237 책임저자:서영록, 130-701, 서울시 동대문구 회기동
경희대학교 의과대학 약리학교실, 기초의과학연구소 Tel: 02-961-0674, Fax: 02-963-0674
E-mail: [email protected]
접수일:2008년 11월 7일, 게재승인일:2008년 11월 21일
Correspondence to:Young Rok Seo
Department of Pharmacology, Institutue for Research of Medical Science, College of Medicine, Kyung Hee University, Hoegi-dong, Dongdaemun- gu, Seoul 130-701, Korea
Tel: +82-2-961-0674, Fax: +82-2-963-0674 E-mail: [email protected]
An Overview on DNA Damage and DNA Repair in Cancer Research
Md. Mujibur Rahman1,3 and Young Rok Seo1,2,3
Departments of 1Biomedical Science, 2Pharmacology, 3School of Medicine, Kyung Hee University, Seoul 130-701, Korea
There have been a significant number of advances in the field of cancer research regarding DNA repair and their impact on cancer diseases attracting the attention of new interest. Unfortunately many aspects of the exciting breakthroughs in our knowledge of DNA damage and repair have yet to be translated into standard care for patients. This would require the cross linking between all the areas of cancer research for fruitful ending. Now more than ever it has become clear that it is necessary to achieve real breakthroughs in improving much needed diagnosis and treatment of cancer. While some genetic variation must occur in order for us to adapt to environmental change and to develop distinguishing characteristics, radical modification of our genetic material can lead to the development of human disease, most notably cancer. Thus, maintaining the overall integrity of our DNA is imperative. These systems must firstly recognize specific forms of DNA damage, remove this damage, and ultimately replace the aberrant segment with a normal piece of DNA. In order to get a clear view our review will focus on the different types of DNA damage and DNA repair due to endogenous and/or environmental attacks. (Cancer Prev Res 13, 237-246, 2008)
Key Words: DNA damage, DNA repair, Cancer
INTRODUCTION
Actually single cell is responsible for the evolutions of tumour formation where thousands of cells engaged which inherits the deviated growth pattern. The reason behind the DNA damage can be environmental or may be transmitted from ancestor. Damages at deoxyribonucleic acid (DNA) embedded at chromosome during the cell division. Loss of their normal growth pattern transmits from cell division to their offspring’s. This kind of changes needs years to produce cancerous cells with the helps of different factors.1)
Human genome consists of numerous genes, but only a few genes are accountable for cell proliferation, differentiation or death, which can be expressed among the tissue or organ of the body. Two types of genes are responsible for cell growth,
which are proto-oncogenes and tumour suppressor. There are many cancer promoting agents like chemicals, viruses, and radiation which are quite capable of mutating proto-oncogenes and/or tumour suppressor genes. When the cell repair mechanisms are defective then these permanent damage to the DNA are passed through daughter cells whenever the cells divide. Significant prolongation of a cell’s life increases the chances that it will accumulate mutations in its DNA that transform the cell. Thus, the failure of a cell to die when it should is another factor that can contribute to carcinogenesis.2) Interestingly, there are many phases in cell cycle which comes in a sequential order and responsible for proper function of the cell, such as it has cell division (M phase) and a lengthy interphase including S phase (DNA synthesis, G1 phase and G2 phase). The proteins are aimed at regular target for cancer mutagenesis.3)
Specially, the non-dividing cells usually enter into dormant G0 phase while mitosis driving cells enter into G1 phase. Here, checkpoint takes part as an important function by controlling the events in the cell cycle simultaneously. DNA repair is done with the cell cycle development. Cyclin-dependent kinases (cdks) control these checkpoints which is responsible for phos- phorylation of main substrates that allocate cell cycle changes to take place, triggering the cyclins proteins and cdk inhibitors such as p15 and p16.4) If there is a problem in cell cycle checkpoint often allows the cells to keep away from quisence are cancer cells5) and disrupts the DNA repair process.6) They follow their own fussy checkpoint, which can be transformed to discriminating (growth) benefit.7)
Raf1 Oncogenes main function is to promote cell prolifera- tion. When these genes are activated, usually by a solitary heterozygous missense mutation, the oncogene products become over-active, inappropriately active, or instead lose their potential to be inactivated. Such “gain-of-function” somatic mutations arising in only one allele of a gene may in some cases be sufficient to alter the phenotype of the cell. By way of example, Ravi et al transformed human small cell lung cancer cells with an activated Raf1 (RAF1) oncogene and reported marked induction of cyclin-dependent kinase inhibitor p27 and a decrease in cdk2 protein kinase activity.8) On the other hand, cancerous cell production occurs through the chromosomal translocations where one segment of a chromosome is broken and joined to other part of another chromosome. The newly fused cells promote tumour develop- ment. Such is the case with the so-called Philadelphia chromo- some, the first translocation to be linked to a human cancer - chronic myelogenous leukemia. Accurate control of genes depends on different gene regulatory target factors to specific intracellular sites.9) ETO chromosomal translocation creates rearranged genes which further encoded the acute myelogenous leukemia fusion protein. Matrix targeting signal doesn’t present these proteins that express the AML1 protein to appropriate gene regulatory sites within the nucleus.10)
Tumour suppressor genes have been defined as “genes that sustain loss-of-function mutations in the development of cancer”11) and their protein products often inhibit cell prolifera- tion. Tumour suppressor genes are however involved in the regulation of a diverse array of different cellular functions including cell cycle checkpoint control, detection and repair of DNA damage, protein ubiquitination and degradation, mito-
genic signaling, cell specification, differentiation and migration, and tumour angiogenesis.12) Thus, tumour suppressor genes often encode proteins with a regulatory role in cell cycle progression (e.g. Rb) but may also encode DNA-binding transcription factors (e.g. p53) and inhibitors of cyclin-depen- dent kinases required for cell cycle progression (e.g. p16).
