Immunotherapy in Pediatric Solid Tumors
Jung Yoon Choi
Department of Pediatrics, Seoul National University College of Medicine, Seoul National University Cancer Research Institute, Seoul, Korea
The survival rates for pediatric patients with solid tumors have improved dramatically in recent decades. However, patients with metastatic disease at diagnosis or pro- gressive solid tumors still have a poor prognosis. Recently, immunotherapy has emer- ged as a novel therapeutic strategy in the pediatric population. Although not widely used in Korea, the anti-GD2 treatment in neuroblastoma is one of the successes of immunotherapy. In addition, many early phase clinical trials for monoclonal and bis- pecific antibodies, immune checkpoint inhibitors (ICIs), chimeric antigen receptor T cell therapy and cancer vaccines are ongoing. According to results reported so far, a majority of pediatric solid tumors showed limited response to ICIs, except Hodgkin lymphoma and hypermutant pediatric tumors. These results indicate that children and adolescents need to be applied different immunotherapeutic approaches from adults. The aim of this review is to understand the immunological environment and immunotherapeutic challenges in pediatric solid tumors.
pISSN 2233-5250 / eISSN 2233-4580 https://doi.org/10.15264/cpho.2020.27.1.22 Clin Pediatr Hematol Oncol 2020;27:22∼31
Received on April 4, 2020 Revised on April 17, 2020 Accepted on April 21, 2020
Corresponding Author: Jung Yoon Choi Department of Pediatrics, Seoul National University College of Medicine, Seoul National University Cancer Research Institute, 101 Daehak-ro, Jongno-gu, Seoul 03080, Korea Tel: +82-2-2072-0211 Fax: +82-2-743-3455 E-mail: [email protected] ORCID ID: orcid.org/0000-0001-8758-3074 Key Words: Immunotherapy, Pediatric, Solid tumor, Immune checkpoint inhibitor,
Chimeric antigen receptor T cell
Copyright ⓒ 2020 Korean Society of Pediatric Hematology-Oncology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/
Introduction
Pediatric solid tumors account for approximately 50%
of pediatric cancers and heterogeneous diseases [1]. The survival rates of patients with childhood cancers have improved dramatically over the decades and 80% of pa- tients achieve long-term remission after treatment. How- ever, treatment outcomes for pediatric patients with re- lapsed/refractory (R/R) solid tumors are still poor. Further- more, the most common treatment for pediatric cancer is non-specific and aggressive cytotoxic chemotherapy, which is related with excessive toxicities, long-term tox- icities and decreased quality of life. Novel therapeutic strategies are desperately needed to improve survival rates and to reduce toxicities. Cancer immunotherapy is
a type of treatment that works by leveraging the patient’s own immune system. Although immunotherapy has been proven to be effective in some pediatric hematologic malignancies and solid tumors, especially neuroblastoma (NBL), there is still a long way to go before it can be ef- fectively used to treat pediatric solid tumors. This review demonstrated the immunological background of why adult-like approaches have limitations in pediatric tumors.
It also summarizes various aspects of immunotherapy currently under investigation and ongoing efforts for pe- diatric solid tumors, focusing on monoclonal and bispe- cific antibodies, checkpoint inhibition, chimeric antigen receptor T cell (CAR-T) therapy, and cancer vaccine therapy.
Hurdles to the Application of Immunotherapy in Pediatric Cancers
1) Tumor microenvironment
There are some similarities and differences between pediatric and adult cancers from an immunological per- spective. Commonly, tumors are composed of infiltrating stromal cells and immune cells in addition to malignant cells. In pediatric cancer, tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) are two of the most prominent infiltrating cells [2]. On the other hand, the tumor active lymphocytes are insuffi- cient, hence the notion of the “cold tumor” [3]. TAMs and MDSCs originate from myeloid precursors in the bone marrow and are major contributors to the tumor micro- environment (TME) [4]. They have various roles that af- fect and regulate tumorigenesis, vasculogenesis, tumor cell growth, extracellular matrix deposition/remodeling and response to therapy [5,6]. MDSCs produce im- munosuppressive factors such as interleukin-10 (IL-10), prostaglandin E2, and TGF-β [7]. CD4+FOXP3+ regu- latory T cells (Tregs) and MDSCs inhibit cytotoxic T lym- phocyte (CTL) proliferation and activation [8]. The ch- ronic cytokine and chemokine production in TME create a contradictory inflamed and immune-suppressive envi- ronment in both adult and pediatric cancers [2]. T cells are exposed to chronic tumor antigen stimulation, mak- ing them exhausted and ineffective [9,10]. In order for immunotherapy to succeed, it is necessary to overcome this TME.
2) Paucity of neo-antigens
Most pediatric cancers originate from embryonic cells rather than from epithelial cells. Most pediatric solid tu- mors have either one or just a few driver mutations [11, 12]. Additionally, compared to adult cancer, pediatric cancer has fewer genotoxic environmental stressors and less cell division, so there are fewer background muta- tions [2]. Instead, 8-10% of childhood cancer patients have germline mutations that predispose to cancer [13].
