유잉육종
주희영
삼성서울병원 소아청소년과
Ewing Sarcoma
Hee Young Ju
Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
Ewing sarcoma is the second most frequently occurring malignant tumor of the bone and soft tissue in adolescents and young adults. Genetically, Ewing sarcoma is charac- terized by balanced chromosomal translocation in which a member of FET gene family is fused with an ETS transcription factor, with the most common fusion being EWSR1-FLI1 (85% of cases). Treatment of Ewing sarcoma is based on multidisciplinary approach (local surgery, radiotherapy and multiagent chemotherapy), which are asso- ciated with chronic late effects that may compromise quality of life of survivors. First line treatment includes combination of drugs incorporating doxorubicin, vincristine, cy- clophosphamide, ifosfamide, etoposide, and dactinomycin. The beneficial role of high dose chemotherapy has been suggested in high-risk localized Ewing sarcoma patients, and the studies are being performed to investigate the role in metastatic disease. The 5-year overall survival for localized Ewing sarcoma has improved to reach 65% to 75%.
But patients with metastatic disease have a 5-year survival rate of <30%, except for those with isolated pulmonary metastasis (approximately 50%). Patients with recurrent tumor have a dismal prognosis. Novel therapeutic strategies based on understanding of molecular mechanisms are needed to improve the outcome of Ewing sarcoma and to lessen the treatment-related late effects.
pISSN 2233-5250 / eISSN 2233-4580 https://doi.org/10.15264/cpho.2019.26.1.27 Clin Pediatr Hematol Oncol 2019;26:27∼34
Received on March 25, 2019 Revised on April 4, 2019 Accepted on April 11, 2019
Corresponding Author: Hee Young Ju Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 06351, Korea
Tel: +82-2-3410-0870 Fax: +82-2-3410-0043 E-mail: [email protected] ORCID ID: orcid.org/0000-0001-6744-0412
Key Words: Ewing sarcoma, Peripheral primitive neuroectodermal tumors, EWS-FLI1 fu- sion protein
Introduction
1) Epidemiology
Ewing sarcoma is the second most common malignant bone or soft tissue sarcoma, mainly occurring in children, adolescents and young adults (AYA). The peak incidence age is 15 years old, which occurs more in male than female
with 3:2 ratio [1]. Ewing sarcoma predominantly affects the
bone of extremity (45%), thorax or abdomen (20%), or the
pelvis (19%). Approximately 20% of cases present the tu-
mor at extraskeletal sites (thigh, pelvis, paraspinal area, and
foot). Among newly diagnosed Ewing sarcoma patients,
20-25% present with metastasis at diagnosis. The incidence
rate of Ewing sarcoma is 1.5 cases per million children and
AYA globally. But the incidence of Ewing sarcoma was re-
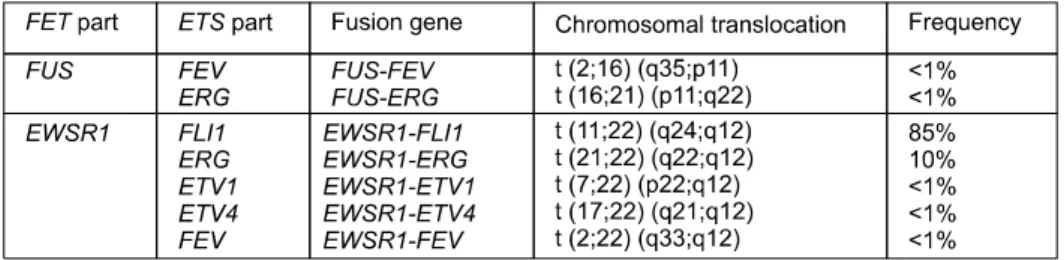
Fig. 1. FET-ETS fusion oncogenes reported to date in Ewing sarcoma. Ewing sarcoma is characterized by recurrent balanced translocation resulting in fusion of the FET gene family (FUS, EWSR1 or TAF15) with the ETS gene family (FLI1, ERG, ETV4 and FEV).
ported to be lower in Asian and African populations, with annual rates of 0.8 and 0.2 cases per million children, re- spectively [1,2]. By recent Korean report [3], age-stand- ardized ratio (ASR) of Ewing sarcoma was 1.4 per million, and highest ASR was observed in 10-14 years old group, which was similar with global report. Epidemiologic associ- ation studies have indicated higher rates of Ewing sarcoma in children of farm workers, but they are not considered as a risk factor [4]. Also, Ewing sarcoma is rarely observed among the cancer predisposition syndromes described to date [5]. Although the etiology of Ewing sarcoma is un- known, it is genetically well characterized.