Several chemical compounds known as to carcinogenic to human cells termed as initiators and promoters. People who worked in the aniline dye and rubber industries have to increase 50-flod increase of urinary bladder cancer that was an evident to exposure to heavy doses of aromatic amine compounds as an initiator of cancer.13) Workers exposed to high levels of vinyl chloride, a hydrocarbon compound from which the widely used plastic polyvinyl chloride is synthesized, have relatively high rates of a rare form of liver cancer called angiosarcoma.14)
Other compounds which act as promoters acts on activating enzymes which involved in conveying signals that stimulates cell division are tetradecanoyl phorbol acetate (TPA), a phorbol ester which lead to prostate cancer.15) Hormones are very strong promoting agents stimulate the replication of cells in target organs. Long term use of the hormone diethylstilbestrol (DES) has been concerned for postmenopausal endometrial carcinoma as well as known to cause vaginal cancer in young women who were in touch with hormone while in the womb.16) The most effective physical damaging agent can cause to cancer is radiation, regarded as one of the main tumour inducing agent in animal including human, is likely that UV radiation acts as a complete carcinogen, which can initiate and promote tumour growth, just as some chemicals are. They form pyrimidine dimmers in DNA between two of the four nucleotide bases that make up DNA - the nucleotides cytosine and thymine. It cause tumour unless it is not repaired immediately. Ionizing radiation also responsible for little amount cancer occurrence Ionizing radiation can lead to a mutation (change) in a cell's DNA, which could contribute to cancer, or to the death of the cell. All cells in the body can be damaged by ionizing radiation.17)
DNA Damage in Cancer
DNA damage, due to environmental factors and normal metabolic processes inside the cell, occurs at a rate of 1,000 to 1,000,000 molecular lesions per cell per day. While this
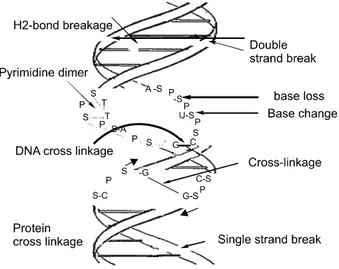
Fig. 1. Overview of DNA damage occur during the genetic stress caused by different stimuli, as radiation, genotoxic drug, endogenous and exogenous stresses. When DNA faced stress from above mentioned stresses then there are several DNA damage happen at the double-helix DNA structure.
constitutes only 0.000165% of the human genome's approximately 6 billion bases (3 billion base pairs), unrepaired lesions in critical genes (such as tumor suppressor genes) can impede a cell's ability to carry out its function and appreciably increase the likelihood of tumor formation.18)
The vast majority of DNA damage affects the primary structure of the double helix; that is, the bases themselves are chemically modified. These modifications can in turn disrupt the molecules' regular helical structure by introducing non-native chemical bonds or bulky adducts that do not fit in the standard double helix.19) Unlike proteins and RNA, DNA usually lacks tertiary structure and therefore damage or disturbance does not occur at that level. DNA is, however, supercoiled and wound around “packaging” proteins called histones (in eukaryotes), and both superstructures are vulner- able to the effects of DNA damage.19)
Classification of DNA Damage
DNA damage is an extremely common event in a cell's lifetime (Fig. 1). The machinery that copies DNA when a cell divides is not 100% efficient. This means that tiny errors accumulate in our cells over our lifetimes. On top of this, the life-sustaining chemical reactions that occur naturally in our cells generate harmful by-products, and these can cause DNA damage. So merely ‘being alive’ can cause DNA damage and,
potentially, cancer. Different categories of DNA damage have already been accounted until now. DNA damage frequencies range from very from time to time, some of the extensive DNA damage can lead severe damage to cells or tissue.
In base loss, understanding the basic chemistry of radicals and DNA bases is an important step in characterizing the potential damage to DNA. The chemical level understanding of such damage has far reaching implications for radiation therapy, health effects of radiation exposure, and aging.
Although there are numerous experimental studies on the initial phases of DNA base oxidation, there are still open questions as to the precise mechanisms of the initial stages of the oxidative process.20) Here, the glycosyl bond linking DNA bases with deoxyribose is labile under physiological conditions.
Within a typical mammalian cell, several thousand purines and several hundred pyrimidines are spontaneously lost per diploid genome per day. Loss of a purine or pyrimidine base creates an apurinic/apyrimidinic (AP) site (also called an abasic site).20) In base modification Amino groups on the bases of DNA may be spontaneously lost by hydrolytic deamination.21,22) Deamination process the primary amino groups of nucleic acid bases are somewhat unstable. They would be converted to keto groups in reactions.21) In a typical mammalian cell, about 100 uracils are generated per haploid genome per day in this fashion. Other deamination reactions include conversion of adenine to ypoxanthine, guanine to xanthine, and 5-methyl cytosine to thymine.
The chemistry and molecular biology of DNA adducts is only one part of the carcinogenic process. Many other factors will determine whether a particular chemical will exert a carcinogenic effect. For example, the size of particles upon which a carcinogenic may be adsorbed will influence whether or not, and if so where, deposition within the lung will occur.