Chromosome rearrangements that can activate proto-
oncogenes or inactivate tumor suppressor genes are more common in pediatric cancers [14]. Thus, pediatric tumors have significantly lower mutation burdens and express less neo-antigens, which cause decreased re- sponse to immunological targeting [15]. However, devel- oping new neo-antigens in pediatric cancer will provide more potential therapeutic targets [16]. When analyzing the mutation spectrum of NBL at diagnosis compared to that at relapse, it was discovered that the overall muta- tion burden was not high, but increased significantly at relapse [17]. These results show that mutation analysis in recurrent solid tumors can help identify treatment targets.
Immunotherapy in Pediatric Solid Tumors
Immunotherapy is functionally classified into two treat- ment categories: direct utilization of the immune system and modulation of the immune system.
1) Direct utilization of the immune system
(1) Monoclonal antibodies
Monoclonal antibody (mAb) therapy is utilized by tar- geting tumor-associated antigens (TAAs). Target cancer cells are removed through Fcγ receptor-mediated anti- body-dependent cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity [18,19]. The most impressive mAb in childhood cancer is the one targeting the GD2 diganglioside, which is universally expressed in NBL. In addition to NBL, GD2 is also expressed in mela- noma, soft tissue sarcoma, osteosarcoma, desmoplastic small round cell tumor (DSRCT), small cell lung cancer, and various other embryonal tumors [20].
Currently, high-risk NBL treatment is composed of in- duction chemotherapy and surgery, consolidation ther- apy with high-dose chemotherapy and autologous stem cell transplantation (ASCT) and radiotherapy, and post- consolidation therapy to treat minimal residual disease.
Dinutuximab (ch 14.18) and naxitamab (humanized 3F8), chimeric anti-GD2 antibodies have improved the survival of high-risk NBL patients. Yu AL, et al. showed that com- bination therapy with isotretinoin, ch14.18, granulocyte- macrophage colony stimulating factor (GM-CSF), and
IL-2 significantly improves event free survival and overall survival than isotretinoin alone, which was the standard treatment after ASCT at the time [21]. Patients enrolled in HR-NBL/SIOPEN trials who have completed induction chemotherapy, ASCT, and radiotherapy, were randomly assigned either to the dinutuximab-β group or the dinu- tuximab-β with IL-2 group. It was found that the addi- tion of IL-2 did not improve the survival rate, but in- creased the risk of side effects such as hypersensitivity, capillary leak, and fever [22]. We may guess that ADCC via natural killer (NK) cells is not the major contributor of anti-GD2 mAbs, given that IL-2 augments NK cell ac- tivity [23,24]. The common adverse effects of anti-GD2 Abs are neuropathic pain, fever, hypersensitivity and on- target off-tumor adverse effects.
Anti-GD2 Ab has been mainly effective in minimal re- sidual disease of NBL. In R/R NBL, a combination treat- ment of chemotherapy and anti-GD2 therapy showed the possibility of improving the survival rate. In a phase 2 study of patients with R/R NBL, patients were randomly assigned to either the temsiroimus or the dinutuximab group with irinotecan and temozolomide. The object re- sponse rate was significantly higher in the dinutuximab group [25].
There are other mAb targets such as insulin growth factor 1 receptor (IGF-1R), human epidermal growth fac- tor receptor 2 (HER2) oncogene and B7-H3, a surface im- munomodulatory glycoprotein B7 homolog 3 protein.
Enoblituzumab, B7-H3-targeting antibody is underway in phase 1 trials for adult refractory solid tumors (NCT 02628535).
(2) Bispecific antibodies
The bi-specific T-cell engagers (BiTE) comprise two binding domains; one is a single-chain variable fragment that binds to the tumor and the other fragment engages an activating receptor on T cells [26]. BiTE technology activates a T cell response by binding to CD3 on T cells.
Blinatumomab, a CD19/CD3 BiTE, showed efficacy and safety in R/R pediatric acute lymphoblastic leukemia (ALL) patients [27]. Although no approved BiTEs are yet available in pediatric solid tumors, research on the de- velopment of BiTE is ongoing. Previously, anti-GD2 mur-
ine 5F11-single-chain variable fragment (scFv) and hu- manized anti-CD3 OKT3-scFv BiTE were developed for patients with NBL [28]. Cheng M, et al substituted the 5F11-scFv into the higher affinity humanized 3F8-scFv, showing increased tumor cell killing in vitro [29]. A phase I/II clinical trial using anti-GD2 BiTE is ongoing (NCT 02173093). A multicenter phase 1 study of solitomab, a bispecific epithelial cell adhesion molecule (EpCAM)/
CD3 T-cell engager, was performed in adult refractory solid tumors. Dose limiting toxicities prevented dose es- calation to potentially therapeutic levels [30].
(3) Immune checkpoint inhibitors
Normal T-cell functions are controlled with T-cell re- ceptor (TCR), co-stimulatory and inhibitory signals. The immune system distinguishes cancer cells from normal cells and activates cytotoxic T cells to lead the apoptosis of the cancer cells. This process requires the regulation of the TCR and co-stimulatory and inhibitory signal, which is called the immune checkpoint [31]. However, cancers often avoid immune surveillance by immuno- suppressive tumor environments and immune tolerance [32]. The development of immune checkpoint inhibitors (ICIs) was a breakthrough in immune-oncology. Immunity checkpoint blockage removes inhibitory signals of T-cell activation that make T cells surmount regulatory mecha- nisms and fight against cancers [33,34].
