저작자표시 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. l 이차적 저작물을 작성할 수 있습니다. l 이 저작물을 영리 목적으로 이용할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다.
Mitochondrial respiratory defects induces
claudin 1 expression via ROS-mediated
HSF1 activation
by
Jong-Hyuk Lee
Major in Molecular Medicine
Department of Biomedical Sciences
The Graduate School, Ajou University
Mitochondrial respiratory defects induces
claudin 1 expression via ROS-mediated
HSF1 activation
by
Jong-Hyuk Lee
A Dissertation Submitted to The Graduate School of
Ajou University in Partial Fulfillment of the Requirements
for the Degree of
Master of Biomedical Sciences
Supervised by
Gyesoon Yoon, Ph.D.
Major in Molecular Medicine
Department of Biomedical Sciences
The Graduate School, Ajou University
This certifies that the dissertation
of Jong-Hyuk Lee is approved.
SUPERVISORY COMMITTEE
Jae-Ho Lee
Gyesoon Yoon
You-sun Kim
The Graduate School, Ajou University
June, 20th, 2014
i
-ABSTRACT-
Mitochondrial respiratory defects induces claudin 1 expression
via ROS-mediated HSF1 activation
Jong-Hyuk Lee
Department of Biomedical Sciences The Graduate School, Ajou University (Supervised by Professor Gyesoon Yoon )
Claudin-1 (Cln-1) are tight junction components that have recently been reported to be involved in cancer metastasis. Interestingly, we found that SNU human hepatoma cells with mitochondrial dysfunction have high level of Cln-1 expression. In those HCC cells, Cln-1 played a key role in regulating invasiveness. Cln-1 expression was also observed in human hepatocellular carcinoma tissues. In this study, we aimed to elucidate how mitochondrial dysfunction is linked with Cln-1 induction. So, we screened whether any specific mitochondrial respiratory defect induces Cln-1 induction. Among diverse respiratory inhibitors, complex I inhibition by rotenone most effectively induced Cln-1 expression at transcriptional level, accompanied by increase in mitochondrial ROS production. Treatment of H2O2 directly induced Cln-1 expression, implying that Cln-1 induction was mediated by
mitochondrial ROS. Interestingly, Cln-1 promoter region (-2700bp ~ +300bp) contained transcription factor binding site Heat Shock Factor1 (HSF1), which are known to be regulated by intracellular redox status. Subsequently, we found, that HSF-1 binds to the Cln-1 promoter. HSFCln-1 knockdown using siRNA decreased Cln-Cln-1 expression and invasive activity in SNU449. These results suggest that Cln-1 induced by HSF1 through respiratory defect mediated ROS in hepatoma cells.
ii
TABLE OF CONTENS
ABSTRACT ··· ⅰ TABLE OF CONTENS ··· ⅱ LIST OF FIGURES ··· ⅲ . Ⅰ INTRODUCTION ··· 1 . Ⅱ MATERIALS AND METHODS ··· 5A. Cell cultures, cell growth rate and tumor sample ··· 5
B. Endogenous cellular oxygen consumption rate ··· 5
C. Cell invasion assay ··· 6
D. Western blot analysis ··· 6
E. Reverse transcription-polymerase chain reaction (RT-PCR) ··· 7
F. Estimation of intracellular and mitochondrial ROS level ··· 7
G. Construction of HSF1 cDNA plasmids and transfection of cDNA plasmids and siRNAs ··· 8
H. ChIP assay ··· 8
I. Total genomic DNA isolation ··· 9
J. Promoter assay ··· 9 . Ⅲ RESULTS ··· 10 . Ⅳ DISCUSSION ··· 29 Ⅴ.CONCLUSION ··· 32 REFERENCES ··· 33 국문요약 ··· 39
iii
LIST OF FIGURES
Fig. 1. Cln-1 is highly expressed in invasive SNU hepatoma cells with
mitochondrial dysfunction. ··· 11
Fig. 2. Cln-1 is associated with mitochondrial dysfunction of hepatoma cell.
··· 12 Fig. 3. Respiratory dysfunction induces Cln-1 expression at transcriptional level.
··· 14
Fig. 4. Respiratory dysfunction by complex I inhibition induces effectively
Cln-1 expression at transcriptional level. ··· 15
Fig. 5. Mitochondrial ROS induced by complex I defect is the mediator to induce Cln-1 expression. ··· 17 Fig. 6. Intracellular ROS is the mediator to induce Cln-1 expression ··· 18
Fig. 7. Analysis of transcription factor binding elements of Cln-1 promoter
sequence. ··· 20 Fig. 8. Respiratory dysfunction by complex I inhibition induces HSF
phosphorylation (activation). ··· 21
Fig. 9. ROS-mediated HSF phosphorylation (activation) is involved in Cln-1
expression. ··· 22
Fig. 10. HSF1 phosphorylation was involved Cln-1 expression through ROS.
iv
Fig. 11. Mitochondrial ROS enhance HSF1 binding to the promoter region of
Cln-1 gene and enhances its activity. ··· 25
Fig. 12. HSF1 phosphorylation was critically involved in hepatoma cell invasion
activity through ROS-mediated Cln-1 expression. ··· 27 Fig. 13. Schematic model for mechanism of Cln-1. ··· 28
1
INTRODUCTION
Aerobically growing eukaryotic cells produce ATP through mitochondrial oxidative phosphorylation (OXPHOS) machinery, highlighted as the power plant, whereas the cells under hypoxic condition depend on glycolytic ATP generation. It is due to the fact that glycolysis is inhibited by the presence of oxygen (Pasteur effect), which allows continuing mitochondrial respiration (Racker, 1974). However, many invasive cancer cells have altered glucose metabolism. They continuously convert glucose to lactate even in the presence of oxygen, known as aerobic glycolysis or the ‘Warburg effect,’ making their growth rely on glycolytic ATP production (Warburg, 1956; Kroemer and Pouyssegur, 2008; Vander Heiden et al., 2009). To maintain effectively aerobic glycolysis, cancer cells have increased glucose uptake activity through upregulated glucose transporters, such as GLUT1 and GLUT3 (Amann et al., 2009; Krzeslak et al., 2012). In addition, expressions of glycolysis-related genes are increased by several oncogenes including Myc, Ras, p53 and Src or hypoxic transducer such as HIF1α (Dang and Semenza, 1999; Gogvadze et al., 2008; Cairns et al., 2011). Based on this tumoral metabolic characteristic, positron emission tomography (PET) is an imaging technique to monitor cellular capacity to uptake [18F] (Vander Heiden et al.,
2009).
Mitochondrial OXPHOS provide an efficient route for ATP synthesis, 36~38 ATP per glucoses by consuming most respired cellular oxygen, which account for 98% of whole cell oxygen consumption (Kowaltowski et al., 2009; Murphy, 2009). However, it is unclear why this efficient mitochondrial OXPHOS is not utilized in the cancer cell in the presence of oxygen. One plausible explanation is based on the fact that mitochondrial dysfunction has often been found to be associated with the development of human cancers (Kwong et al., 2007; Wu et al., 2010), implying that aerobic glycolysis is also linked with mitochondrial defects. Recent studies explain that it may be due to functional inability by hypoxia following fast tumor growth (Chang et al., 2011) or genetic alteration for mitochondrial protein expressions by oncogene or tumor suppressor, such as Ras and p53 (Dang and Semenza, 1999). This is further supported by the findings that mutations or deletions in mitochondrial DNA (mtDNA) (Wu et al., 2010) and in nuclear gene coding electron transport chain proteins (Kwong et al., 2007) have often been detected in diverse cancers,
2
such as hepatocellular carcinoma (Tamori et al., 2004), colorectal cancer (Lievre et al., 2005), lung cancer (Matsuyama et al., 2003), breast cancer (Tseng et al., 2006), renal cell carcinoma (Simonnet et al., 2002) and gastric cancer (Wu et al., 2005; Hung et al., 2010).