The simultaneous exposure to several different agents may enhance or inhibit the metabolism of a chemical to its ultimate carcinogenic form (Rice et al, 1984; Smolarek and Baird, 1984).
The ultimate carcinogenic metabolites may be influenced in their ability to react with DNA by a number of factors such as internal levels of detoxifying enzymes, the presence of other metabolic intermediates such as glutathione with which they could react either enzymatically or non- enzymatically, and the state of DNA which is probably most heavily influenced by whether or not the cell is undergoing replication or particular sequences being expressed.23)
In chemical modification, the nucleic acid bases are suscep- tible to numerous modifications by a wide variety of chemical agents. For example, several types of hyper-reactive oxygen (singlet oxygen, peroxide radicals, hydrogen peroxide and hydroxyl radicals) are generated as byproducts during normal oxidative metabolism and also by ionizing radiation (X-rays, gamma rays). These are frequently called Reactive Oxygen Species (ROS). ROS can modify DNA bases. A common product of thymine oxidation is thymineglycol. Many environ- mental chemicals, including “natural” ones (frequently in the food we eat) can also modify DNA bases, frequently by addition of a methyl or other alkyl group (alkylation). In addition, normal metabolism frequently leads to alkylation. It has been shown that S-adenosylmethionine, the normal biological methyl group donor, reacts accidentally with DNA to produce alkylated bases like 3-methyladenine at a rate of several thousand per day per mammalian diploid genome.
Alkylation occurs most readily at the nucleophilic positions.24) In photodamage, Ultraviolet light is absorbed by the nucleic acid bases, and the resulting influx of energy can induce chemical changes. The most frequent photoproducts are the consequences of bond formation between adjacent pyrimidines within one strand, and, of these, the most frequent are cyclobutane pyrimidine dimers (CPDs). T-T CPDs are formed most readily, followed by T-C or C-T; C-C dimers are least abundant. One can obtain an idea of the extent of distortion of DNA chain structure caused by CPDs by noting that, in the diagram of a T-T CPD below, the cyclobutane ring, shaded in light blue, should have sides of approximately equal length.
Thus the two adjacent pyrimidines must be pulled closer to each other than in normal DNA.25) Dimers can also be produced by formation of a single covalent bond between the 6 position of one pyrimidine and the 4 position of the adjacent pyrimidine on the 3’ side. The order of abundance of such pyrimidine (6-4) pyrimidone photoproducts (6-4PPs) is T- C>>C-C>T-T>C-T. Although only one bond attaches the adjacent pyrimidines, there is nevertheless extensive distortion of the normal DNA structure. In the diagram below of the T-C 6-4PP, notice that the amino group, originally from the 4 position of the cytosine, ends up at the 5 position of the thymine.26)
In replication errors, another major source of potential alterations in DNA is the generation of mismatches or small insertions or deletions during DNA replication. Although DNA
polymerases are moderately accurate, and most of their mistakes are immediately corrected by polymerase-associated proofreading exonucleases, nevertheless the replication machi- nery is not perfect. As we shall see later, efficient repair mechanisms correct most of these problems.27)
In inter-strand crosslinks, bases attach on both strands, bifunctional alkylating agents such as the psoralens can cross-link both strands. Cross-links can also be generated by UV and ionizing radiation.28) In DNA-protein crosslinks, DNA topoisomerases generate covalent links between themselves and their DNA substrates during the course of their enzymatic action. Usually these crosslinks are transient and are reversed as the topoisomerase action is completed. Occasionally something interferes with reversal, and a stable topoisomerase- DNA bond is established. Bifunctional alkylating agents and radiation can also create crosslinks between DNA and protein molecules. All of these lesions must be repaired.28)
In strand breaks, Single-strand and double-strand breaks are produced at low frequency during normal DNA metabolism by topoisomerases, nucleases, replication fork “collapse”, and repair processes. Breaks are also produced by ionizing radiation.
In fact, Hermann Mller's discovery in 1927 that X-rays can cause mutations (as a consequence of occasional failure to properly repair damage induced by ionizing radiation) was the first experimental demonstration that environmental factors can affect genome stability.29,30)
Classification of DNA Repair
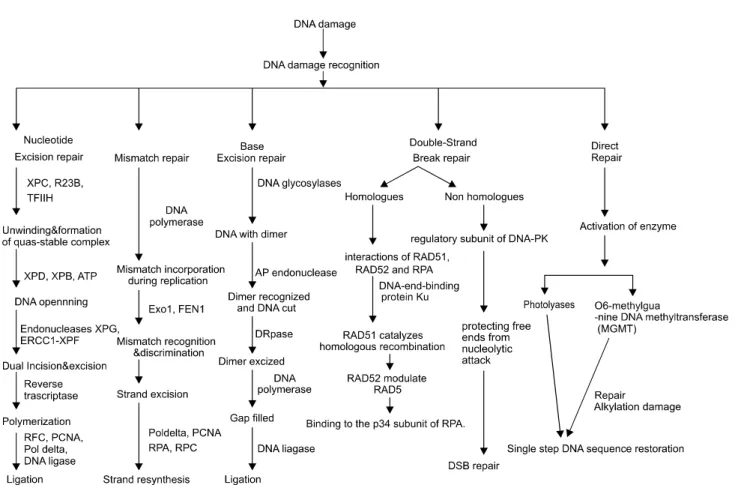
DNA damage is followed by different kinds of DNA repair (Fig. 2), which help to reinstall cell normal functions into the cells. If there are any difficulties in the repair system, mutation or cancer occurs in the host cell. Nucleotide excision repair, deals with the DNA damage that is bulky and forms a block to DNA replication and transcription (i.e., UV induced dimmers and some kinds of chemical adducts). It probably recognizes not a specific structure but a distortion in the double helix. The mechanism consists of cleavage of the DNA strand containing the damage by endonucleases on either side of damage followed by exonuclease removal of a short segment containing the damaged region.29) Carcinogenic effects of ultraviolet light comes from the sun, different chemicals or some heavy metal like nickel, cadmium, zinc etc and pose a great DNA damage to cell. In human, NER is the main means
Fig. 2. Diagrammatical presentation total DNA repair happen after DNA damage. DNA responds immediately to recover the damage by different DNA repair pathway responsible for normal functions of the cells.