2) Group of disease
Historically, the group ‘Ewing sarcoma family of tu- mours’ included the tumors based on morphological and immunophenotypical features and the presence of chromo- somal translocations. This group included extraosseous Ewing sarcoma, peripheral primitive neuroectodermal tu- mours and Askin tumours. However, the 2013 WHO classi- fication of sarcomas uniformly defined these tumours as
‘Ewing sarcoma’ [6,7], characterized by pathognomonic FET-ETS gene fusions [8,9]. The WHO classification also in- cludes the term ‘Ewing-like sarcomas’, which are small round cell sarcomas with morphologically similar appear- ances to Ewing sarcoma but are characterized by different fusion genes and clinical, pathological features. Ewing sar- coma is characterized by its driver mutations, specific chro- mosomal translocations that fuse a number of the FET fam- ily of protein (encoded by FUS, EWSR1 and TAF15), which are RNA-binding proteins involved in transcription and splicing, with different members of the ETS (E25-specific) family of transcriptional factors which are involved in cell
proliferation, cell differentiation, cell-cycle control, angio- genesis and apoptosis-most commonly FLI1 (85% of cases) [10]. The resulting chimeric fusion proteins act as aberrant transcription factors, causing tumor by deregulation genes involved in cell-cycle regulation, cell migration and pro- liferation.
Well-known genetic translocation of Ewing sarcoma is a recurrent balanced chromosomal translocation, most com- monly t(11;22)(q12;q24). This translocation results in the fusion gene of EWSR1 with FLI1. Among the 10 to 15%
of Ewing sarcoma cases negative for EWSR1-FLI1 fusions, other fusions occur, mainly between EWSR1 and ERG. With a lower frequency, ETV1, ETV4, FEV fusions to EWSR1 were identified (Fig. 1).
Cytogenetic and genomic studies identified recurrent
chromosomal abnormalities in Ewing sarcoma [11]. Chro-
mosome 1q gain and possibly chromosome 16q loss was
reported to be related with poor clinical outcome of Ewing
sarcoma [11,12]. Aberrant transcription can be induced by
various FET-ETS gene fusions. A recent study has shown
that gene transcription mediated by EWSR1-FLI1 leads to
the frequent formation of R loops, which are three-standard
structures composed of a DNA:RNA hybrid and an non-
template single-stranded DNA. These R loops might sensi-
tize Ewing sarcoma cells to poly (ADP-ribose) polymerase
(PARP) inhibitors, by synthetic lethality [13]. In addition,
epigenome profiling showed that EWSR1-FLI1 drives epi-
genetic reprogramming by inducing de novo enhancers and
by repressing enhancers in many mesenchymal origin cells
[14].
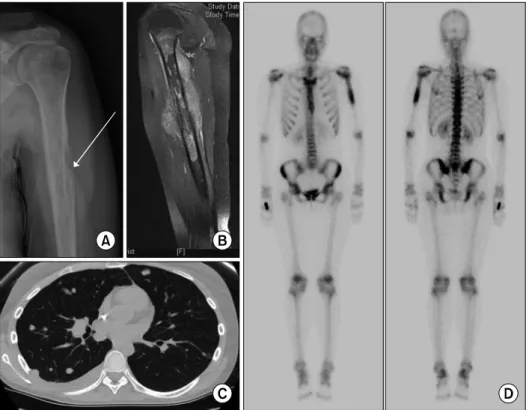
Fig. 2. Radiological presentation of Ewing sarcoma. X-ray shows osteo- lytic lesion of bone and involve- ment of the periosteal tissue (A).
MRI scan showing primary tumor in the humerus and accompanying soft tissue edema (B). CT scan of the lungs show lung metastasis with pleural effusion of right lung (C).
Bone scintigraphy highlighting Ewing sarcoma in the humerus (D).
Diagnosis
1) Clinical features
Majority of patients present with a history of locoregional pain, which may be intermittent. In a substantial number of patients, pain is followed by a palpable soft-tissue mass.