Although the contribution of mitochondrial gene mutation and consequent respiratory defects to cancer promotion has been confirmed (Petros et al., 2005; Shidara et al., 2005), it is unclear whether mitochondrial impairment is causative or correlative factors and how such defects are involved in cancer development. Interestingly, accumulating studies suggest that cancer cells harboring mitochondrial dysfunction display more invasive and metastatic properties (Chang et al., 2009; Hung et al., 2012; He et al., 2013; Ma et al., 2013). For example, stable down-modulation of GRIM-19 or NDUFS3 decreased complex I activity and reactive oxygen species (ROS) production, resulting in enhanced cell adhesion, migration, invasion, and spheroid formation (He et al., 2013). Mitochondrial dysfunction-mediated ROS increase is involved in enhanced cell migration activity of SC-M1 human gastric cancer cells (Hung et al., 2010). These observations lead to the hypothesis that cancer cells with mitochondrial dysfunction harbor augmented intrinsic reactive oxygen species (ROS) level partly derived from inefficient electron transfer process in the defective respiratory chains (Pelicano et al., 2006; Moreno-Sanchez et al., 2007) and the increased ROS may in turn activate certain redox-sensitive signal pathways and, thereby, stimulate cellular proliferation, cell migration and invasion activities, eventually contributing to carcinogenesis (Ishikawa et al., 2008; Bauer, 2012). However, the underlying molecular mechanisms by which mitochondrial ROS activate cancer cell invasion activity are not fully understood.
Claudins are a family of proteins that are the critical components of the tight junctions, which controls paracellular permeability of cells and the flow of molecules between cells. Claudins have four transmembrane domains with two extracellular loops (EL1 and EL2) and their N- and C-terminal ends are located in the cytoplasm (Escudero-Esparza et al., 2011). The C-terminal domain of claudins serves as a binding site of diverse PDZ domain proteins, ZO-1, ZO-2, ZO-3, multi- PDZ domain protein 1, that are involved in intracellular signaling (Itoh et al., 1999; Hamazaki et al., 2002). Unexpectedly, aberrant up-regulation of claudins has recently been reported in diverse human cancers, including hepatocellular carcinoma (HCC) and primary colon carcinoma (Michl et al., 2003; Rangel et al., 2003). Claudin-1 was
3
both necessary and sufficient to induce invasive behavior in human liver cells via activation of c-Abl-PKC signaling pathway (Yoon et al., 2010). Direct evidences for the roles of claudins in cancer cell invasion activities have also been described. Claudin-1 (Cln-1) overexpression increased invasiveness of oral carcinoma cells (Dos Reis et al., 2008) and transient transfection of melanoma cells with Cln-1 increased metalloproteinase 2 (MMP-2) secretion and activation, contributing to melanoma cell invasion (Leotlela et al., 2007). More promising evidence was reported that claudin-1, -2, -3, and -5 recruit and promote the activation of pro–MMP-2 (Miyamori et al., 2001; Ichiyasu et al., 2004), well supporting the involvement of Cln-1 in cancer cell invasion and metastasis. On the other hand, hepatocellular carcinoma with mitochondrial dysfunction display increased expression of Cln-1 and high invasion activity, implying the close link between mitochondrial defects and Cln-1-mediated tumor invasion. However, it is not clearly understood how Cln-1 expression is increased in cancer and linked with the metabolic hallmark of cancer, mitochondrial dysfunction.
HSF1 is a multifaceted transcription factor that controls the cellular response to disrupted protein homeostasis. To protect and maintain protein homeostasis under diverse proteotoxic stresses including heat stress, HSF1 activates transcriptions of classic heatshock proteins (HSPs), such as HSP27, HSP70, and HSP90, that are major cellular protein chaperones, allowing repair and refolding of damaged proteins (Santagata et al., 2011). HSF1 is constitutively expressed in most tissues and its activation is mostly regulated by post-translational modification, e.g. phosphorylation at S326 (Guettouche et al., 2005). Active HSF1 trimerizes and binds HSF sequence-binding elements (HSEs), leading upregulation of target genes (Kroeger and Morimoto, 1994). Recently, enhanced activation of HSF1 was reported to be associated with a poor prognosis in several cancer types (Santagata et al., 2011; Mendillo et al., 2012; Ciocca et al., 2013). Elevated level of nuclear HSF1 was
observed in ∼80% of in situ and invasive breast carcinomas (Santagata et al., 2011). High
expression of HSF1 was also found in invasive carcinoma and associated with high histologic grade, larger tumor size, and nodal involvement at diagnosis (Santagata et al., 2011). HSF1 promoted HCC cell migration and invasion in vitro and in vivo by activating the expression and phosphorylation of heat shock protein 27 (Fang et al., 2012). More
4
interestingly, HSF1 can be activated by redox regulation (Jacquier-Sarlin and Polla, 1996; Christians et al., 2002; Yan et al., 2002). Collectively, these observations suggest that HSF1 may play a potential role to connect between mitochondrial ROS-mediated oxidative stress and cancer invasion activity.
5
MATERIALS AND METHODS
A. Cell cultures, cell growth rate and tumor samples.Human hepatoma cells (SNU-354, SNU-387, SNU-423, and SNU-449) were purchased from Korean Cell Line Bank (Seoul, Korea) and were cultured in GIBCO® RPMI1640
medium (Invitrogen, Carlsbad, CA) supplemented with 10% GIBCO® fetal bovine serum
(FBS) (Invitrogen) and GIBCO® antibiotics (Invitrogen) at 37°C in a humidified incubator
with 5% CO2. Chang cell was obtained from American Tissue Culture Collections (ATCC,
Rockville, MD) and Chang cell clone, denoted as Chang-L, which has higher hepatic characteristics (albumin production and liver-specific carbamoyl-phosphate synthase-1 expression) were isolated by single cell dilution and expansion, and used for this study (Kim et al., 2011). Chang-L clones were cultured in GIBCO® Dulbecco’s modified Eagle’s
medium (DMEM) (Invitrogen) supplemented with 10% FBS. Cell growth rates were monitored by counting the trypan blue-negative viable cells.
HCC tumor samples and surrounding control tissues were obtained from 6 patients with hepatocellular carcinoma during the period of 2003 to 2005 at Ajou University Hospital after surgical resection with informed consent through Ajou Institutional Review Board. No patient in the current study received chemotherapy or radiation therapy before the surgery.
B. Endogenous cellular oxygen consumption rate.
Endogenous cellular oxygen consumption rate (OCR) was measured by using Mitocell equipped with Clark oxygen electrode (782 Oxygen Meter, Strathkelvin Instrument, Glasgow, UK) or XF-24 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA) according to the protocol provided. Measurement of endogenous cellular respiration with Mitocell was performed as described previously with slight modification (Villani and Attardi, 2001; Yoon et al., 2003). Briefly, at indicated times, cells were washed with TD buffer (0.137 M NaCl, 5 mM KCl, 0.7 mM Na2HPO4, 25 mM Tris–HCl, pH 7.4), and
6
cells were transferred to the chamber of Mitocell equipped with Clark oxygen electrode (782
Oxygen Meter, Strathkelvin Instrument) for measurement of endogenous cellular O2
consumption rate.
OCR was also measured in situ with cultured cells using XF-24 Extracellular Flux Analyzer (Seahorse Bioscience) according to the protocol provided. Briefly, cells were seeded on XF24 cell culture microplates (Seahorse Bioscience) at a density of 10,000 cells per well and preincubated with XF assay medium (Seahorse Bioscience) containing 1 mM pyruvate and 5 mM glucose. Its mitochondrial specificity was confirmed by adding 5 mM KCN.
C. Cell invasion assay.
Assays were basically performed with Transwell® Permeable Supports (Corning, Acton, MA, USA) according to the manufacturer's instructions. Briefly, cells (2×104) pre-starved
with serumfree RPMI for 16 h were placed into the upper chamber with 0.2 ml of serum-free RPMI. RPMI (0.8 ml) supplemented with 10% fetal bovine serum was placed in the lower chamber as a chemo-attractant. Uncoated porous filters (8-μm pore size) were pre-coated with 7% Growth Factor Reduced BD Matrigel™ Matrix (Becton Dickinson Labware, Franklin Lakes, NJ, USA) for the cell invasion assay. The cells invaded into the lower surface were fixed by 100% methanol for 1 min, stained with Hematoxylin Solution (Sigma-Aldrich, Saint Louis, MI) and Eosin Y Solution (Sigma-Aldrich), and counted. All experiments were performed in independent triplicate experiments.