The most frequently studied molecules in relation to immune checkpoint blockade are cytotoxic T-lympho- cyte-associated antigen-4 (CTLA-4) and programmed cell death receptor (PD-1) and its ligands PD-Ls. CTLA-4 is expressed on the surface of activated T cells and damp- ens T-cell activation. CTLA-4 (providing inhibitory co- stimulatory signal) and CD28 (providing positive co-stim- ulatory signal) share ligands (CD80 and CD86) with each other. However, because CTLA-4 has higher avidity and affinity than CD28, it becomes possible to competitively attenuate T-cell activation [33,35]. PD-1 mainly regulates effector T-cell activity within tissues and tumors and maintains peripheral tolerance [36]. PD-1 is more broad- ly expressed than CTLA-4. CLTA-4 is mainly present in T cells, but PD-1 is present in B cells and NK cells as well. CTLA-4 and PD-1 are highly expressed on Tregs.
Fig. 1. Schematic diagrams of the mechanisms of action of CTLA-4, PD-1 and PD-L1 blockade with or without chimeric antigen receptor T cell (CAR-T) therapy [36,58]. (A) T-cell activation, attenuation by normal inhibitory signals, and negative regulation using anti-CTLA-4, anti-PD-1 or anti-PD-L1 antibody therapy. (B) CAR-mediated activation, attenuation by tumor and checkpoint mediated inhibitory signals. Checkpoint receptor binding and checkpoint ligand blocking antibodies may synergize with chimeric antigen receptor therapy.
APC, antigen presenting cell; CTLA-4, cytotoxic T-lymphocyte-associated antigen-4; PD-1, programmed cell death receptor; PD-L1, programmed death-ligand 1; TAA, tumor-associated antigen; TCR, T-cell receptor.
Tregs may promote the proliferation by the ligand bind- ing [33]. PD-1 modulates T-cell activation through bind- ing with PD-L1 and PD-L2 [37,38]. PD-L1 is known to be upregulated in many cancers. PD-Ls inhibit cytokine pro- duction and anti-tumor lymphocytes in the TME. The molecular mechanisms of checkpoint inhibitors are shown in Fig. 1.
Immunohistochemistry is commonly used to check PD-L1 upregulation of the tumor cells. A study was con- ducted on PD-L1 expression in pediatric cancers, and 39 (9%) of the 451 evaluable tumors were identified to show PD-L1 expression in at least 1% of tumor cells. The high- est incidence of PD-L1 expressing tumors included Burkitt lymphoma (80%; 8/10 tumors), glioblastoma multiforme
(GBM, 36%; 5/14 tumors), and NBL (14%; 17/118 tumors) [39].
There are several studies that evaluate the safety and efficacy of ICIs in pediatric populations. In the first phase I/II pediatric trial of nivolumab, a PD-1 inhibitor was evaluated in patients aged 1-18 years with R/R solid tumors and lymphomas [40]. Of the total 85 patients, 63 (74.1%) had solid tumors (22 NBL, 13 osteosarcoma, 12 rhabdomyosarcoma, 11 Ewing sarcoma, 2 epithelioid sar- coma, 2 other sarcomas). The recommended phase 2 dose for pediatric patients was 3 mg/kg every 14 days of nivolumab. There were no objective responses in solid tumors, while 30% of Hodgkin lymphomas and 10% of non-Hodgkin lymphomas showed complete or partial re-
sponse (PR). Stable disease was the best response in 33%
of sarcoma and 50% of NBL patients. The common im- mune-related adverse events were hepatic toxicity and pleural or pericardial effusions. PD-L1-positivity was re- ported in 7-50% of solid tumors, 88% of non-Hodgkin lymphoma and 100% of Hodgkin lymphoma cohorts.
Another PD-1 inhibitor trial in a pediatric population was the pembrolizumab study for advanced melanoma;
or PD-L1-positive, advanced, R/R solid tumor, or lym- phoma [41]. A total of 863 patients were enrolled. The recommended phase 2 dose for pediatric patients was 2 mg/kg every 3 weeks of pembrolizumab. Hodgkin lym- phoma showed the highest objective response of 60%, while 5.8% (8 of 136) solid tumor and other lymphoma patients had PR. The diagnoses of patients who had PR were adrenocortical carcinoma, mesothelioma, malig- nant ganglioglioma, epithelioid sarcoma, lymphoepithe- lial carcinoma and malignant rhabdoid tumor. Except for melanoma, most tumor samples showed PD-L1 expres- sion. Nevertheless, the low response rate is presumed to be due to the low mutation burden of pediatric cancer, low expression of major histocompatibility complex (MHC), the immaturity of the immune system in young children, and so on [42-44].
