Yeungnam Univ J Med 2017;34(1):19-28
The relationship between muscle mitochondrial nutritional overloading and insulin resistance
Jae-Han Jeon
1, Jun-Sung Moon
2, Kyu-Chang Won
2, In-Kyu Lee
11
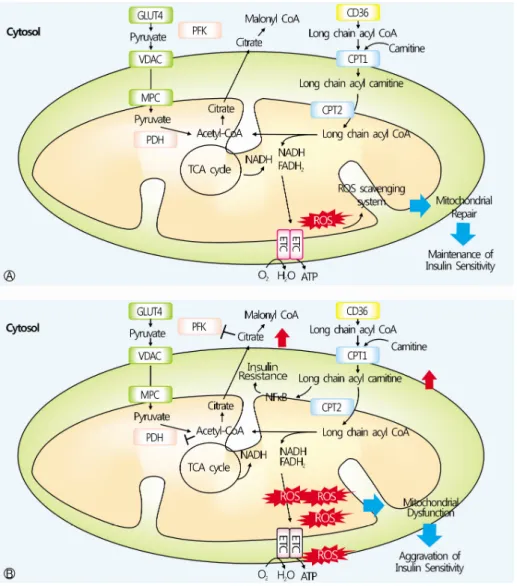
전체 글
Yeungnam Univ J Med 2017;34(1):19-28
1
수치
관련 문서
Regarding block-generating nodes, CTO claimed that " The DApp representative will play a representative role as a node, rather than in the form of a block producer
Based on the results, a study model was developed and a hypothesis that transformational and transactional leadership would significantly affect organizational
Injury is a burden on athletes, but the rehabilitation exercise of this study suggests that the improvement of knee muscle function and knee function
The purpose of this study is to analyze the relationship between dance performances, which is a part of social contribution activities of local governments,
When a symmetry operation has a locus, that is a point, or a line, or a plane that is left unchanged by the operation, this locus is referred to as the symmetry
In this study, we found that Pin1 plays a pivotal role in insulin- induced AP-1 activation through its interaction with p70S6K and prolongs activity of
The rectifier is a circuit that acts as a dc voltage source by converting the ac voltage from a standard wall outlet to a dc voltage. This voltage is effectively applied
* When a symmetry operation has a locus, that is a point, or a line, or a plane that is left unchanged by the operation, this locus is referred to as the symmetry element..