of DNA repair in responses of these damage29) for the removal of UV-induced photoleisons, cyclobutane pyrimidine dimers and (6-4) photoproducts. NER is also known as to repair other lesions adducts arise from polyclic carcinogens, cisplatine, psoralens, and other chemical agents that form bulky, helix distorting lesions.30,31) Nucleotide excision repair (NER) holds a special place among DNA repair systems for several reasons.
For one thing, it is highly versatile and able to repair lesions as varied as UV-induced pyrimidine dimers, intra-strand crosslinks, protein DNA crosslinks, and a wide range of bulky chemical adducts. The secret of this versatility probably lies in the fact the NER does not attempt to ascertain the nature of a DNA lesion, but rather senses distortions in the structure of the double helix.29) Of course, different lesions induce various degrees of distortion and therefore constitute stronger or weaker substrates. For lesions that cause very little distortion, such as UV-induced cyclobutane pyrimidine dimers (CPD), the damage DNA binding complex DDB consisting of DDB1 and DDB2 (encoded be the XPE gene) comes to the rescue by
binding to the lesion and inducing a strong kink in the DNA, thereby enhancing its recognition by NER.32) Mutants that are defective in NER have been isolated in many organisms and are sensitive to killing and mutagenesis by UV and chemicals which act like UV. Humans with the hereditary disease xeroderma pigmentosum are sunlight-sensitive; they have very high risks of skin cancers on sun-exposed areas of the body and have defects in genes homologous to those required for NER in simple eukaryotes. DNA is the major target of direct or indirect UV-induced cellular damage. Low pigmentation capacity in white Caucasians and rare congenital defects in DNA repair are mainly responsible for protection failures. The important function of nucleotide excision DNA repair (NER) to protect against skin cancer becomes obvious by the rare genetic disease xeroderma pigmentosum, in which diverse NER genes are mutated.33) NER mutants also in lower organisms are UV-sensitive and have elevated levels of mutation and recombination induced by UV (because they are unable to use the accurate NER method to remove pyrimidine dimers and
must use mutagenic or recombinogenic systems).
The integrity of genetic information depends on the fidelity of DNA replication and on the efficiency of several different DNA repair processes. Among many types of DNA repair, the general DNA mismatch repair (MMR) pathway is responsible for correcting base substitution mismatches and insertion- deletion mismatches (IDLs) generated during DNA replication in organisms from bacteria to mammals.34)
Mismatch repair proteins are ubiquitous players in a diverse array of important cellular functions. In its role in post-replica- tion repair, MMR safeguards the genome correcting base mispairs arising as a result of replication errors. Loss of MMR results in greatly increased rates of spontaneous mutation in organisms ranging from bacteria to humans. Mutations in MMR genes cause hereditary nonpolyposis colorectal cancer, and loss of MMR is associated with a significant fraction of sporadic cancers. Given its prominence in mutation avoidance and its ability to target a range of DNA lesions, MMR has been under investigation in studies of ageing mechanisms. Each year approximately 150,000 people in the United States and half a million worldwide are diagnosed with colon cancer. Of these, approximately 3∼4% occur in familial cancer syndromes of which Hereditary Nonpolyposis Colorectal Cancer (HNPCC) or Lynch Syndrome is the most common.35)
The genetic hallmark of HNPCC is the occurrence of mutations in a set of genes responsible for the specific mismatch repair (MMR) system. It is not rare to observe some errors during base pair arrangements that take place during the process of DNA replication. This phenomenon does not usually result in cell damage because of the MMR system, which comprises a unit of at least 6 proteins, encoded by the fol- lowing genes: MLH1, MSH2, MSH3, MSH6, PMS1, and PMS2. The proteins of the MMR system act in a coordinated manner to identify minor reading errors on the replicated DNA strand, proceeding to the elimination of damaged DNA and substitution of a sequence that is perfectly paired with the mother strand.36) Most of HNPCC cases are due to mutations that inactivate the MSH2 and MLH1 genes, the former being responsible for 50%, the latter for 30% of the cases.37) Cases of HNPCC involving other genes of the MMR complex have been described, but are much less common. Great interest has been aroused by studies which observed that mutations of the MSH6 gene increase susceptibility to endometrial carcinoma in families with a history of HNPCC.45,46)
Work on DNA repair in bacteria and fungi had already revealed that loss of MMR conferred a mutator phenotype in which base substitutions as well as frame shift mutations were greatly elevated. Petes and colleagues tested the stability of poly (dGT) tracts in S. cerevisiae cells harbouring single and double mutations in MMR genes MSH2, MLH1, and PMS1.38) Loss of any one MMR gene was sufficient to elevate the frequency of tract instability two orders of magnitude.