About 20% of patients present fever, which increase the risk of tumor mistaken for osteomyelitis [15]. Median length of symptom before diagnosis is 3-9 months, however, time to diagnosis is not associated with presence of metastasis or outcome [16]. Ewing sarcoma most commonly involves the lower extremity, with the pelvis being the next most com- mon site. In long bones, Ewing sarcoma is generally of dia- physeal origin. Additionally, tumor sites vary with age; re- cent study showed that older AYA patients (20-24 years) had more pelvic and axial primary tumors, larger tumors and worse outcomes than children (0-9 years) [17]. In addi- tion, Ewing sarcoma in older patients tend to occur in soft tissues [18].
2) Diagnostic evaluation
Tumor imaging and metastatic evaluation of Ewing sarco- ma include initial radiological evaluation of affected bone, and successive CT of the lungs and bone scintigraphy for detection of metastases (Fig. 2). On radiographs, Ewing sar- coma show tumor-related osteolysis as a destructive mass of diaphyseal-metaphyseal bone with a multilayered ap- pearance. MRI provides better images to evaluate the extent of disease. 18F-fluorodeoxyglucose (FDG) PET-CT can be used to determine the presence of metastasis, including the metastases in the bone marrow [19]. Some studies sug- gested that FDG PET-CT may be sufficient for initial screen- ing of bone marrow metastases in Ewing sarcoma [20,21].
3) Pathology
Histologically, Ewing sarcoma has a solid pattern of
growth and is composed of monomorphic small round cells
with round nuclei [7]. The chromatin is stippled, and nucle-
oli are usually not observable. CD99 is a cell-surface glyco-
protein which considered a relevant diagnostic marker for
Ewing sarcoma [22]. Ninety-five percent of Ewing sarcoma
specimen present strong, diffuse membranous expression of CD99, but CD99 is not specific for Ewing sarcoma and occurs in a large group of normal tissues, other small round cell tumors and lymphoblastic lymphoma. Currently, diag- nosis of Ewing sarcoma can be confirmed only by molec- ular pathology, with fluorescence in situ hybridization (FISH) and/or reverse transcription PCR (RT-PCR). In some patients, next-generation sequencing (NGS) or RNA se- quencing for rare gene fusions can be needed [23].
4) Risk classification
Metastatic status at diagnosis is the strongest prognostic factor of Ewing sarcoma. Approximately 10-30% of patients present metastases at the time of diagnosis. Common meta- stasis sites are lung (38%), bone (31%), and bone marrow (11%). Five-year overall survival (OS) remains <30% for patients with metastatic disease at diagnosis. However, those with isolated pulmonary metastasis shows better clin- ical outcome with OS of ∼50% [24-27]. According to a Korean single center retrospective study, 5-year OS rate of adult and children Ewing sarcoma patients was 33% [28].
Other single center study showed the 5-year OS rate of lo- calized Ewing sarcoma (N=76) was 58.9% [29].
The EUROEWING 99 R3 study of primary disseminated multifocal Ewing sarcoma identified additional prognostic factors (age at diagnosis >14 years, primary tumor volume
>200 mL, presence and number of bone lesions, additional pulmonary metastases, and bone marrow involvement) [30].
In localized Ewing sarcoma, initial tumor size (>8 cm) or volume (>200 mL) is considered as a strong prognostic factor [31]. However, for localized tumors resected after in- duction chemotherapy, histologic response is the strongest independent prognostic factor [32-35]. Other reported prog- nostic factors include age, fever, and baseline lactate de- hydrogenase level [25,27,36].
Type of EWS-FLI family fusion has been reported as a prognostic factor at retrospective studies [37,38], but pro- spective evaluation found no prognostic impact of fusion transcript type [39,40]. Recurrent copy number variations (gain of chromosomes 1q, 8q, 20, and loss of chromosome 16q) has been reported as a possible prognostic relevance [41]. Also, poor prognostic value of concurrent STAT2 and
TP53 mutations has been reported [11].
Management
1) Treatment
Before the introduction of systemic chemotherapy, Ewing sarcoma had a cure rate of less than 10%, even in patients with localized disease [42]. Currently, patients with newly diagnosed Ewing sarcoma are treated with a combination of multi-agent cytotoxic chemotherapy and local control meas- ures (surgery and/or radiotherapy). Induction chemothe- rapy is given before local treatment to reduce the size of the primary tumor and address micrometastatic disease [43].