D. Western blot analysis.
Western blotting was performed using standard procedures. Antibodies against Claudin-1
(717800) were purchased from Invitrogen, Inc (Carlsbad, CA). Antibodies against HSF1 (4356S), β-actin (sc-1616) and α-tubulin (sc-5286) were obtained from Cell Signaling Technology, Inc (Danvers, MA) and Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA)
7
Antibodies against for NDUFA9 of complex I (A21344), flavoprotein (A11142) of complex II, UQCRC2 of complex III (A11143), and ATP5A1 of complex V (A21350) were from Molecular Probes Corp. (Eugene, OR) and labeled as Comp I, II, III, IV, and V, respectively, in the figures.
E. Reverse transcription-polymerase chain reaction (RT-PCR).
Total RNA was isolated using Trizol (Invitrogen) and cDNA was prepared using avian myeloblastosis virus (AMV) reverse transcriptase (Promega, Madison, WI). PCR was performed with 25-30 cycles of the reaction involving 95°C for 30 seconds, 53-58°C for 30 seconds, and 72°C for 70-90 seconds. The PCR primer sets were produced by Bioneer
(Seoul, Korea) to detect all variants ofClaudin-1 (5’-GAGCGAGTCATGGCCAACGCG-3’
and 5’-GCCTCTGTGTCACACGTAGTC-3’) and β-actin (5'-CCTTCCTGGGCATGGAGT CCTGT-3’ and 5'-GGAGCAATGATCTTGATCTTC-3’).
F. Measurement of intracellular and mitochondrial ROS level.
To determine intracellular and mitochondrial ROS levels, dichlorofluorescin diacetate
(DCFH-DA) (Molecular probe, Eugene, OR) and mitochondrial specific MitoSOX®
(Invitrogen) fluorogenic probes were used, respectively (Yu and Kim, 2011). Briefly, cells
were incubated in media containing DCFH-DA (20 μM) and MitoSOX® (25 μM) for 20 min
at 37oC. Stained cells were washed and resuspended in PBS, and analyzed by flow cytometry
(FACS Vantage, Becton Dickinson Corp.). Mean values of arbitrary fluorescence units of 10,000 cells were used and expressed as percentage of negative control.
8
G. Construction of HSF1 cDNA plasmids and transfection of cDNA plasmids and siRNAs.
To generate a cDNA plasmid, pcDNA-HSF1-HA, conventional cloning procedures were applied. Briefly, pcDNA-HSF1-HA plasmid was constructed by TA cloning into pGEMT-easy (Promega) with HSF1 cDNA fragment amplified by PCR using total Chang cell cDNAs and the primer set (5’-AGAATTCATGGATCTGCCCG-3’ and 5’-TGAGCTCGGAGACAG TGGG-3’). The HSF1 cDNA was inserted into EcoRI and XhoI sites of the pcDNA3-HA vector previously constructed (Seo et al., 2008).
To introduce plasmids and small interfering RNAs (siRNAs) into cells, cells were transfected with the plasmids and siRNA duplexes using FuGENE HD (Promega) and OligofectamineTM Reagent (Invitrogen), respectively, according to the manufacturer’s
instructions. HSF1 siRNAs (#1, 5'- ACUGUAGAUUGCUUCUGUA-3' and 5'- UACAGA AGCAAUCUACAGU-3') (#2, 5'- GAACUAAAGCCAAGGGUAU-3' and 5'- AUACCCU UGGCUUUAGUUC -3') were obtained from Bioneer Co. Negative control siRNAs (5’-CC UACGCCACCAAUUUCGU-3’ and 5’-ACGAAAUUGGUGGCGUAGG-3’) were obtained from Bioneer.
H. ChIP assay.
A chromatin immunoprecipitation (ChIP) assay was performed with a ChIP kit (Upstate Biotechnology, Lake Placid. NY), according to the manufacturer’s instructions, using antibodies against HSF1 (4356S) or control IgG (Santa Cruz). The precipitated DNA was subjected to PCR amplification with specific primers for the Claudin-1 promoter region containing HSF1-binding sites (Song et al., 2013). The following primers were used for
PCR:HSF-1 (5’- TTTGTCTGAGGGTACATAGCAGA-3’ and 5’-GGCAGCACTGAGAC
9
I. Total genomic DNA isolation.
Total genomic DNA was isolated as described previously with slight modification (Yoon et al., 2006). Briefly, cell lysates were incubated at 37 °C for 1 h with 0.1 mg/ml RNase A, and then at 55 °C for 3 h with 0.1 mg/ml Proteinase K and 1 % SDS. Phenol/chloroform/iso amyl alchol were treated for several times. Genomic DNA (gDNA) was precipitated by addition of a 2.5 volume of absolute ethanol and 1/10 volume of 3 M sodium acetate (pH 5.2), pelleted by centrifugation at 13,000 rpm for 20 min, and dissolved in 100 μl of TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0).
J. Promoter assay.
For transfection of reporter plasmids, Chang cells were plated on 6-well plates at a density
of 1×104 cells/well. Plasmid DNA was mixed with FuGENE HD transfection reagent
(Promega) and transfected into the cells following the manufacturer’s protocol. After 48 hours of transfection, the cells were washed twice with PBS and then lysed in reporter lysis buffer (Promega). Luciferase activity was measured with a Dual luciferase reporter assay system (Promega) according to the manufacturer’s instructions. Luciferase activity was measured in triplicate, averaged, and relative promoter activity was computed by normalizing the Firefly luciferase activity against that of the Renilla luciferase (Song et al., 2013).
10
RESULTS
Cln-1 is upregulated in hepatoma cell with mitochondrial dysfunction and high invasion activity.
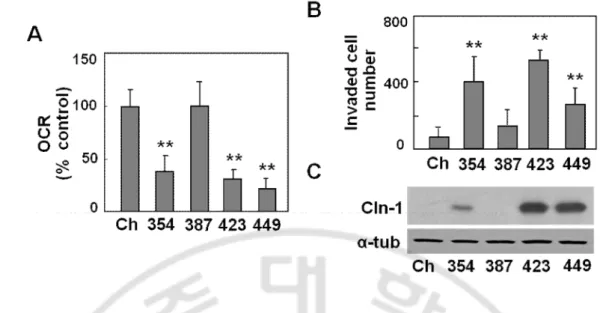
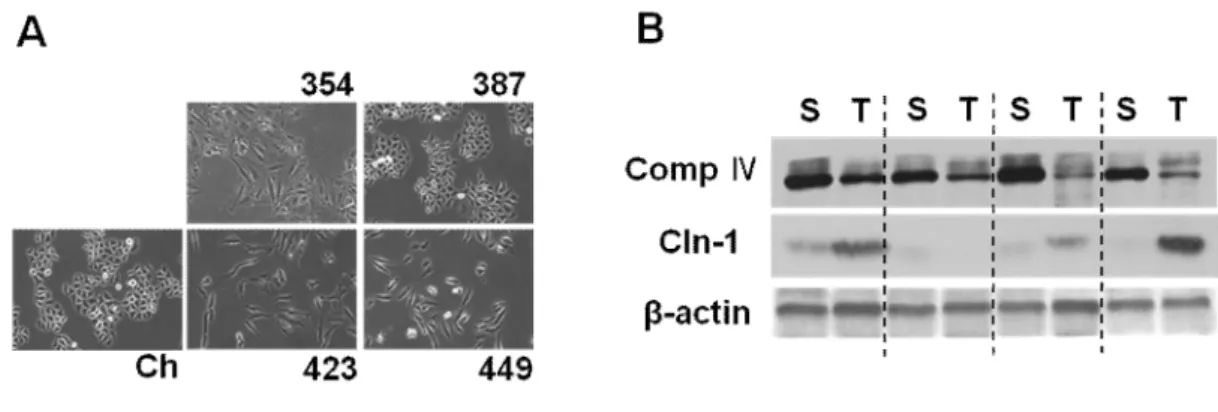
First, I have investigated the relationship between mitochondrial dysfunction and Cln-1 expression in hepatoma cells, by employing four types of SNU hepatoma cells (SNU-354, SNU-387, SNU-423, and SNU449) derived from human hepatocellular carcinomas (Park et al., 1995; Ku and Park 2005), and compared these levels to those of Chang-L clone, which was derived from Chang cell (immortalized human hepatocyte) and were previously characterized as having certain liver-characteristics and active mitochondrial respiration (Kim et al, 2011). Interestingly, SNU354, SNU423, and SNU449 cells, which have defective mitochondrial respiration and increased cell invasion activity, showed increased Cln-1 expression (Fig. 1). Cellular morphology of these three cells with high Cln-1 expression displayed more flattened and extended like fibroblast whereas Chang and SNU387 look like typical epithelial octagonal shape (Fig. 2A). Moreover, increased Cln-1 expression was also observed in 80% of HCC tumor samples with mitochondrial defects (Fig. 2B). These results imply that Cln-1 is associated with mitochondrial dysfunction of hepatoma cells with high invasion activity.