Base excision repair has several DNA damaging agents which present as environmental contaminants, the majority of DNA damage has an endogenous origin and it is mainly due to: (i) oxidation of DNA by reactive oxygen species (ROS) which are generated in the cells by normal aerobic metabolism; (ii) spontaneous deamination of DNA bases, and (iii) methylation of DNA bases by S-adenosylmethionine. The majority of these lesions is blocking lesions or constitutes a pause for the progression of replicative DNA polymerases (Pols) thus leading to cytotoxicity. This repair pathway can be subdivided in five steps: (i) base removal by a specific DNA glycosylase; (ii) incision at the resulting abasic site by an AP-endonuclease; (iii) processing of the produced blocked termini; (iv) filling in of the gap; and (v) resealing of the damaged DNA strand.39) The major pathway to remove oxidized DNA bases is base excision repair (BER). BER is initiated by the recognition and excision of damaged base by the specific DNA glycosylase. In humans about 10 DNA glycosylases of different, but partially over- lapping substrate specificities are known.40) Some of them are bifunctional enzymes, with endowed AP-lyase activity and cleave phosphodiester bonds at 3' side of AP site by β- elimination or at 3' and 5' side by β/δ-elimination, creating
*/β-unsaturated aldehyde at the 3' end of cleaved DNA strand and phosphate groups at 5' and 3' end, respectively.
Monofunctional DNA glycosylases need the assistance of AP-endonucleases, which hydrolyse phosphodiester bond at the 5' end of the AP site. This yields DNA single strand break (SSB) with the 5' end bearing baseless deoxyribose (5'dRP) and the 3' hydroxyl group. “Dirty” 3' ends, bearing phosphogly- colates or phosphates are further processed and prepared for DNA synthesis. The major mammalian AP-endonuclease, APE1 has 3' phosphodiesterase activity and efficiently removes 3' phosphoglycolate groups, but has a very weak 3' phosphatase activity.41) Phosphate groups at 3' hydroxyls are most probably removed by polynucleotide kinase.42) Human AP-endonuclease plays also two additional roles: (i) stimulates excision of
damaged bases, 8-oxoG and εC by OGG1 and TDG glyco- sylases by increasing enzymes’ turnover on damaged DNA;43) (ii) may initiate nucleotide incision repair (NIR) pathway by the cleavage of phosphodiester bond adjacent to nucleotide bearing damaged base without its prior excision by DNA glycosylase. Several major oxidized bases are repaired via this pathway, namely 8-oxoG,44) 5-OHC45) and other.46) There are indications that multiple glycosylases of overlapping specificity are not essential for normal development.47) In contrast, defects in components of the latter steps of BER, such as Pol β48) and ligase, are incompatible with normal development in mice.
Animal models of BER exploiting complete knockouts of these latter genes are unlikely to be viable, but tissue-specific knock- outs using recombinase systems,48) are likely to be informative.
In direct repair, the simplest response to DNA damage is to remove or reverse the lesion in a single-step reaction, restoring the local sequence to its original state. In bacteria and yeast, for example, several photolyases can directly reverse DNA damage resulting from UV or cisplatin treatment in a light-dependent reaction.31) It is not surprising that human homologs of, e.g., the E. coli (6-4) photolyase have been found,49) since photolyases probably appearedeven before eukaryotes in evolutionary history. Nevertheless, the expression of this enzyme in all human tissues,49) is paradoxical, because visible light is not available in most tissues. Photolyase has been shown to bind to cisplatin-damaged DNA.50) In addition, it was recently demonstrated that photolyase activity may be induced by oxidative stress. In Saccharomyces, mutation of photolyase is associated with resistance to MNNG, 4NQO, and cis-diamminechloroplatinum.50) Perhaps the binding of photo- lyase with chemical or UV-damaged DNA51) serves as a regula- tory signal for other repair pathways52) or some yet unidentified source for energy exists.
A second direct repair enzyme in human is O6-methyl- guanine DNA methyltransferase (MGMT). MGMT is impor- tant in the repair of alkylation damage. The alkyl group from the lesion is transferred to a cysteine residue in the active site of MGMT,53) in an irreversible reaction which is not strictly catalytic. In addition to O6-methylguanine, MGMT repairs larger alkylation lesions, including O6-ethylguanine, O6-butyl- guanine, and O4-methylthymine, but with lower efficiency.54) In double-strand break repair, Double-strand breaks (DSB) in DNA arise under physiological conditions, including somatic recombination or the overlapping of extensive excision repair
tracts.55) They are also produced directly by ionizing radiation and oxidative insults.56) Unresolved DSBs block replication and transcription of involved sequences. Further, exposed ends of such fragments are prone to nuclease attack with subsequent destruction.57) Thus, efficient repair of DSBs is necessary for local and overall genomic integrity and for maintenance of gene expression. Although work with radiation-sensitive cell lines suggests that mammalian cells primarily rejoin DSBs by nonhomologous mechanisms, allelic recombinational repair is also found in mammalian cells increased at least two orders of magnitude by the induction of chromosomal DSB.58) One of the possible consequences of this is loss of heterozygosity (LOH). Homologous DSB repair involves physical interactions of RAD51, RAD52, and RPA.59) RAD51 protein catalyzes homologous recombination through its homologous pairing and strandexchange activities. RAD52 may modulate these activities through its RAD51-interacting region (amino acids 291-330).