Currently, surgical resection is considered superior to de- finitive radiotherapy for local control [31,44,45]. Thus, tu- mor resection is performed whenever a marginal or wide resection seems possible. Definitive radiotherapy is only recommended for inoperable lesions, with a recommended dose of 54 to 55 Gy to the tumor with a 2 cm security mar- gin [46]. Postoperative radiation therapy is advised in cases of incomplete surgical resection; however, in Europe, pa- tients with poor histologic response to induction chemo- therapy also receive postoperative radiation therapy al- though tumor is completely resected [33,47].
An intergroup study conducted by the Pediatric Oncolo-
gy Group and the Children’s Cancer Group demonstrated
that the addition of ifosfamide and etoposide to cyclo-
phosphamide, doxorubicin, and vincristine significantly im-
proved outcomes for patients with localized Ewing sarcoma
[27]. In a subsequent trial, the Children’s Oncology Group
studied the value of dose intensity by comparing 3-weekly
versus 2-weekly (compressed) regimens of vincristine, dox-
orubicin and cyclophosphamide alternating with ifosfamide
and etoposide. The 5-year event free survival (EFS) was
better in patients who received the compressed regimen
(73% versus 65%) [48]. Another cooperative group clinical
trial, Euro-Ewing 99 showed that intense induction chemo-
therapy with VIDE (vincristine, ifosfamide, doxorubicin and
etoposide) followed by cyclophosphamide-based or ifosfa-
mide-based consolidation chemotherapy resulted in no dif-
ference of survival [49]. Alternation of both agents may be
a means to reduce cumulative doses of each and their late
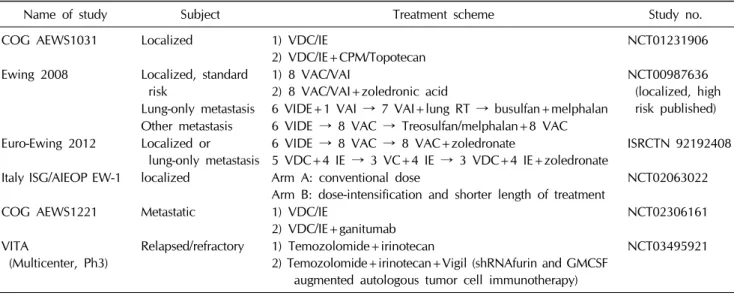
Table 1. Currently ongoing randomized trials for Ewing sarcoma
Name of study Subject Treatment scheme Study no.
COG AEWS1031 Localized 1) VDC/IE
2) VDC/IE+CPM/Topotecan NCT01231906
Ewing 2008 Localized, standard risk
1) 8 VAC/VAI
2) 8 VAC/VAI+zoledronic acid
NCT00987636 (localized, high risk published) Lung-only metastasis 6 VIDE+1 VAI → 7 VAI+lung RT → busulfan+melphalan
Other metastasis 6 VIDE → 8 VAC → Treosulfan/melphalan+8 VAC Euro-Ewing 2012 Localized or
lung-only metastasis
6 VIDE → 8 VAC → 8 VAC+zoledronate
5 VDC+4 IE → 3 VC+4 IE → 3 VDC+4 IE+zoledronate
ISRCTN 92192408 Italy ISG/AIEOP EW-1 localized Arm A: conventional dose
Arm B: dose-intensification and shorter length of treatment
NCT02063022
COG AEWS1221 Metastatic 1) VDC/IE
2) VDC/IE+ganitumab
NCT02306161 VITA
(Multicenter, Ph3)
Relapsed/refractory 1) Temozolomide+irinotecan
2) Temozolomide+irinotecan+Vigil (shRNAfurin and GMCSF augmented autologous tumor cell immunotherapy)
NCT03495921
effects, which is tested by ongoing long-term comparison
study.
High dose busulfan and melphalan (BuMel) improved EFS and OS when given after vincristine, ifosfamide, doxor- ubicin, and etoposide induction in localized ES with predefi ned high-risk factors. Recent Euro-Ewing 99 and EWING- 2008 trial showed benefit of high dose busulfan and mel- phalan chemotherapy followed by autologous hemato- poietic stem cell transplantation compared to VAI (vincris- tine, dactinomycin, and ifosfamide for 7 cycles) in high-risk localized Ewing sarcoma patients (poor histologic response after 6 cycles of VIDE, large tumor >200 mL) [50].