11
Fig. 1. Cln-1 is highly expressed in invasive SNU hepatoma cells with mitochondrial dysfunction. Chang cell clone (Ch-L) and four different SNU hepatoma cell lines (SNU354, SNU387, SNU423, and SNU449) were cultured for 2 days to maintain exponentially growing state. A) Cellular oxygen consumption rate (OCR) was measured using XF analyzer as described in ‘Materials and methods.’ B) Cell invasion activity was assessed using MatrigelTM-coated traswell as described in ‘Materials and methods.’ Invaded cell numbers were counted. C) Western blot analysis for Cln1 expression. **, p<0.01 vs. Chang clone (Ch) by student t-test.
12
Fig. 2. Cln-1 is associated with mitochondrial dysfunction of hepatoma cell. A) Cellular morphology of Chang cell clone (Ch-L) and four different SNU hepatoma cell lines (SNU354, SNU387, SNU423, and SNU449). B) human HCC tumor sample (T) and their surrounding tissues (S) were applied to western blot analysis.
13
Complex I inhibition by rotenone effectively induce Cln-1 expression at transcriptional level.
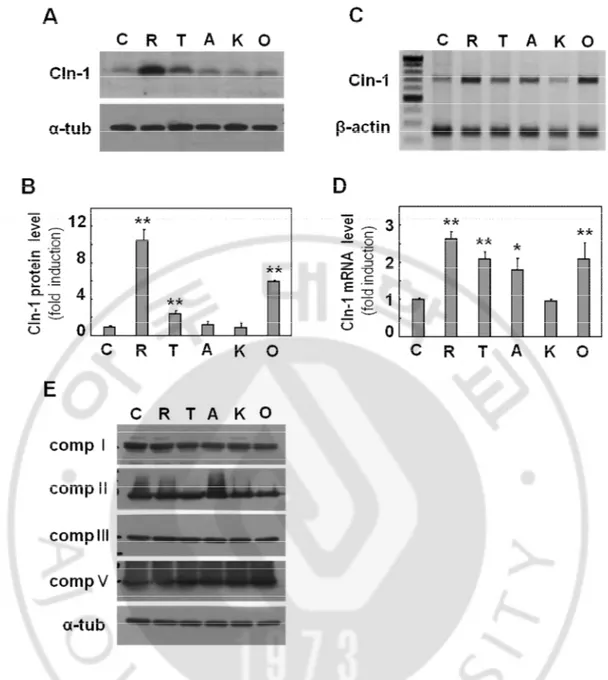
Next, I have examined whether mitochondrial respiratory defects may directly induce Cln-1 expression. When Chang cell was treated with subcytotoxic doses of respiratory inhibitors, rotenone (complex I inhibitor), TTFA (complex II inhibitor), antimycin A (complex III inhibitor), KCN (complex IV inhibitor), and oligomycin (complex V inhibitor) for 12h as previously reported (Byun et al., 2008), Cln-1 protein expression was increased significantly in the cells treated with rotenone, TTFA and oligomycin (Fig. 3A and 3B). The effect of rotenone was the most effective. Similar increases were observed at mRNA levels as evidenced by RT-PCR (Fig. 3C and 3D). These results were not accompanied with any alteration of respiratory protein expressions (Fig. 3E).
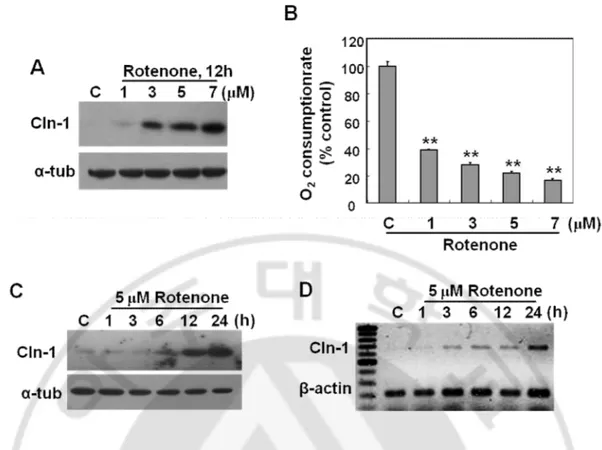
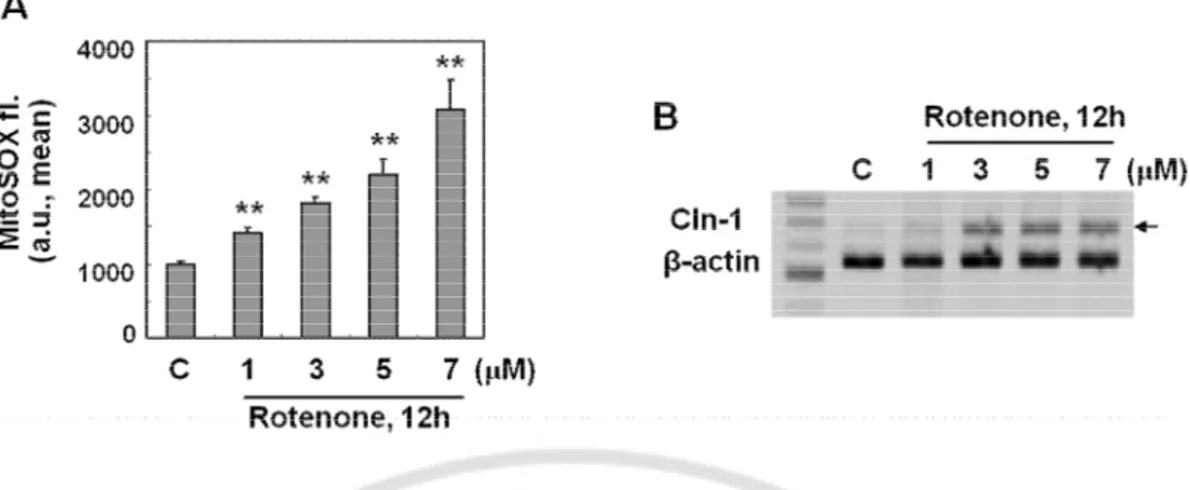
I have further examined the effect of rotenone in detail. Complex I inhibition by rotenone increased Cln-1 expression in a dose-dependent manner and the mRNA induction started 3h after treatment (Fig. 4). These results indicate that direct inhibition of respiratory activity effectively induce Cln-1 expression at transcriptional level.
14
Fig. 3. Respiratory dysfunction induces Cln-1 expression at transcriptional level. A-D) Chang clone was challenged with 5 μM Rotenone (R), 200 μM TTFA (T), 5 μM antimycin A (A), 5 mM KCN (K) or 5 μM oligomycin (O) for 12h. A) Expression levels of Cln-1 protein (A) and mRNA (C) were monitored by Western blot and RT-PCR analysis, respectively. Quantitative analyses of the expression levels are shown (B,D). E) Western blot analysis for mitochondrial respiratory complex expression. *, p<0.05; **, p<0.01 vs. control by student t-test.
15
Fig. 4. Respiratory dysfunction by complex I inhibition induces effectively Cln-1 expression at transcriptional level. A) Chang clone was treated with the indicated concentration of rotenone for 12h. Western blot analysis for Cln-1 expression. B) Endogenous cellular oxygen consumption rate was measured and its specificity for mitochondrial respiration was confirmed by adding rotenone. C-D) Chang clone was challenged with 5 μM rotenone for the indicated time periods. Western blot (C) and RT-PCR (D) analyses for Cln-1 expression are shown. **, p<0.01 vs. control by student t-test.