The ability of RAD52 to induce homologous recombination requires its binding to the p34 subunit of RPA. This RPA binding domain is at amino acids 221-280. Recall that RPA is also involved in NER - this may provide a linkage between these two repair systems.
Nonhomologous end-joining pathway of DSB repair begins with binding of the free ends by DNA-dependent protein kinase (DNA-PK). DNA-PK is also involved in the regulation of transcription, in apoptosis, and in viral infection of human cells.60) The catalytic subunit (p350) is actually XRCC7 (X-ray cross complementing group 7).61) DNA-end-binding protein Ku serves as the regulatory subunit of DNA-PK,61,62) enhanc- ing repair accuracy, possibly by protecting free ends from nucleolytic attack.63) Ku recruits DNA-PK p350 to DNA double strand breaks.
Importance of DNA Repair
The importance of DNA repair is evident from the large investment that cells make in DNA repair enzymes. For example, analysis of the genomes of bacteria and yeasts has revealed that several percent of the coding capacity of these organisms is devoted solely to DNA repair functions. The importance of DNA repair is also demonstrated by the increased rate of mutation that follows the inactivation of a DNA repair gene. Many DNA repair pathways and the genes that encode them - which we now know operate in a wide
variety of organisms, including humans - were originally identified in bacteria by the isolation and characterization of mutants that displayed an increased mutation rate or an increased sensitivity to DNA-damaging agents. Cellular DNA is confronting damage regularly and as a result to ubiquitous environmental agents, most organisms have an instinct to repair their DNA and the macromolecular DNA is repaired by cells through different mechanisms.64) Organisms have evolved to efficiently respond to DNA insults that result from either endogenous sources (cellular metabolic processes) or exogenous sources (environmental factors). Endogenous sources of DNA damage include hydrolysis, oxidation, alkylation, and mismatch of DNA bases; sources for exogenous DNA damage include ionizing radiation (IR), ultraviolet (UV) radiation, and various chemicals agents. At the cellular level, damaged DNA that is not properly repaired can lead to genomic instability, apoptosis, or senescence, which can greatly affect the organism’s development and ageing process. An alternative route of discovery of human DNA repair genes has been through searching for the causative defects in human disease. The first human repair deficiency disease identified was xeroderma pigmentosum (XP),65) a hereditary deficiency in any of several nucleotide excision repair genes. Other human disorders related to defective DNA repair or defective cellular responses to DNA damage include trichothiodystrophy,66) Cockayne syndrome, Fanconi’s anemia,67) ataxia telangiectasia,68) Bloom’s syndrome69) and hereditary nonpolypopsis colorectal cancer (HNPCC).70) Clinical disease arising from defects in DNA repair is complex and often severe. Interpreting the relationship between DNA repair and disease has been facilitated by cell lines and animal models carrying mutations or knock-outs of these repair genes, resulting in chromosomal mapping, cloning, and sequencing of DNA repair genes, as well as biochemical studies of specific pathways and responses. Therefore, it is essential for cells to efficiently respond to DNA damage through coordinated and integrated DNA-damage checkpoints and repair pathways.71) In conclusion, it is universal truth that our genetic material is exposed to a number of physical and chemical agents, both environmental and intracellular, that introduce, either directly or indirectly, a wide range of DNA modifications. These agents include sunlight (ultraviolet radiation), X-rays, food mutagens, and reactive chemical species [e.g. oxygen free radicals that are formed as by-products of energy (ATP) production]. In addition, our DNA being a chemical is susceptible to spontane-
ous decay. Furthermore, errors can be introduced during DNA replication (i.e. the copying of our DNA) and imperfect exchanges can take place between our chromosomes, the DNA elements that contain genetic information. In all, modifications to DNA can lead to permanent changes in our genetic material. By inactivating or deregulating control proteins, such mutagenic events can have a detrimental effect on normal biological processes, leading to cell death or cellular dysfunc- tion, and ultimately human disease, most notably cancer. In order to figure out al the reasons behind DNA repair, DNA damage, and their consequences was cancer or any other diseases would be a great area to do further research as many of the thing still to invented.
REFERENCES
1) Wang E, Lenferink A, O'Connor-McCourt M. Cancer systems biology: exploring cancer-associated genes on cellular networks. Cellular and Molecular Life Sciences 64, 1752-1762, 2007.
2) Vogt PK. Cancer genes. West J Med 158, 273-278, 1993.
3) Molinari M. Cell cycle checkpoints and their inactivation in human cancer. Cell Prolif 33, 261-274, 2000.
4) Hunter T, Pines J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell 79, 573-582, 1994.
5) Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 1, 222-2231, 2001.
6) Hall M, Peters G. Genetic alterations of cyclins, cyclin-depen- dent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res 68, 67-108, 1996.
7) Kaufmann WK, Paules RS. DNA damage and cell cycle checkpoints. FASEB J 10, 238-247, 1996.
8) Ravi D, Ramadas K, Mathew BS, Nalinakumari KR, Nair MK, Pillai MR. Angiogenesis during tumor progression in the oral cavity is related to reduced apoptosis and high tumor cell proliferation. Oral Oncol 34, 543-548, 1998.
9) McNeil S, Zeng C, Harrington KS, Hiebert S, Lian JB, Stein JL, van Wijnen AJ, Stein GS. The t(8;21) chromosomal trans- location in acute myelogenous leukemia modifies intranuclear targeting of the AML1yCBFalfa2 transcription factor. PNAS 96, 14882-14887, 1999.