Early studies in Ewing sarcoma with lung metastasis showed that whole lung irradiation (WLI) could improve the 5-year EFS when compared to no WLI (49% versus 36%) [51]. Later, a collaborative European and US trial com- pared treatment with chemotherapy of vincristine, dactino- mycin and ifosfamide plus WLI and high-dose BuMel che- motherapy by randomization. The trial showed no differ- ence between the two treatment, with 3-year EFS of 51%
and 55%, respectively [25].
When Ewing sarcoma recurs after initial therapy, the prognosis is very poor, with less than 5% of patients re- maining alive more than 2 years following recurrence [52,53]. Several chemotherapy regimens showed activity in relapsed Ewing sarcoma; cyclophosphamide with top- otecan, irinotecan with temozolomide, gemcitabine with
docetaxel, and high dose ifosfamide [54-57].
Strategies under clinical investigation in Ewing sarcoma include the use of PARP inhibitors as enhancers of drug sensitivity in combination with agents such as trabectedin and/or temozolomide [58,59]. Insulin-like growth factor-1 receptor (IGF1R)-blocking antibodies or small-molecule in- hibitors produced some transient report in few patients, but was dropped because of failure to prolong the effectiveness as a single-agent treatment. Children’s Oncology Group is processing (recruiting) on a randomized trial with and with- out IGFR1 antibody, in combination with VDC-IE chemo- therapy in metastatic Ewing sarcoma. (NCT02306161) More- over, targeting the epigenetic deregulation by inhibitors of lysine-specific histone deacetylase 1A, histone deacetylase, and DNA methyltransferase are on studies [60-62]. These new trials are potentially expected to advance the outcome of Ewing sarcoma (Table 1).
2) Quality of life
Patients with Ewing sarcoma are at risk of substantial dis- ease-related and therapy-related late sequelae. Long term follow up report by Childhood Cancer Survivor Study (CCSS) described chronic health impairment in 70% of sur- vivors of Ewing sarcoma at 35 years after diagnosis [63].
All local treatment modalities can increase the risk of
long-term neuromusculoskeletal complications, reduced
functional capacity [64-66]. However, recent study showed
that, on average, recommended level of active lifestyle was achieved in Ewing sarcoma survivors [67]. Chemotherapy regimens used in Ewing sarcoma usually adopts anthracy- clines, alkylating agents and etoposide. Cardiomyopathy, renal impairment, renal Fanconi syndrome and infertility have been described [63]. Furthermore, second malignancy is a concern, which was reported to occur in ∼9% of Ewing sarcoma survivors [68]. Etoposide and high-dose chemotherapy were identified as risk factors for second ma- lignancy [68]. Radiotherapy is also well known as risk factor for second malignancy, especially osteosarcoma, acute myeloid leukemia, breast cancer and thyroid cancer [69].
Conclusion
Multimodality treatment improved the outcome of Ewing sarcoma, but survival rate of patients with metastatic or re- current Ewing sarcoma still remain poor. Risk-directed ther- apy and efforts for developing novel therapeutic ap- proaches based on understanding of molecular mechanisms are needed to improve the outcome of Ewing sarcoma and to lessen the treatment-related late effects.
References
1. Jawad MU, Cheung MC, Min ES, Schneiderbauer MM, Koniaris LG, Scully SP. Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome:
an analysis of 1631 cases from the SEER database, 1973-2005.
Cancer 2009;115:3526-36.
2. Worch J, Cyrus J, Goldsby R, Matthay KK, Neuhaus J, DuBois SG. Racial differences in the incidence of mesenchymal tu- mors associated with EWSR1 translocation. Cancer Epidemiol Biomarkers Prev 2011;20:449-53.
3. Park HJ, Moon EK, Yoon JY, et al. Incidence and survival of childhood cancer in Korea. Cancer Res Treat 2016;48:869-82.
4. Valery PC, McWhirter W, Sleigh A, Williams G, Bain C. Farm exposures, parental occupation, and risk of Ewing's sarcoma in Australia: a national case-control study. Cancer Causes Con- trol 2002;13:263-70.