① Biomarkers: Several biomarkers are being presented to predict the response to ICIs. These include the ex- pression of PD-L1 and PD-L2 on tumor cells, RNA ex- pression signatures, lymphocytic infiltrates, and tumor mutation burden (TMB) [45-49]. Researchers discovered a small group in colorectal cancer showing highly acti- vated Th1- and CTL-rich environments. Through the ge- netic analysis, it was discovered that the group has the loss of DNA mismatch repair activity, which is called mi- crosatellite instability (MSI) [50]. MSI-high cancer means there is a high number of mutations within microsatel- lites. MSI-high tumors in this environment may survive due to a dramatic overexpression of immune checkpoint- related proteins such as CTLA-4, PD-1 and lympho- cyte-activation gene 3 (LAG-3) [47]. They also have a higher Foxp3+ Tregs and PD-L1 expression. Colorectal cancer and other solid tumors with mismatch repair defi- ciency showed a greater response to pembrolizumab
[51]. Le DT, et al. prospectively evaluated the efficacy of PD-1 blockade in different subtypes of mismatch re- pair-deficient cancers. A total of 86 patients were en- rolled, the object response rate was 53%, complete re- sponse rate was 21%, and 11 patients were in CR two years after the treatment [52]. These results signify that patients with a mismatch repair deficiency, regardless of the tumor type, can get benefit from PD-1 blockade.
TMB, another important biomarker, is defined by the total number of mutations per coding area of a tumor genome. Hypermutation is defined as >10 mut/Mb [15].
MSI-high tumors have a high mutation burden, therefore potentially more neo-antigens. Palles C, et al identified specific heterozygous POLE or POLD1 germline variants in multiple-adenoma and/or early-onset colorectal can- cer cause replication errors and coupled repair of base pair-level mutations, thus contain high mutation burden without MSI [53]. Rizvi NA, et al. revealed that a high mu- tation burden in non-small cell lung cancer was related with improved survival when treated with pembroli- zumab [49]. Noskova H, et al. reported that TMB along with tumor progression was increased in pediatric solid tumor samples [54]. Clinical trials are ongoing to clarify the safety and effectiveness of PD-1 blockade in pedia- tric cancer patients with MSI-high and high tumor muta- tion burden tumors. Efforts to discover various bio- markers in pediatric cancers will be continued. Early- phase trials involving ICIs are presented in Table 1.
2) Modulation of the immune system
(1) Chimeric antigen receptor T cell therapy
CAR-T usually contain a single-chain variable frag- ment from a monoclonal antibody that can identify TAA;
a transmembrane hinge region; and a signaling domain such as CD28, CD3z, or 4-1BB [55]. MHC independent TAA recognition induces tumor-directed cytotoxicity. As CAR-T has demonstrated its effectiveness in pediatric ALL [56], CAR-T studies are now being conducted in pe- diatric solid tumors. Incorporation of synthetic immuno- therapy such as CAR-T is an option to overcome limi- tation of ICIs in pediatric cancers lack of tumor reactive effector lymphocytes [57].
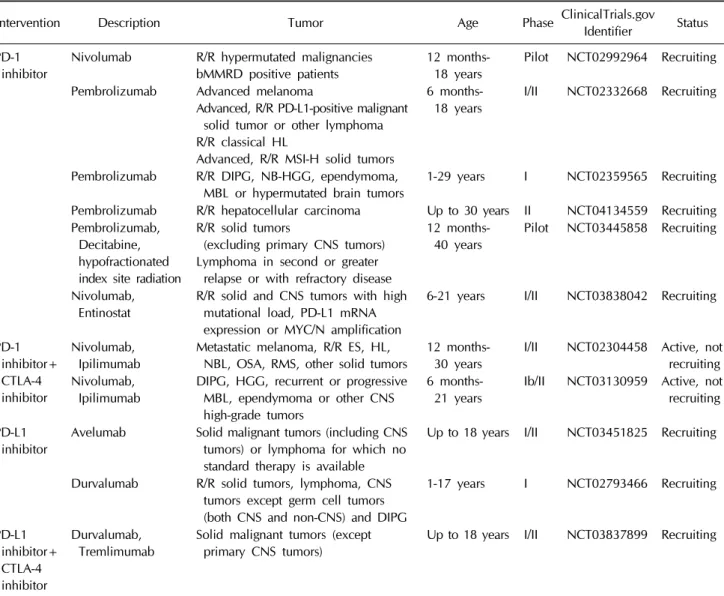
Table 1. Clinical trials involving immune checkpoint inhibitors monotherapy or combination therapy for pediatric solid tumors Intervention Description Tumor Age Phase ClinicalTrials.gov Identifier Status PD-1
inhibitor Nivolumab R/R hypermutated malignancies
bMMRD positive patients 12 months-
18 years Pilot NCT02992964 Recruiting Pembrolizumab Advanced melanoma
Advanced, R/R PD-L1-positive malignant solid tumor or other lymphoma R/R classical HL
Advanced, R/R MSI-H solid tumors
6 months-
18 years I/II NCT02332668 Recruiting
Pembrolizumab R/R DIPG, NB-HGG, ependymoma,
MBL or hypermutated brain tumors 1-29 years I NCT02359565 Recruiting Pembrolizumab R/R hepatocellular carcinoma Up to 30 years II NCT04134559 Recruiting Pembrolizumab,
Decitabine, hypofractionated index site radiation
R/R solid tumors
(excluding primary CNS tumors) Lymphoma in second or greater
relapse or with refractory disease
12 months-