16
Mitochondrial dysfunction by rotenone induce Cln-1 expression through mitochondrial ROS generation.
I further aimed to investigate the molecular mechanism involved in the mitochondrial defect-mediated Cln-1 induction. It is well known that mitochondrial respiratory defect is closely linked with mitochondrial ROS generation. As expected, mitochondrial ROS level was significantly increased in a dose dependent manner after cells were exposed to rotenone, (Fig. 5A). In the same condition, Cln-1 mRNA level was also increased (Fig. 5B). These results implied that mitochondrial ROS may be involved in the Cln-1 mRNA expression.
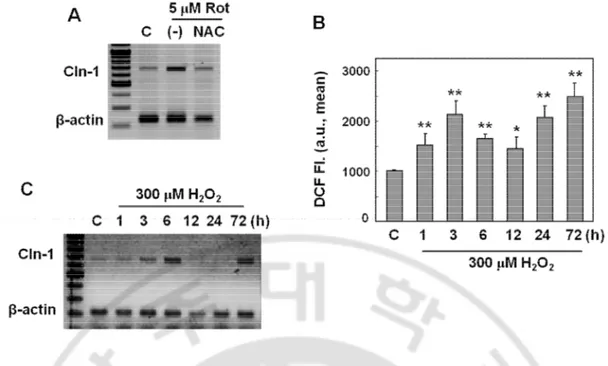
Pretreatment of N-acetyl cysteine blocked the rotenone-induced Cln-1 expression (Fig. 6A). In addition, treatment with exogenous H2O2 induced Cln-1 mRNA expression in a
biphasic pattern, well corresponded with the intracellular ROS profile (Fig. 6B and 6C). These results clearly indicated that mitochondrial ROS induced by rotenone was one of the important factors involved in Cln-1 expression.
17
Fig. 5. Mitochondrial ROS induced by complex I defect is the mediator to induce Cln-1 expression. A-B) Chang clone was treated with the indicated concentrations of rotenone for the indicated periods. A) Mitochondrial ROS levels were monitored by flow cytometric analysis after staining cells with MitoSOX fluorescence dye. B) mRNA expression levels for Cln-1 were examined by RT-PCR analysis. **, p<0.01 vs. control by student t-test.
18
Fig. 6. Intracellular ROS is the mediator to induce Cln-1 expression. A) Chang clone was challenged with 5 μM rotenone for 12h with or without pretreatment of NAC for 6h. mRNA expression levels for Cln-1 were examined by RT-PCR analysis. B-C) Chang clone was treated with 300 μM H2O2 for the indicated time periods. Intracellular ROS levels (B)
and mRNA expression levels (C) are shown. *, p<0.05; **, p<0.01 vs. control by student t-test.
19
ROS-mediated HSF1 phosphorylation is an important event involved in Cln-1 expression.
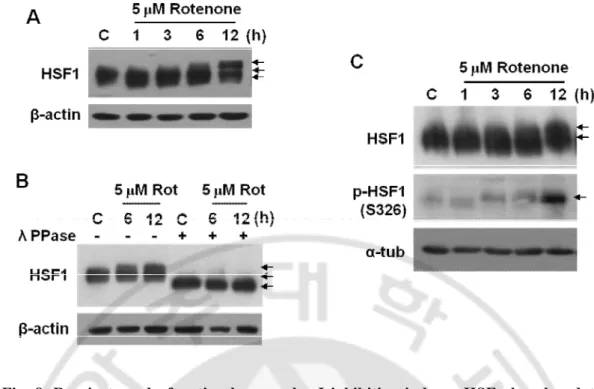
To investigate how mitochondrial ROS modulate Cln-1 expression, I have first analyzed potential transcription factor binding sites in the promoter region of Cln-1 gene by using TFSEARCH program. Several HSF1 binding sites with 100% matched score were found within -2730 base upstream from transcription start site, implying its potential involvement (Fig. 7). Upon exposure to rotenone, HSF1 protein band was shifted with delayed mobility on SDS-PAGE (Fig. 8A) and the delayed band mobility by rotenone was recovered when incubated with λ phosphatase, indicating that the delay in band mobility is due to phosphorylation of HSF1 (Fig. 8B). HSF1 has several phosphorylation sites and is activated by phosphorylation on S326 residue (Guettouche et al., 2005). Phosphorylation status of HSF1 was further confirmed to be increased in response to rotenone treatment by using phospho-specific antibody against serine 326 residue (S326) of HSF1 (Fig. 8C).
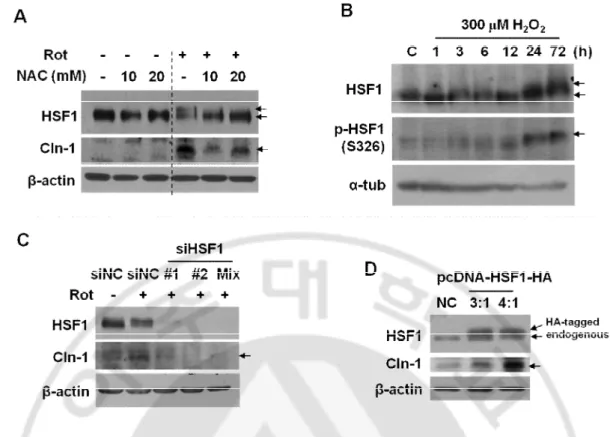
In addition, I have examined whether HSF1 phosphorylation is modulated by ROS. Both delayed migration of HSF1 and Cln-1 induction by rotenone was restored by NAC
pretreatment (Fig. 9A). And treatment with exogenous H2O2 induced Cln-1 mRNA
expression in a biphasic pattern (Fig. 9B). The rotenone-mediated HSF1 band shift was restored by siRNA-mediated HSF1 knockdown and overexpression of HSF1 increased Cln-1 expression (Fig. 9C and 9D). These results suggest that respiratory defects induced by rotenone activate HSF1 by phosphorylation through mitochondrial ROS, thereby inducing Cln-1 expression.
20
Fig. 7. Analysis of transcription factor binding elements of Cln-1 promoter sequence. Identification of putative HSEs within the human Cln-1 gene promoter. TFSearch, a web-based search engine for transcription factor binding sites, was used to search for HSEs within the Cln-1 promoter.
21
Fig. 8. Respiratory dysfunction by complex I inhibition induces HSF phosphorylation (activation). A-C) Chang clone was treated with 5 μM rotenone for the indicated time periods. A) HSF1 protein expression by Western blot analyses. B) Western blot analyses after cell lysates were incubated with lambda phosphatase for 2h. C) HSF1 phosphorylation status on ser326 were monitored.
22
Fig. 9. ROS-mediated HSF phosphorylation (activation) is involved in Cln-1 expression. Western blot analysis. A) Chang clone was challenged with 5 μM rotenone for 12h with or
without pretreatment of NAC for 6h. B) Chang clone was treated with 300 μM H2O2 for the
indicated time periods. C) Chang clone was treated with 5 μM rotenone 36 h after the cells were transfected with siRNAs for HSF1. D) Chang clone was transfected with pcDNA-HSF1-HA to overexpress HSF1.
23
Mitochondrial ROS enhance HSF1 binding to the promoter region of Cln-1 gene and enhances its activity.
Next, I being monitored phosphorylation status of HSF1 in SNU hepatoma cells. SNU hepatoma cells with high Cln-1 expression and mitochondrial defects (SNU354, SNU423 and SNU449) has delayed migration of HSF1 and increased phosphorylation status on S326 as shown in Fig. 10A. Among the three SNU cells, SNU449 showed highest expression of phosphorylated HSF1, well corresponded with Cln-1 expression level. These three SNU cells with mitochondrial defects showed increase in intracellular ROS level (Fig. 10B). When SNU449 cell was exposed to NAC, HSF1 protein band migrated faster and Cln-1 expression decreased (Fig. 11A). Moreover, HSF1 binding to the promoter region of Cln-1 was decreased (Fig. 11B). These results imply that mitochondrial ROS enhance HSF1 binding to the promoter region of Cln-1 gene and enhances its activity.