5. Rahman N. Realizing the promise of cancer predisposition genes. Nature 2014;505:302-8.
6. Doyle LA. Sarcoma classification: an update based on the 2013 World Health Organization classification of tumors of soft tissue and bone. Cancer 2014;120:1763-74.
7. Fletcher CD, Bridge JA, Hogendoorn PC, Mertens F. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon:
IARC, 2013;305-10.
8. Watson S, Perrin V, Guillemot D, et al. Transcriptomic defi- nition of molecular subgroups of small round cell sarcomas.
J Pathol 2018;245:29-40.
9. Sankar S, Lessnick SL. Promiscuous partnerships in Ewing's sarcoma. Cancer Genet 2011;204:351-65.
10. Delattre O, Zucman J, Plougastel B, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome trans- location in human tumours. Nature 1992;359:162-5.
11. Tirode F, Surdez D, Ma X, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 2014;4:1342-53.
12. Mackintosh C, Ordonez JL, Garcia-Dominguez DJ, et al. 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene 2012;
31:1287-98.
13. Gorthi A, Romero JC, Loranc E, et al. EWS-FLI1 increases tran- scription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018;555:387-91.
14. Riggi N, Knoechel B, Gillespie SM, et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly acti- vate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014;26:668-81.
15. Pizzo PA, Poplack DG. Principles and practice of pediatric oncology. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2015.
16. Widhe B, Widhe T. Initial symptoms and clinical features in osteosarcoma and Ewing sarcoma. J Bone Joint Surg Am 2000;82:667-74.
17. Worch J, Ranft A, DuBois SG, Paulussen M, Juergens H, Dirksen U. Age dependency of primary tumor sites and meta- stases in patients with Ewing sarcoma. Pediatr Blood Cancer 2018;65:e27251.
18. Rochefort P, Italiano A, Laurence V, et al. A retrospective mul- ticentric study of Ewing sarcoma family of tumors in patients older than 50: management and outcome. Sci Rep 2017;7:
17917.
19. Völker T, Denecke T, Steffen I, et al. Positron emission to- mography for staging of pediatric sarcoma patients: results of a prospective multicenter trial. J Clin Oncol 2007;25:5435-41.
20. Newman EN, Jones RL, Hawkins DS. An evaluation of [F-18]-fluorodeoxy-D-glucose positron emission tomography, bone scan, and bone marrow aspiration/biopsy as staging in- vestigations in Ewing sarcoma. Pediatr Blood Cancer 2013;60:1113-7.
21. Kopp LM, Hu C, Rozo B, et al. Utility of bone marrow aspira- tion and biopsy in initial staging of Ewing sarcoma. Pediatr Blood Cancer 2015;62:12-5.
22. Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specific marker for Ewing's sar-
coma and peripheral primitive neuroectodermal tumors.
Evidence for a common histogenesis of Ewing's sarcoma and peripheral primitive neuroectodermal tumors from MIC2 ex- pression and specific chromosome aberration. Cancer 1991;67:1886-93.
23. Chen S, Deniz K, Sung YS, Zhang L, Dry S, Antonescu CR.
Ewing sarcoma with ERG gene rearrangements: A molecular study focusing on the prevalence of FUS-ERG and common pitfalls in detecting EWSR1-ERG fusions by FISH. Genes Chromosomes Cancer 2016;55:340-9.
24. Luksch R, Tienghi A, Hall KS, et al. Primary metastatic Ewing's family tumors: results of the Italian Sarcoma Group and Scandinavian Sarcoma Group ISG/SSG IV Study including myeloablative chemotherapy and total-lung irradiation. Ann Oncol 2012;23:2970-6.
25. Oberlin O, Rey A, Desfachelles AS, et al. Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors: a study by the Societe Francaise des Cancers de l'Enfant. J Clin Oncol 2006;24:3997-4002.
26. Paulussen M, Craft AW, Lewis I, et al. Results of the EICESS-92 Study: two randomized trials of Ewing's sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol 2008;26:4385-93.
27. Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003;348:694-701.
28. Lim SM, Yoo CJ, Han JW, et al. Incidence and survival of pediatric soft tissue sarcomas: comparison between adults and children. Cancer Res Treat 2015;47:9-17.