40 years Pilot NCT03445858 Recruiting
Nivolumab,
Entinostat R/R solid and CNS tumors with high mutational load, PD-L1 mRNA expression or MYC/N amplification
6-21 years I/II NCT03838042 Recruiting
PD-1 inhibitor+
CTLA-4 inhibitor
Nivolumab,
Ipilimumab Metastatic melanoma, R/R ES, HL,
NBL, OSA, RMS, other solid tumors 12 months-
30 years I/II NCT02304458 Active, not recruiting Nivolumab,
Ipilimumab DIPG, HGG, recurrent or progressive MBL, ependymoma or other CNS high-grade tumors
6 months-
21 years Ib/II NCT03130959 Active, not recruiting PD-L1
inhibitor Avelumab Solid malignant tumors (including CNS tumors) or lymphoma for which no standard therapy is available
Up to 18 years I/II NCT03451825 Recruiting
Durvalumab R/R solid tumors, lymphoma, CNS tumors except germ cell tumors (both CNS and non-CNS) and DIPG
1-17 years I NCT02793466 Recruiting
PD-L1 inhibitor+
CTLA-4 inhibitor
Durvalumab,
Tremlimumab Solid malignant tumors (except
primary CNS tumors) Up to 18 years I/II NCT03837899 Recruiting
bMMRD, biallelic mismatch repair deficiency; CNS, central nervous system; DIPG, diffuse intrinsic pontine glioma; ES, Ewing sarcoma; HL, hodgkin lymphoma; MBL, medulloblastoma; MSI-H, microsatellite-instability-high; NBL, neuroblastoma; NB-HGG, non- brainstem high-grade gliomas; PD-L1, programmed death-ligand 1; RMS, rhabdomyosarcoma; R/R, recurrent/refractory.
① GD-2: Clinical trials of GD2-CAR-T in NBL were the first experiments where CAR-T cells have been tested in pediatric solid tumors [58]. In a phase 1 clinical trial of first-generation GD2-CAR-T in refractory NBL, 3 (27%) of 11 patients achieved complete remission. In addition, two of them have sustained remission for more than 5 years [59]. In 2017, a study regarding third-generation GD2-CAR-T incorporating the CD28 and the OX40 (CD134) costimulatory domains was reported. In this study, pa- tients received CAR-T alone, CAR-T plus cyclophos- phamide and fludarabine, or CAR-T plus cyclophos-
phamide, fludarabine and pembrolizumab. They demon- strated the third-generation GD2-CAR-T was safe and antitumor responses were modest. Lymphodepletion by cyclophosphamide and fludarabine helped to increase GD2-CAR-T expansion, but PD-1 inhibitor did not fur- ther increase GD2-CAR-T expansion [60].
② HER2: HER2 is a member of the epidermal growth factor receptor family with tyrosine kinase activity. HER2/
neu is highly expressed in medulloblastoma, osteosarco- ma, Wilms tumor and GBM [61]. Mineo JF, et al. reported that humanized anti-HER2/neu antibody have the ability
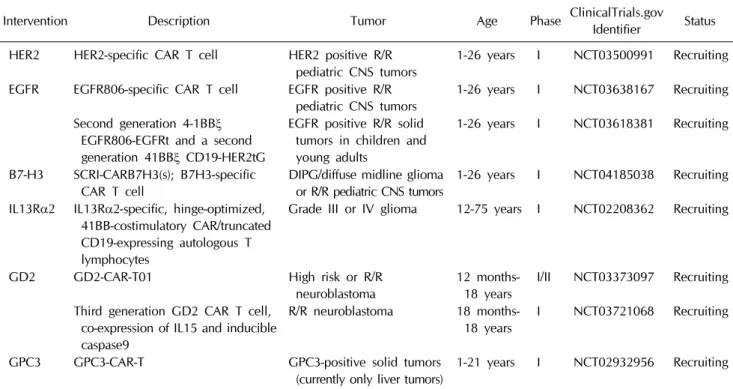
Table 2. Clinical trials involving chimeric antigen receptor T-cells for pediatric solid tumors
Intervention Description Tumor Age Phase ClinicalTrials.gov Identifier Status HER2 HER2-specific CAR T cell HER2 positive R/R
pediatric CNS tumors 1-26 years I NCT03500991 Recruiting EGFR EGFR806-specific CAR T cell EGFR positive R/R
pediatric CNS tumors 1-26 years I NCT03638167 Recruiting Second generation 4-1BBξ
EGFR806-EGFRt and a second generation 41BBξ CD19-HER2tG
EGFR positive R/R solid tumors in children and young adults
1-26 years I NCT03618381 Recruiting
B7-H3 SCRI-CARB7H3(s); B7H3-specific
CAR T cell DIPG/diffuse midline glioma
or R/R pediatric CNS tumors 1-26 years I NCT04185038 Recruiting IL13Rα2 IL13Rα2-specific, hinge-optimized,
41BB-costimulatory CAR/truncated CD19-expressing autologous T lymphocytes
Grade III or IV glioma 12-75 years I NCT02208362 Recruiting
GD2 GD2-CAR-T01 High risk or R/R
neuroblastoma 12 months-
18 years I/II NCT03373097 Recruiting Third generation GD2 CAR T cell,
co-expression of IL15 and inducible caspase9
R/R neuroblastoma 18 months-
18 years I NCT03721068 Recruiting GPC3 GPC3-CAR-T GPC3-positive solid tumors
(currently only liver tumors) 1-21 years I NCT02932956 Recruiting CAR, chimeric antigen receptor; CNS, central nervous system; DIPG, diffuse intrinsic pontine glioma; EGFR, epidermal growth factor receptor; EGFRt, truncated EGFR; GPC3, glypican-3; HER2, human epidermal growth factor receptor 2; HER2tG, truncated HER2 extracellular protein; IL13Rα2, interleukin-13 receptor alpha 2; R/R, recurrent/refractory.