24
Fig. 10. HSF1 phosphorylation was involved Cln-1 expression through ROS. A-B) Chang cell clone (Ch-L) and four different SNU hepatoma cell lines (SNU354, SNU387, SNU423, and SNU449) were cultured for 2 days to maintain exponentially growing state. Western blot analysis (A), and Intracellular ROS levels (B) were monitored. **, p<0.01 vs. Chang clone (Ch) by student t-test.
25
Fig. 11. Mitochondrial ROS enhance HSF1 binding to the promoter region of Cln-1 gene and enhances its activity. A and B) SNU449 cell was treated with NAC for 6h. Western blot analysis (A) and ChIP assay for HSF1 binding on Cln-1 promoter region (B) are shown. C) luciferase activity was assayed after 2 days in the presence of the indicated rotenone. Luciferase activity was normalized to Renilla activity as expressed by the cotransfected plasmid. **, p<0.01 vs. control by student t-test.
26
HSF1 is a key transcription factor to control hepatoma cell invasion activity through inducing Cln-1 expression.
Finally, I was tested whether HSF1 is involved in hepatoma cell invasion activity. Previous reports indicated that the three hepatoma cells with mitochondrial defects (SNU354, SNU423, and SNU449 cells) has high invasion activity (Kim et al., 2011). When HSF1 expression was decreased by siRNA-mediated knockdown in SNU449 cell (highest HSF1 phosphorylation), cell invasion activity was significantly diminished, together with decrease in Cln-1 expression (Fig. 12A and 12B). Taken together, HSF1 phosphorylation was critically involved in hepatoma cell invasion activity through ROS-mediated Cln-1 expression.
27
Fig. 12. HSF1 phosphorylation was critically involved in hepatoma cell invasion activity through ROS-mediated Cln-1 expression. A-B) SNU449 cell was transfected with siRNA for HSF1 for 24h. Western blot analysis (A) and cell invasion activity (B) are shown. *, p<0.05 vs. control by student t-test.
28
Fig. 13. Schematic model for mechanism of Cln-1. These results suggest that Cln-1 induced by HSF1 activation through respiratory defect mediated ROS in hepatoma cells.
29
DISCUSSION
Mitochondrial defects are considered to be hallmarks of cancer cells and importantly involved in cancer cell metastatic properties, including invasion activity (Chang et al., 2009; Hung et al., 2012; He et al., 2013; Ma et al., 2013). It is unclear how mitochondrial respiratory defects control cancer cell invasion activity. However, according to previous study, it was reported that mitochndrial dysfunction is linked with the claudin-1 induction.
Claudins are a family of proteins that are the critical components of the tight junctions, which controls paracellular permeability of cells and the flow of molecules between cells. According to several recent reports Cln-1 overexpression increases cancer cell invasion and metastasis (Dhawan et al., 2005; Leotlela et al., 2007; Dos Reis et al., 2008). For example, hepatocellular carcinoma with mitochondrial dysfunction display increased expression Cln-1 and high invasion activity, implying the close link between mitochondrial defects and Cln-1-mediated tumor invasion (Kim et al., 2011). However, it is not clearly understood how Cln-1 expression is increased in cancer and linked with the metabolic hallmark of cancer, mitochondrial dysfunction. So, it is aimed to elucidate “How does mitochondrial defects induce Cln-1 expression?” I am screened whether any specific mitochondrial respiratory defect induces Cln-1 induction. Among diverse respiratory inhibitors, complex I inhibition by rotenone most effectively induced Cln-1 expression at transcriptional level, accompanied by increase in mitochondrial ROS production.
The role of mitochondria in tumorigenesis has been linked to their ROS production. Under normal physiological conditions, about 1 to 3% of total mitochondrial oxygen consumption is incompletely reduced and leads to ROS production. As mitochondrial respiratory chain is a permanent source of ROS, cell has an effective antioxidant systems to nullify the toxic effects of the ROS and other free radicals (Gao et al., 2007). Excessive ROS production can cause mtDNA mutations, which may lead to mitochondrial respiratory chain dysfunction that could contribute to the onset of many diseases including neoplasia. When mitochondria function is affected, mitochondrial ROS production can be increased (He et al., 2013). So, it has been confirmed treatment of H2O2 directly induced 1 expression, implying that
30
More recent studies have revealed that mitochondria are also engaged in retrograde regulation, in which cells respond to changes in the functional state of the organelle via changes in nuclear gene expression. Retrograde regulation encompasses a wide assortment of cellular activities, including nutrient sensing, growth control, aging, and other signaling processes that function in metabolic and organelle homeostasis (Liu and Butow, 2006; Barbour and Turner, 2014). Interestingly, Cln-1 promoter region (-2700bp ~ +300bp) contained HSF1 binding site, This transcription factor is known to be regulated by intracellular redox status (Jacquier-Sarlin and Polla, 1996; Christians et al., 2002; Yan et al., 2002). Also HSF1 promoted cancer migration and invasion (Fang et al., 2012; Xi et al., 2012). So, it has been confirmed that activation of HSF1 through mitochondrial ROS induces Cln-1 expression and HSF1 knockdown using siRNA decreased Cln-1 expression and invasive activity in SNU449.
A prominent feature of HSF1 is conversion into a transcriptionally active trimer occurs concurrently with extensive hyperphosphorylation of serine residues, most of which reside within the regulatory domain. In previously study, the analysis of HSF1 phosphorylation sites was performed by Guettouche and colleagues, who purified exogenously expressed human HSF1 from heat-shocked HeLa cells and identified phosphorylation target sites by means of mass spectrometry. The analysis, covering >90% of the HSF1 amino acid sequence, demonstrated the phosphorylation of HSF1 on at least 12 serin residues (S121, S230, S292, S303, S307, S314, S319, S326, S344, S363, S419, and S444), with no detectable amount of threonine or tyrosine phosphorylation (Guettouche et al., 2005; Anckar and Sistonen, 2011). Among these residue, S230, S326 and S419 is associated with regulation of HSF1 activity (Holmberg et al., 2001; Guettouche et al., 2005; Kim et al., 2005; Neef et al., 2011). The phosphorylation of S326, previously had been shown to facilitate the association between HSF1 and the coactivator Daxx (Kim et al., 2005). In addition, stress-inducible phosphorylation of S230 by the calcium/calmodulin-dependent kinase CaMKII contribute to HSF1 activation as both mutation of S230 and pharmacological inhibition of CaMKII lead to a reduced heat shock response (Holmberg et al., 2001). Phosphorylation of HSF1 on S419 by polo-like kinase 1 (PLK1) is an essential step for HSF1 nuclear translocation by heat stress (Guettouche et al., 2005).
31
In this study, it is aimed to elucidate how mitochondrial dysfunction is linked with Cln-1 induction and if it does,what the underlying mechanisms are.These data lead to three major
findings. (ⅰ) Mitochondrial defects can affect cancer invasion activityby Cln-1 induction.
(ⅱ) Complex I deficiency-induced ROS generation may play a critical role in these processes by regulating HSF1 activation. (ⅲ) Mitochondrial ROS enhances HSF1 binding activity to the promoter of Cln-1 gene and enhances its activity.This study demonstrated that mitochondrial ROS activates HSF1 through phosphorylation of S326 residue and enhances its binding to Cln-1 promoter.
32
CONCLUSION
Mitochondrial dysfunction is hallmarks of cancer cells and importantly involved in cancer cell metastatic properties, including invasion activity. However, it is unclear how mitochondrial respiratory defects control cancer cell invasion activity. This study clearly demonstrate that mitochondrial dysfunction increase mitochondrial ROS and activates (phosphorylates) HSF1, thereby inducing Cln-1 expression is a key molecule to enhance hepatoma cell invasion activity. This study emphasize that mitochondrial ROS activates HSF1 through phosphorylation of S326 residue and enhances its binding to Cln-1 promoter.
33
REFERENCES
1.
Amann T, Maegdefrau U, Hartmann A, Agaimy A, Marienhagen J, Weiss TS,
Stoeltzing O, Warnecke C, Scholmerich J, Oefner PJ, Kreutz M, Bosserhoff
AK, Hellerbrand C: GLUT1 expression is increased in hepatocellular
carcinoma and promotes tumorigenesis. Am J Pathol 174: 1544-1552, 2009
2.