29. Lee JA, Kim DH, Cho J, et al. Treatment outcome of Korean patients with localized Ewing sarcoma family of tumors: a sin- gle institution experience. Jpn J Clin Oncol 2011;41:776-82.
30. Ladenstein R, Pötschger U, Le Deley MC, et al. Primary dis- seminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 2010;28:3284-91.
31. Krasin MJ, Rodriguez-Galindo C, Davidoff AM, et al. Efficacy of combined surgery and irradiation for localized Ewings sar- coma family of tumors. Pediatr Blood Cancer 2004;43:229-36.
32. Bacci G, Ferrari S, Bertoni F, et al. Prognostic factors in non- metastatic Ewing's sarcoma of bone treated with adjuvant che- motherapy: analysis of 359 patients at the Istituto Ortopedico Rizzoli. J Clin Oncol 2000;18:4-11.
33. Oberlin O, Deley MC, Bui BN, et al. Prognostic factors in lo- calized Ewing's tumours and peripheral neuroectodermal tu- mours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 2001;85:1646-54.
34. Göbel V, Jürgens H, Etspüler G, et al. Prognostic significance of tumor volume in localized Ewing's sarcoma of bone in chil- dren and adolescents. J Cancer Res Clin Oncol 1987;113:187-91.
35. Gaspar N, Rey A, Berard PM, et al. Risk adapted chemo- therapy for localised Ewing's sarcoma of bone: the French EW93 study. Eur J Cancer 2012;48:1376-85.
36. Hense HW, Ahrens S, Paulussen M, Lehnert M, Jürgens H.
Factors associated with tumor volume and primary metastases in Ewing tumors: results from the (EI)CESS studies. Ann Oncol 1999;10:1073-7.
37. Zoubek A, Dockhorn-Dworniczak B, Delattre O, et al. Does expression of different EWS chimeric transcripts define clin- ically distinct risk groups of Ewing tumor patients? J Clin Oncol 1996;14:1245-51.
38. de Alava E, Kawai A, Healey JH, et al. EWS-FLI1 fusion tran- script structure is an independent determinant of prognosis in Ewing's sarcoma. J Clin Oncol 1998;16:1248-55.
39. Le Deley MC, Delattre O, Schaefer KL, et al. Impact of EWS-ETS fusion type on disease progression in Ewing's sarco- ma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 2010;28:1982-8.
40. van Doorninck JA, Ji L, Schaub B, et al. Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 2010;28:1989-94.
41. Kovar H, Alonso J, Aman P, et al. The first European inter- disciplinary ewing sarcoma research summit. Front Oncol 2012;2:54.
42. Meyers PA. Systemic therapy for osteosarcoma and Ewing sarcoma. Am Soc Clin Oncol Educ Book 2015:e644-7.
43. Pappo AS, Dirksen U. Rhabdomyosarcoma, Ewing sarcoma, and other round cell sarcomas. J Clin Oncol 2018;36:168-79.
44. DuBois SG, Krailo MD, Gebhardt MC, et al. Comparative eval- uation of local control strategies in localized Ewing sarcoma of bone: a report from the Children's Oncology Group.
Cancer 2015;121:467-75.
45. Bacci G, Forni C, Longhi A, et al. Long-term outcome for pa- tients with non-metastatic Ewing's sarcoma treated with ad- juvant and neoadjuvant chemotherapies. 402 patients treated at Rizzoli between 1972 and 1992. Eur J Cancer 2004;40:73-83.
46. Donaldson SS, Torrey M, Link MP, et al. A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: end re- sults of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 1998;42:125-35.
47. Schuck A, Ahrens S, Paulussen M, et al. Local therapy in lo- calized Ewing tumors: results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int J Radiat Oncol Biol Phys 2003;55:168-77.
48. Womer RB, West DC, Krailo MD, et al. Randomized con- trolled trial of interval-compressed chemotherapy for the treat- ment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 2012;30:4148-54.
49. Le Deley MC, Paulussen M, Lewis I, et al. Cyclophosphamide compared with ifosfamide in consolidation treatment of stand-
ard-risk Ewing sarcoma: results of the randomized non- inferiority Euro-EWING99-R1 trial. J Clin Oncol 2014;32:2440-8.
