitinib responsiveness in human bladder cancer cells. Mol Cancer Ther. 2007. 6: 277-285.
Sirotnak FM. Studies with ZD1839 in preclinical model. Semin Oncol. 2003. 1: 12-20.
Turenne GA, Price BD. Glycogen synthase kinase 3β phosphorylates serine 33 of p53 and activated p53' transcriptional activity.
BMC Cell Biol. 2001. 2: 12.
Van Erp NP, Gelderblom H, Guchelaar HJ. Clinical Pharmaco- kinetics of tyrosin kinase inhibitors. Cancer Treat Rev. 2009.
35: 692-706.
Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH. ZD1839 (Iressa): an orally activive inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002. 62: 5749-5754.
Zhang Z, Wang H, Li M, Agrawal S, Chen X, Zhang R. MDM2 is a negative regulator of p21WAF1/CIP1,independent of p53.
J Bio Chem. 2004. 16: 16000-16006.