10) Holth LT, Chadee DN, Spencer VA, Samuel SK, Safneck JR, Davie JR. Chromatin, nuclear matrix and the cytoskeleton :Role of cell structure in neoplastic transformation. Inter- national Journal of Oncology 13, 827-837, 1998.
11) Haber D, Harlow E. Tumour-suppressor genes: evolving definitions in the genomic age. Nat Genet 16, 320-322, 1997.
12) Sherr CJ. Principles of tumor suppression. Cell 116, 235-246, 2004.
13) Ward EM, Sabbioni G, Debord DG, Teass AW, Brown KK,
Talaska GG, Roberts DR, Ruder AM, Streicher RP.
Monitoring of aromatic amine exposures in workers at a chemical plant with a known bladder cancer excess. J Natl Cancer Inst 88, 1046-1052, 1996.
14) Hsieh HI, Chen PC, Wong RH, Wang JD, Yang PM, Cheng TJ. Effect of the CYP2E1 genotype on vinyl chloride monomer-induced liver fibrosis among polyvinyl chloride workers. Toxicology 239, 34-44, 2007.
15) Lorenzo PI, Saatcioglu F. Inhibition of apoptosis in prostate cancer cells by androgens is mediated through downregulation of c-Jun N-terminal kinase activation. Neoplasia 10, 418-428, 2008.
16) Retha R, Newbold MS. Prenatal exposure to diethylstilbestrol (DES). Fertil Steril 89, 55-56, 2008.
17) Glover D, Little JB, Lavin MF, Gueven N. Low dose ionizing radiation-induced activation of connexin 43 expression. Int J Radiat Biol 79, 955-964, 2003.
18) Medina PP, Slack FJ. microRNAs and cancer: an overview.
Cell Cycle 7, 2485-2492, 2008.
19) Liu LS, Wang X, Yang BY, Sun Y. Mechanism of damage of DNA induced by carbaryl and heavy metal ions. Guang Pu Xue Yu Guang Pu Fen Xi 28, 1353-1355, 2008.
20) Mundy CJ, Wu Y, Colvin ME, Car R, Quong AA. DNA base damage: a car-parrinello molecular dynamics study of guanine and thymine with an OH radical. Nanotech 3, 380-381, 2003.
21) Lindahl T, Nyberg B. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry 13, 3405-3410, 1974.
22) Karran P, Lindahl T. Hypoxanthine in deoxyribonucleic acid:
generation by heat-induced hydrolysis of adenine residues and release in free form by a deoxyribonucleic acid glycosylase from calf thymus. Biochemistry 19, 6005-6011, 1980.
23) Jeffrey AM. DNA modification by chemical carcinogens.
Pharmacol Ther 28, 237-272, 1985.
24) Strauss BS, Karran P, Higgins NP. Alkylation damage and DNA excision repair in mammalian cells. J Toxicol Environ Health 2, 1395-1414, 1977.
25) Lippke JA, Gordon LK, Brash DE, Haseltine WA. Distri- bution of UV light-induced damage in a defined sequence of human DNA: detection of alkaline-sensitive lesions at pyrimidine nucleoside-cytidine sequences. Proc Natl Acad Sci USA 78, 3388-3392, 1981.
26) Villanueva A, Canete M, Hazen MJ. Uptake and DNA photodamage induced in plant cells in vivo by two cationic porphyrins. Mutagenesis 4, 157-159, 1989
27) Reid TM, Fry M, Loeb LA. Endogenous mutations and cancer.
Princess Takamatsu Symp 221-229, 1991.
28) Vos JM, Wauthier EL. Differential introduction of DNA damage and repair in mammalian genes transcribed by RNA polymerases I and II. Mol Cell Biol 11, 2245-2252, 1991.
29) Wood RD. DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie 81, 39-44, 1999.
30) Wood RD. DNA repair in eukaryotes. Annu Rev Biochem 65,
135-167, 1996.
31) Sancar A. DNA excision repair. Annu Rev Biochem 65, 43-81, 1996.
32) Tang J, Chu G. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair (Amst) 1, 601-616, 2002.
33) Rass K, Reichrath J. UV damage and DNA repair in malignant melanoma and nonmelanoma skin cancer. Adv Exp Med Biol 624, 162-178, 2008.
34) Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem 74, 681-710, 2005.
35) Lynch HT, Lynch JF. Hereditary nonpolyposis colorectal cancer (Lynch syndromes I and II): a common genotype linked to oncogenes? Med Hypotheses 18, 19-28, 1985.
36) Henry T. Lynch MD, Albert de la Chapelle. Hereditary colorectal cancer. N Engl J Med 348, 919-932, 2003.
37) Chung DC, Rustgi AK. DNA mismatch repair and cancer.
Gastroenterology 109, 1685-1699, 1995.
38) Strand M, Prolla TA, Liskay RM, Petes TD. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 365, 274-276, 1993.
39) Fortini P, Pascucci B, Parlanti E, D'Errico M, Simonelli V, Dogliotti E. The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie 85, 1053-1071, 2003.
40) Wood RD, Mitchell M, Lindahl T. Human DNA repair genes.
Mutat Res 577, 275-283, 2005.
41) Kelley MR, Parsons SH. Redox regulation of the DNA repair function of the human AP endonuclease Ape1/ref-1. Anti- oxidant Redox Signaling 3, 671-683, 2001.
42) Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-indepen- dent DNA base excision repair in human cells. Molecular Cell 15, 209-220, 2004.
43) Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Research 29, 430-438, 2001.