50. Whelan J, Le Deley MC, Dirksen U, et al. High-dose chemo- therapy and blood autologous stem-cell rescue compared with standard chemotherapy in localized high-risk Ewing sarcoma:
results of Euro-E.W.I.N.G.99 and Ewing-2008. J Clin Oncol 2018:JCO2018782516.
51. Paulussen M, Ahrens S, Craft AW, et al. Ewing's tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing's Sarcoma Studies patients. J Clin Oncol 1998;16:3044-52.
52. Stahl M, Ranft A, Paulussen M, et al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer 2011;57:549-53.
53. Leavey PJ, Mascarenhas L, Marina N, et al. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence fol- lowing multi-modality therapy: a report from the Children's Oncology Group. Pediatr Blood Cancer 2008;51:334-8.
54. Casey DA, Wexler LH, Merchant MS, et al. Irinotecan and te- mozolomide for Ewing sarcoma: the Memorial Sloan-Kettering experience. Pediatr Blood Cancer 2009;53:1029-34.
55. Hunold A, Weddeling N, Paulussen M, Ranft A, Liebscher C, Jurgens H. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer 2006;47:795-800.
56. Fox E, Patel S, Wathen JK, et al. Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarco- ma, osteosarcoma, or unresectable or locally recurrent chon- drosarcoma: results of Sarcoma Alliance for Research Through Collaboration Study 003. Oncologist 2012;17:321.
57. Ferrari S, del Prever AB, Palmerini E, et al. Response to high-dose ifosfamide in patients with advanced/recurrent Ewing sarcoma. Pediatr Blood Cancer 2009;52:581-4.
58. Engert F, Schneider C, Weibeta LM, Probst M, Fulda S. PARP inhibitors sensitize Ewing sarcoma cells to temozolomide-in- duced apoptosis via the mitochondrial pathway. Mol Cancer Ther 2015;14:2818-30.
59. Ordonez JL, Amaral AT, Carcaboso AM, et al. The PARP in- hibitor olaparib enhances the sensitivity of Ewing sarcoma to trabectedin. Oncotarget 2015;6:18875-90.
60. Theisen ER, Pishas KI, Saund RS, Lessnick SL. Therapeutic op- portunities in Ewing sarcoma: EWS-FLI inhibition via LSD1 targeting. Oncotarget 2016;7:17616-30.
61. Hurtubise A, Bernstein ML, Momparler RL. Preclinical evalua- tion of the antineoplastic action of 5-aza-2'-deoxycytidine and different histone deacetylase inhibitors on human Ewing's sar- coma cells. Cancer Cell Int 2008;8:16.
62. Sonnemann J, Dreyer L, Hartwig M, et al. Histone deacetylase inhibitors induce cell death and enhance the apoptosis-induc- ing activity of TRAIL in Ewing's sarcoma cells. J Cancer Res Clin Oncol 2007;133:847-58.
63. Ginsberg JP, Goodman P, Leisenring W, et al. Long-term sur- vivors of childhood Ewing sarcoma: report from the childhood cancer survivor study. J Natl Cancer Inst 2010;102:1272-83.
64. Diller L, Chow EJ, Gurney JG, et al. Chronic disease in the Childhood Cancer Survivor Study cohort: a review of pub- lished findings. J Clin Oncol 2009;27:2339-55.
65. Geenen MM, Cardous-Ubbink MC, Kremer LC, et al. Medical assessment of adverse health outcomes in long-term survivors of childhood cancer. JAMA 2007;297:2705-15.
66. Oeffinger KC, Mertens AC, Sklar CA, et al. Chronic health con- ditions in adult survivors of childhood cancer. N Engl J Med 2006;355:1572-82.
67. Ranft A, Seidel C, Hoffmann C, et al. Quality of survivorship in a rare disease: clinicofunctional outcome and physical ac- tivity in an observational cohort study of 618 long-term survi- vors of Ewing sarcoma. J Clin Oncol 2017;35:1704-12.
68. Paulussen M, Ahrens S, Lehnert M, et al. Second malignancies after ewing tumor treatment in 690 patients from a cooperative German/Austrian/Dutch study. Ann Oncol 2001;12:1619-30.
69. Longhi A, Ferrari S, Tamburini A, et al. Late effects of chemo- therapy and radiotherapy in osteosarcoma and Ewing sarcoma patients: the Italian Sarcoma Group Experience (1983-2006).
Cancer 2012;118:5050-9.