to induce apoptosis and cellular-dependent cytotoxicity of HER2/neu-expressing GBM cell lines [62]. The first study evaluating HER2-CAR-T cells in pediatric and young adult sarcoma including osteosarcoma, primitive neuroectodermal tumor and DSRCT was reported in 2015. This phase I/II study showed persistence of HER2- CAR-T cells and tolerable safety. Three osteosarcoma and one DSRCT patients showed stable disease for 3 to 14 months [63]. HER2-CAR-modified autologous virus- specific T cells (HER2-CAR VSTs) were evaluated in adult and pediatric relapsed or progressive GBM patients. In this phase I study, HER2-CAR VSTs were safe and showed some clinical benefits. Maximal response was PR in a 17-year-old male with right thalamic GBM [64]. A pre- clinical model of HER2-CAR-T showed potential efficacy in osteosarcoma [65].
③ IL13Rα2: Interleukin-13 receptor alpha 2 (IL13Rα2), a glioma-associated antigen, is associated with a reduced rate of survival of glioma patients [66]. One adult re- lapsed GBM patient was enrolled in IL13Rα2-CAR-T cell
therapy. After CAR-T cell intracranial infusions, com- plete tumor regression was observed and the response continued for 7.5 months [67].
④ B7-H3: B7-H3 is one of the checkpoint molecule highly expressed on pediatric sarcomas and brain tumor cells. CAR-T targeting B7-H3 represented potent pre- clinical activity in various pediatric solid tumors and brain tumors [68].
Recently, it was reported that CAR-T can be exhausted by immunosuppressive cells, including MDSC cells or Treg cells [69,70]. Researches to overcome T-cell ex- haustion and restore immune memories are being in- vestigated. Combination therapy with ICIs and CAR-T may synergize the CAR-T activity (Fig. 1) [57]. Cytokine release syndrome, CAR-T-related encephalopathy syn- drome and sustained on-target off-tumor effects are also considered. Early-phase trials using second and third- generation CAR-T are ongoing (Table 2).
(2) Cancer vaccines
Dendritic cells (DCs) are the most powerful antigen presenting cells and are typically used as anti-cancer vaccines. They play a role as a bridge between the adap- tive and the innate immune responses and allow to gen- erate immune responses against tumors [71-73]. A phase I trial combining decitabine/dendritic cell vaccine for children aged 2.5-15 years with relapsed NBL, Ewing sar- coma, osteosarcoma and rhabdomyosarcoma was con- ducted. The DC vaccines were tolerated, and six of the 10 patients had a T cell response; one of the 10 patients had a complete tumor response [74,75].
Conclusion
Cancer treatment is entering a new era through im- munotherapy that stimulates the patient’s own immune system. As with adult cancer, innovative immunotherapy is also being tried in pediatric solid tumors. However, there are still very few patients with pediatric solid tu- mors who can benefit from immunotherapy. Pediatric solid tumor is less likely to respond to ICIs because it has a lower mutation burden than adult cancer. Several ICIs are in clinical trials for testing the potentiality for MSI- high and high mutation burden pediatric solid tumors.
Furthermore, to overcome the limitations of monotherapy, studies are also being conducted on combination therapy such as checkpoint inhibitors with CAR-T. Further re- search is needed on the TME of children and adolescents who may be different from that of adults but are not yet known. Immunotherapy in pediatric solid tumors is still in the early stages, but it is encouraging that various at- tempts are being made. The future treatment is expected to optimize the selection of therapeutic strategies, and ultimately improve patient outcome and quality of life.
Conflict of Interest Statement
The author has no conflict of interest to declare.
References
1. Park HJ, Moon EK, Yoon JY, et al. Incidence and survival of childhood cancer in Korea. Cancer Res Treat 2016;48:869- 82.
2. Hutzen B, Ghonime M, Lee J, et al. Immunotherapeutic chal- lenges for pediatric cancers. Mol Ther Oncolytics 2019;15:
38-48.
3. Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 2017;14:717-34.
4. Marayati R, Quinn CH, Beierle EA. Immunotherapy in pediat- ric solid tumors-A systematic review. Cancers (Basel) 2019;
11.
5. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-as- sociated macrophages (TAM) as major players of the can- cer-related inflammation. J Leukoc Biol 2009;86:1065-73.
6. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol 2010;22:231-7.
7. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenviron- ment. Trends Immunol 2016;37:208-20.
8. Spranger S, Spaapen RM, Zha Y, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor micro- environment is driven by CD8(+) T cells. Sci Transl Med 2013;
5:200ra116.
9. Snell LM, McGaha TL, Brooks DG. Type I interferon in chron- ic virus infection and cancer. Trends Immunol 2017;38:542- 57.
10. Wei F, Zhong S, Ma Z, et al. Strength of PD-1 signaling differ- entially affects T-cell effector functions. Proc Natl Acad Sci U S A 2013;110:E2480-9.
11. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr., Kinzler KW. Cancer genome landscapes. Science 2013;
339:1546-58.
12. Lawrence MS, Stojanov P, Polak P, et al. Mutational hetero- geneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214-8.
13. Zhang J, Walsh MF, Wu G, et al. Germline mutations in pre- disposition genes in pediatric cancer. N Engl J Med 2015;373:
2336-46.
14. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad- Massade L. Relevance of fusion genes in pediatric cancers:
Toward precision medicine. Mol Ther Nucleic Acids 2017;6:
315-26.
15. Campbell BB, Light N, Fabrizio D, et al. Comprehensive analysis of hypermutation in human cancer. Cell 2017;171:1042-56 e10.
16. Cheever MA, Allison JP, Ferris AS, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res 2009;15:5323-37.
17. Schramm A, Koster J, Assenov Y, et al. Mutational dynamics between primary and relapse neuroblastomas. Nat Genet
2015;47:872-7.
18. Wilson NS, Yang B, Yang A, et al. An Fcγ receptor-de- pendent mechanism drives antibody-mediated target-re- ceptor signaling in cancer cells. Cancer Cell 2011;19:101-13.
19. Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun 2012;12:14.
20. Dobrenkov K, Cheung NK. GD2-targeted immunotherapy and radioimmunotherapy. Semin Oncol 2014;41:589-612.
21. Yu AL, Gilman AL, Ozkaynak MF, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma.
N Engl J Med 2010;363:1324-34.
22. Ladenstein R, Potschger U, Valteau-Couanet D, et al. Inter- leukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/
SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol 2018;19:1617-29.
23. Park JA, Cheung NV. Targets and antibody formats for immu- notherapy of neuroblastoma. J Clin Oncol 2020:JCO1901410.
24. Henney CS, Kuribayashi K, Kern DE, Gillis S. Interleukin-2 augments natural killer cell activity. Nature 1981;291:335-8.
25. Mody R, Naranjo A, Van Ryn C, et al. Irinotecan-temozolo- mide with temsirolimus or dinutuximab in children with re- fractory or relapsed neuroblastoma (COG ANBL1221): an open- label, randomised, phase 2 trial. Lancet Oncol 2017;18:946- 57.
26. Wu Z, Cheung NV. T cell engaging bispecific antibody (T- BsAb): From technology to therapeutics. Pharmacol Ther 2018;
182:161-75.
27. von Stackelberg A, Locatelli F, Zugmaier G, et al. Phase I/
phase II study of blinatumomab in pediatric patients with re- lapsed/refractory acute lymphoblastic leukemia. J Clin Oncol 2016;34:4381-9.
28. Cheng M, Ahmed M, Xu H, Cheung NK. Structural design of disialoganglioside GD2 and CD3-bispecific antibodies to re- direct T cells for tumor therapy. Int J Cancer 2015;136:476- 86.
29. Cheng M, Santich BH, Xu H, Ahmed M, Huse M, Cheung NK.
Successful engineering of a highly potent single-chain varia- ble-fragment (scFv) bispecific antibody to target disialog- anglioside (GD2) positive tumors. Oncoimmunology 2016;5:
e1168557.
30. Kebenko M, Goebeler ME, Wolf M, et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/
CD3 T-cell engager (BiTE(R)) antibody construct, in patients with refractory solid tumors. Oncoimmunology 2018;7:e1450710.
31. Ring EK, Markert JM, Gillespie GY, Friedman GK. Checkpoint proteins in pediatric brain and extracranial solid tumors:
Opportunities for immunotherapy. Clin Cancer Res 2017;23:
342-50.
32. Vinay DS, Ryan EP, Pawelec G, et al. Immune evasion in can- cer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol 2015;35 Suppl:S185-S98.
33. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252-64.
34. Sharma P, Allison JP. The future of immune checkpoint ther- apy. Science 2015;348:56-61.
35. Schneider H, Downey J, Smith A, et al. Reversal of the TCR
stop signal by CTLA-4. Science 2006;313:1972-5.
36. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018;8:
1069-86.
37. Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001;2:261-8.
38. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000;192:1027-34.
39. Majzner RG, Simon JS, Grosso JF, et al. Assessment of pro- grammed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer 2017;123:
3807-15.
40. Davis KL, Fox E, Merchant MS, et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): a multicentre, open-label, single- arm, phase 1-2 trial. Lancet Oncol 2020;21:541-50.
41. Geoerger B, Kang HJ, Yalon-Oren M, et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1- positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): interim analysis of an open-la- bel, single-arm, phase 1-2 trial. Lancet Oncol 2020;21:121- 33.