44) Ishchenko AA, Yang X, Ramotar D, Saparbaev M. The 3'→
5' exonuclease of Apn1 provides an alternative pathway to repair 7,8-dihydro-8-oxodeoxyguanosine in Saccharomyces cerevisiae. Molecular and Cellular Biology 25, 6380-6390, 2005.
45) Daviet S, Couve-Privat S, Gros L, Shinozuka K, Ide H, Saparbaev MK, Ishchenko AA. Major oxidative products of cytosine are substrates for the nucleotide incision repair pathway. DNA Repair (Amst) 6, 8-18, 2007.
46) Gros L, Ishchenko AA, Ide H, Elder RH, Saparbaev MK. The major human AP endonuclease (Ape1) is involved in the nucleotide incision repair pathway. Nucleic Acids Research 32, 73-81, 2004.
47) Engelward BP, Weeda G, Wyatt MD, Broekhof JL, de Wit J, Donker I, Allan JM, Gold B, Hoeijmakers JH, Samson LD.
Base excision repair deficient mice lacking the Aag alkyla- denine DNA glycosylase. Proc Natl Acad Sci USA 94, 13087- 13092, 1997.
48) Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K.
Deletion of a DNA polymerase beta gene segment in T cells using cell typespecific gene targeting. Science 265, 103-106, 1994.
49) van der Spek PJ, Kobayashi K, Bootsma D, Takao M, Eker AP, Yasui A. Cloning, tissue expression, and mapping of a human photolyase homolog with similarity to plant blue-light receptors. Genomics 37, 177-182, 1996.
50) Fox ME, Feldman BJ, Chu G. A novel role for DNA photolyase:Binding to DNA damaged by drugs is associated with enhanced cytotoxicity in Saccharomyces cerevisiae. Mol Cell Biol 14, 8071-8077, 1994.
51) Sibghat-Ullah, Sancar A. Substrate overlap and functional competition between human nucleotide excision repair and Escherichia coli photolyase and (a)BC excision nuclease.
Biochemistry 29, 5711-5718, 1990.
52) Ozer Z, Reardon JT, Hsu DS, Malhotra K, Sancar A. The other function of DNA photolyase: Stimulation of excision repair of chemical damage to DNA. Biochemistry 34, 15886- 15889, 1995.
53) Hazra TK, Roy R, Biswas T, Grabowski DT, Pegg AE, Mitra S. Specific recognition of O6-methylguanine in DNA by active site mutants of human O6-methylguanine-DNA methyl- transferase. Biochemistry 36, 5769-5776, 1997.
54) Sancar A. DNA repair in humans. Annu Rev Genet 29, 69-105, 1995.
55) Lieber MR. Site-specific recombination in the immune system.
FASEB J 5, 2934-2944, 1991.
56) Dizdaroglu M. Oxidative damage to DNA in mammalian chromatin. Mutat Res 275, 331-342, 1992.
57) Rufer JT, Morgan WF. Potentiation of DNA damage by inhibition of poly(ADP-ribosyl)ation: a test of the hypothesis for random nuclease action. Exp Cell Res 200, 506-512, 1992.
58) Moynahan ME, Jasin M. Loss of heterozygosity induced by a chromosomal double-strand break. Proc Natl Acad Sci U S A 94, 8988-8993, 1997.
59) Park MS, Ludwig DL, Stigger E, Lee SH. Physical interaction
between human RAD52 and RPA is required for homologous recombination in mammalian cells. J Biol Chem 271, 18996- 19000, 1996.
60) Lees-Miller SP. The DNA-dependent protein kinase, DNA- PK: 10 years and no ends in sight. Biochem Cell Biol 74, 503- 512, 1996.
61) Smider V, Chu G. The end-joining reaction in V(D)J recombination. Semin Immunol 9, 189-197, 1997.
62) Yaneva M, Kowalewski T, Lieber MR. Interaction of DNA-dependent protein kinase with DNA and with Ku:
biochemical and atomic-force microscopy studies. EMBO J 16, 5098-5112, 1997.
63) Boulton SJ, Jackson SP. Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res 24, 4639-4648, 1996.
64) Yu Z, Chen J, Ford BN, Brackley ME, Glickman BW.
Human DNA repair systems: an overview. Environ Mol Mutagen 33, 3-20, 1999.
65) Stich HF. Response of homozygous and heterozygous xero- derma pigmentosum cells to several chemical and viral carcinogens. Basic Life Sci 5, 773-784, 1975.
66) Rebora A, Crovato F. PIBI(D)S syndrome--trichothiodystrophy with xeroderma pigmentosum (group D) mutation. J Am Acad Dermatol 6, 940-947, 1987.
67) Poon PK, Parker JW, O'Brien RL. Faulty DNA repair following ultraviolet irradiation in Fanconi's anemia. Basic Life Sci 5, 821-824, 1975.
68) Vincent RA Jr, Sheridan RB 3rd, Huang PC. DNA strained breakage repair in ataxia telangiectasia fibroblast-like cells.
Mutat Res 33, 357-366, 1975.
69) Inoue T, Hirano K, Yokoiyama A, Kada T, Kato H. DNA repair enzymes in ataxia telangiectasia and Bloom's syndrome fibroblasts. Biochim Biophys Acta 479, 497-500, 1977.
70) Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpoly- posis colon cancer. Cell 75, 1027-1038, 1993.
71) Hakem R. DNA-damage repair; the good, the bad, and the ugly. EMBO J 27, 589-605, 2008.