Anckar J, Sistonen L: Regulation of HSF1 function in the heat stress
response: implications in aging and disease. Annu Rev Biochem 80:
1089-1115, 2011
3.
Barbour JA, Turner N: Mitochondrial Stress Signaling Promotes Cellular
Adaptations. Int J Cell Biol 2014: 156020, 2014
4.
Bauer G: Tumor cell-protective catalase as a novel target for rational
therapeutic approaches based on specific intercellular ROS signaling.
Anticancer Res 32: 2599-2624, 2012
5.
Byun HO, Kim HY, Lim JJ, Seo YH, Yoon G: Mitochondrial dysfunction by
complex II inhibition delays overall cell cycle progression via reactive
oxygen species production. J Cell Biochem 104: 1747-1759, 2008
6.
Cairns RA, Harris IS, Mak TW: Regulation of cancer cell metabolism. Nat
Rev Cancer 11: 85-95, 2011
7.
Chang CJ, Yin PH, Yang DM, Wang CH, Hung WY, Chi CW, Wei YH, Lee
HC: Mitochondrial dysfunction-induced amphiregulin upregulation mediates
chemo-resistance and cell migration in HepG2 cells. Cell Mol Life Sci 66:
1755-1765, 2009
8.
Chang Q, Jurisica I, Do T, Hedley DW: Hypoxia predicts aggressive growth
and spontaneous metastasis formation from orthotopically grown primary
xenografts of human pancreatic cancer. Cancer Res 71: 3110-3120, 2011
9.
Christians ES, Yan LJ, Benjamin IJ: Heat shock factor 1 and heat shock
proteins: Critical partners in protection against acute cell injury. Crit Care
Med 30: S43-S50, 2002
10.
Ciocca DR, Arrigo AP, Calderwood SK: Heat shock proteins and heat shock
factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol
87: 19-48, 2013
11.
Dang CV, Semenza GL: Oncogenic alterations of metabolism. Trends
Biochem Sci 24: 68-72, 1999
12.
Dhawan P, Singh AB, Deane NG, No Y, Shiou SR, Schmidt C, Neff J,
Washington MK, Beauchamp RD: Claudin-1 regulates cellular
transformation and metastatic behavior in colon cancer. J Clin Invest 115:
1765-1776, 2005
13.
Dos Reis PP, Bharadwaj RR, Machado J, Macmillan C, Pintilie M, Sukhai
MA, Perez-Ordonez B, Gullane P, Irish J, Kamel-Reid S: Claudin 1
overexpression increases invasion and is associated with aggressive
34
histological features in oral squamous cell carcinoma. Cancer 113:
3169-3180, 2008
14.
Escudero-Esparza A, Jiang WG, Martin TA: The Claudin family and its role
in cancer and metastasis. Front Biosci (Landmark Ed) 16: 1069-1083, 2011
15.
Fang F, Chang R, Yang L: Heat shock factor 1 promotes invasion and
metastasis of hepatocellular carcinoma in vitro and in vivo. Cancer 118:
1782-1794, 2012
16.
Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, Bhujwalla ZM,
Felsher DW, Cheng L, Pevsner J, Lee LA, Semenza GL, Dang CV:
HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 12:
230-238, 2007
17.
Gogvadze V, Orrenius S, Zhivotovsky B: Mitochondria in cancer cells: what
is so special about them? Trends Cell Biol 18: 165-173, 2008
18.
Guettouche T, Boellmann F, Lane WS, Voellmy R: Analysis of
phosphorylation of human heat shock factor 1 in cells experiencing a stress.
BMC Biochem 6: 4, 2005
19.
Hamazaki Y, Itoh M, Sasaki H, Furuse M, Tsukita S: Multi-PDZ domain
protein 1 (MUPP1) is concentrated at tight junctions through its possible
interaction with claudin-1 and junctional adhesion molecule. J Biol Chem
277: 455-461, 2002
20.
He X, Zhou A, Lu H, Chen Y, Huang G, Yue X, Zhao P, Wu Y: Suppression
of mitochondrial complex I influences cell metastatic properties. PLoS One 8:
e61677, 2013
21.
Holmberg CI, Hietakangas V, Mikhailov A, Rantanen JO, Kallio M,
Meinander A, Hellman J, Morrice N, MacKintosh C, Morimoto RI, Eriksson
JE, Sistonen L: Phosphorylation of serine 230 promotes inducible
transcriptional activity of heat shock factor 1. EMBO J 20: 3800-3810, 2001
22.
Hung WY, Huang KH, Wu CW, Chi CW, Kao HL, Li AF, Yin PH, Lee HC:
Mitochondrial dysfunction promotes cell migration via reactive oxygen
species-enhanced beta5-integrin expression in human gastric cancer SC-M1
cells. Biochim Biophys Acta 1820: 1102-1110, 2012
23.
Hung WY, Wu CW, Yin PH, Chang CJ, Li AF, Chi CW, Wei YH, Lee HC:
Somatic mutations in mitochondrial genome and their potential roles in the
progression of human gastric cancer. Biochim Biophys Acta 1800: 264-270,
2010
24.
Ichiyasu H, McCormack JM, McCarthy KM, Dombkowski D, Preffer FI,
Schneeberger EE: Matrix metalloproteinase-9-deficient dendritic cells have
impaired migration through tracheal epithelial tight junctions. Am J Respir
Cell Mol Biol 30: 761-770, 2004
25.
Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi
H, Nakada K, Honma Y, Hayashi J: ROS-generating mitochondrial DNA
mutations can regulate tumor cell metastasis. Science 320: 661-664, 2008
35
26.
Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S: Direct binding
of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the
COOH termini of claudins. J Cell Biol 147: 1351-1363, 1999
27.
Jacquier-Sarlin MR, Polla BS: Dual regulation of heat-shock transcription
factor (HSF) activation and DNA-binding activity by H2O2: role of
thioredoxin. Biochem J 318 ( Pt 1): 187-193, 1996
28.
Kim JH, Kim EL, Lee YK, Park CB, Kim BW, Wang HJ, Yoon CH, Lee SJ,
Yoon G: Decreased lactate dehydrogenase B expression enhances claudin
1-mediated hepatoma cell invasiveness via mitochondrial defects. Exp Cell Res
317: 1108-1118, 2011
29.
Kim SA, Yoon JH, Lee SH, Ahn SG: Polo-like kinase 1 phosphorylates heat
shock transcription factor 1 and mediates its nuclear translocation during heat
stress. J Biol Chem 280: 12653-12657, 2005
30.
Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE:
Mitochondria and reactive oxygen species. Free Radic Biol Med 47: 333-343,
2009
31.
Kroeger PE, Morimoto RI: Selection of new HSF1 and HSF2 DNA-binding
sites reveals difference in trimer cooperativity. Mol Cell Biol 14: 7592-7603,
1994
32.
Kroemer G, Pouyssegur J: Tumor cell metabolism: cancer's Achilles' heel.
Cancer Cell 13: 472-482, 2008
33.
Krzeslak A, Wojcik-Krowiranda K, Forma E, Jozwiak P, Romanowicz H,
Bienkiewicz A, Brys M: Expression of GLUT1 and GLUT3 glucose
transporters in endometrial and breast cancers. Pathol Oncol Res 18: 721-728,
2012
34.
Kwong JQ, Henning MS, Starkov AA, Manfredi G: The mitochondrial
respiratory chain is a modulator of apoptosis. J Cell Biol 179: 1163-1177,
2007
35.
Leotlela PD, Wade MS, Duray PH, Rhode MJ, Brown HF, Rosenthal DT,
Dissanayake SK, Earley R, Indig FE, Nickoloff BJ, Taub DD, Kallioniemi
OP, Meltzer P, Morin PJ, Weeraratna AT: Claudin-1 overexpression in
melanoma is regulated by PKC and contributes to melanoma cell motility.
Oncogene 26: 3846-3856, 2007
36.
Lievre A, Chapusot C, Bouvier AM, Zinzindohoue F, Piard F, Roignot P,
Arnould L, Beaune P, Faivre J, Laurent-Puig P: Clinical value of
mitochondrial mutations in colorectal cancer. J Clin Oncol 23: 3517-3525,
2005
37.