42. Grobner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;
555:321-7.
43. Haworth KB, Leddon JL, Chen CY, Horwitz EM, Mackall CL, Cripe TP. Going back to class I: MHC and immunotherapies for childhood cancer. Pediatr Blood Cancer 2015;62:571-6.
44. Simon AK, Hollander GA, McMichael A. Evolution of the im- mune system in humans from infancy to old age. Proc Biol Sci 2015;282:20143085.
45. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443-54.
46. Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune mi- croenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014;20:5064-74.
47. Llosa NJ, Cruise M, Tam A, et al. The vigorous immune mi- croenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 2015;5:43-51.
48. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in can- cer patients. Nature 2014;515:563-7.
49. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology.
Mutational landscape determines sensitivity to PD-1 block- ade in non-small cell lung cancer. Science 2015;348:124-8.
50. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010;138:2073-87 e3.
51. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509-20.
52. Le DT, Durham JN, Smith KN, et al. Mismatch repair defi- ciency predicts response of solid tumors to PD-1 blockade.
Science 2017;357:409-13.
53. Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 pre- dispose to colorectal adenomas and carcinomas. Nat Genet 2013;45:136-44.
54. Noskova H, Kyr M, Pal K, et al. Assessment of tumor muta- tional burden in pediatric tumors by real-life whole-exome sequencing and in silico simulation of targeted gene panels:
How the choice of method could affect the clinical decision?
Cancers (Basel) 2020;12.
55. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor- expressing T cells. Immunol Rev 2014;257:107-26.
56. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leuke- mia. N Engl J Med 2018;378:439-48.
57. Pearson ADJ, Scobie N, Norga K, et al. ACCELERATE and European Medicine Agency Paediatric Strategy Forum for medicinal product development for mature B-cell malig- nancies in children. Eur J Cancer 2019;110:74-85.
58. Schultz LM, Majzner R, Davis KL, Mackall C. New develop- ments in immunotherapy for pediatric solid tumors. Curr Opin Pediatr 2018;30:30-9.
59. Louis CU, Savoldo B, Dotti G, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011;118:6050-6.
60. Heczey A, Louis CU, Savoldo B, et al. CAR T cells administer- ed in combination with lymphodepletion and PD-1 inhibi- tion to patients with neuroblastoma. Mol Ther 2017;25:2214- 24.
61. Orentas RJ, Lee DW, Mackall C. Immunotherapy targets in pediatric cancer. Front Oncol 2012;2:3.
62. Mineo JF, Bordron A, Quintin-Roue I, et al. Recombinant humanised anti-HER2/neu antibody (Herceptin) induces cel- lular death of glioblastomas. Br J Cancer 2004;91:1195-9.
63. Ahmed N, Brawley VS, Hegde M, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2- positive sarcoma. J Clin Oncol 2015;33:1688-96.
64. Ahmed N, Brawley V, Hegde M, et al. HER2-specific chimeric
antigen receptor-modified virus-specific T cells for progres- sive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol 2017;3:1094-101.
65. Rainusso N, Brawley VS, Ghazi A, et al. Immunotherapy tar- geting HER2 with genetically modified T cells eliminates tu- mor-initiating cells in osteosarcoma. Cancer Gene Ther 2012;
19:212-7.
66. Brown CE, Warden CD, Starr R, et al. Glioma IL13Ralpha2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS One 2013;8:e77769.
67. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblas- toma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016;375:2561-9.
68. Majzner RG, Theruvath JL, Nellan A, et al. CAR T cells target- ing B7-H3, a pan-cancer antigen, demonstrate potent pre- clinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res 2019;25:2560-74.
69. Caruana I, Savoldo B, Hoyos V, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med 2015;21:524-9.
70. Long AH, Haso WM, Shern JF, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015;21:581-90.
71. Elster JD, Krishnadas DK, Lucas KG. Dendritic cell vaccines:
A review of recent developments and their potential pediatric application. Hum Vaccin Immunother 2016;12:2232-9.
72. Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nat Med 2004;10:475-80.
73. Lee HJ, Kim SK, Cho D, Lee JJ. Cellular immunotherapy as a beacon of hope for hematological malignancies. Blood Res 2015;50:126-8.
74. Krishnadas DK, Shusterman S, Bai F, et al. A phase I trial combining decitabine/dendritic cell vaccine targeting MAGE- A1, MAGE-A3 and NY-ESO-1 for children with relapsed or therapy-refractory neuroblastoma and sarcoma. Cancer Immunol Immunother 2015;64:1251-60.
75. Krishnadas DK, Shapiro T, Lucas K. Complete remission fol- lowing decitabine/dendritic cell vaccine for relapsed neuro- blastoma. Pediatrics 2013;131:e336-41.
![Fig. 1. Schematic diagrams of the mechanisms of action of CTLA-4, PD-1 and PD-L1 blockade with or without chimeric antigen receptor T cell (CAR-T) therapy [36,58]](https://thumb-ap.123doks.com/thumbv2/123dokinfo/5446908.236767/4.892.78.810.506.1013/schematic-diagrams-mechanisms-blockade-chimeric-antigen-receptor-therapy.webp)