Liu Z, Butow RA: Mitochondrial retrograde signaling. Annu Rev Genet 40:
159-185, 2006
38.
Ma J, Zhang Q, Chen S, Fang B, Yang Q, Chen C, Miele L, Sarkar FH, Xia J,
36
and invasion through HIF1alpha accumulation via increased production of
reactive oxygen species. PLoS One 8: e69485, 2013
39.
Matsuyama W, Nakagawa M, Wakimoto J, Hirotsu Y, Kawabata M, Osame
M: Mitochondrial DNA mutation correlates with stage progression and
prognosis in non-small cell lung cancer. Hum Mutat 21: 441-443, 2003
40.
Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel
E, Ince TA, Whitesell L, Lindquist S: HSF1 drives a transcriptional program
distinct from heat shock to support highly malignant human cancers. Cell
150: 549-562, 2012
41.
Michl P, Barth C, Buchholz M, Lerch MM, Rolke M, Holzmann KH, Menke
A, Fensterer H, Giehl K, Lohr M, Leder G, Iwamura T, Adler G, Gress TM:
Claudin-4 expression decreases invasiveness and metastatic potential of
pancreatic cancer. Cancer Res 63: 6265-6271, 2003
42.
Miyamori H, Takino T, Kobayashi Y, Tokai H, Itoh Y, Seiki M, Sato H:
Claudin promotes activation of pro-matrix metalloproteinase-2 mediated by
membrane-type matrix metalloproteinases. J Biol Chem 276: 28204-28211,
2001
43.
Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E:
Energy metabolism in tumor cells. FEBS J 274: 1393-1418, 2007
44.
Murphy MP: How mitochondria produce reactive oxygen species. Biochem J
417: 1-13, 2009
45.
Neef DW, Jaeger AM, Thiele DJ: Heat shock transcription factor 1 as a
therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov 10:
930-944, 2011
46.
Pelicano H, Martin DS, Xu RH, Huang P: Glycolysis inhibition for anticancer
treatment. Oncogene 25: 4633-4646, 2006
47.
Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, Lim S,
Issa MM, Flanders WD, Hosseini SH, Marshall FF, Wallace DC: mtDNA
mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S
A 102: 719-724, 2005
48.
Racker E: History of the Pasteur effect and its pathobiology. Mol Cell
Biochem 5: 17-23, 1974
49.
Rangel LB, Agarwal R, D'Souza T, Pizer ES, Alo PL, Lancaster WD,
Gregoire L, Schwartz DR, Cho KR, Morin PJ: Tight junction proteins
claudin-3 and claudin-4 are frequently overexpressed in ovarian cancer but
not in ovarian cystadenomas. Clin Cancer Res 9: 2567-2575, 2003
50.
Santagata S, Hu R, Lin NU, Mendillo ML, Collins LC, Hankinson SE,
Schnitt SJ, Whitesell L, Tamimi RM, Lindquist S, Ince TA: High levels of
nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in
breast cancer. Proc Natl Acad Sci U S A 108: 18378-18383, 2011
37
51.
Seo YH, Jung HJ, Shin HT, Kim YM, Yim H, Chung HY, Lim IK, Yoon G:
Enhanced glycogenesis is involved in cellular senescence via GSK3/GS
modulation. Aging Cell 7: 894-907, 2008
52.
Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G,
Oda H, Ohta S: Positive contribution of pathogenic mutations in the
mitochondrial genome to the promotion of cancer by prevention from
apoptosis. Cancer Res 65: 1655-1663, 2005
53.
Simonnet H, Alazard N, Pfeiffer K, Gallou C, Beroud C, Demont J, Bouvier
R, Schagger H, Godinot C: Low mitochondrial respiratory chain content
correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis
23: 759-768, 2002
54.
Song I, Kim K, Kim JH, Lee YK, Jung HJ, Byun HO, Yoon G, Kim N:
GATA4 negatively regulates osteoblast differentiation by downregulation of
Runx2. BMB Rep, 2013
55.
Tamori A, Nishiguchi S, Nishikawa M, Kubo S, Koh N, Hirohashi K, Shiomi
S, Inoue M: Correlation between clinical characteristics and mitochondrial
D-loop DNA mutations in hepatocellular carcinoma. J Gastroenterol 39:
1063-1068, 2004
56.
Tseng LM, Yin PH, Chi CW, Hsu CY, Wu CW, Lee LM, Wei YH, Lee HC:
Mitochondrial DNA mutations and mitochondrial DNA depletion in breast
cancer. Genes Chromosomes Cancer 45: 629-638, 2006
57.
Vander Heiden MG, Cantley LC, Thompson CB: Understanding the Warburg
effect: the metabolic requirements of cell proliferation. Science 324:
1029-1033, 2009
58.
Villani G, Attardi G: In vivo measurements of respiration control by
cytochrome c oxidase and in situ analysis of oxidative phosphorylation.
Methods Cell Biol 65: 119-131, 2001
59.
Warburg O: On the origin of cancer cells. Science 123: 309-314, 1956
60.
Wu CW, Yin PH, Hung WY, Li AF, Li SH, Chi CW, Wei YH, Lee HC:
Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric
cancer. Genes Chromosomes Cancer 44: 19-28, 2005
61.
Wu YT, Wu SB, Lee WY, Wei YH: Mitochondrial respiratory
dysfunction-elicited oxidative stress and posttranslational protein modification in
mitochondrial diseases. Ann N Y Acad Sci 1201: 147-156, 2010
62.
Xi C, Hu Y, Buckhaults P, Moskophidis D, Mivechi NF: Heat shock factor
Hsf1 cooperates with ErbB2 (Her2/Neu) protein to promote mammary
tumorigenesis and metastasis. J Biol Chem 287: 35646-35657, 2012
63.
Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ: Mouse heat
shock transcription factor 1 deficiency alters cardiac redox homeostasis and
increases mitochondrial oxidative damage. EMBO J 21: 5164-5172, 2002
64.
Yoon CH, Kim MJ, Park MJ, Park IC, Hwang SG, An S, Choi YH, Yoon G,
38
signaling and has a causal role in the acquisition of invasive capacity in
human liver cells. J Biol Chem 285: 226-233, 2010
65.
Yoon YS, Byun HO, Cho H, Kim BK, Yoon G: Complex II defect via
down-regulation of iron-sulfur subunit induces mitochondrial dysfunction and cell
cycle delay in iron chelation-induced senescence-associated growth arrest. J
Biol Chem 278: 51577-51586, 2003
66.
Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung HY, Rojo M, Malka F, Jou
MJ, Martinou JC, Yoon G: Formation of elongated giant mitochondria in
DFO-induced cellular senescence: involvement of enhanced fusion process
through modulation of Fis1. J Cell Physiol 209: 468-480, 2006
67.
Yu JS, Kim AK: Wogonin induces apoptosis by activation of ERK and p38
MAPKs signaling pathways and generation of reactive oxygen species in
human breast cancer cells. Mol Cells 31: 327-335, 2011
39
-국문요약-미토콘드리아
기능손상으로 생성된 활성산소가
Claudin-1 의 발현에 미치는 영향 연구
아주대학교 대학원 의생명과학과 이 종 혁 (지도교수: 윤 계 순) 미토콘드리아는 세포가 필요로 하는 에너지 생성을 관장하는 세포 내 소기관이다. 정상세포는 산소가 존재하는 환경에서는 미토콘드리아의 산화적 인산화 과정을 통해 에너지를 생성하고, 산소가 존재하지 않는 경우 해당과정만을 이용해 에너지를 생성한다. 그러나 많은 암세포의 경우, 미토콘드리아의 ATP 생성능력이 저하되어 있는 것이 관찰되어 있고, 산소의 존재 유무와 상관없이 해당과정에 의존적으로 ATP 를 합성한다. 이와 같이 암세포에서 관찰되는 미토콘드리아 기능의 저하가 어떠한 기전에 의해 나타나며, 암세포의 증식능력과 전이활성 (invasion)에 어떻게 연관되어 있는지에 대한 연구는 미흡한 실정이다. 이전보고에서 간암에서 미토콘드리아 기능손상이 밀착연접 단백질의 하나인 Claudin-1 (Cln-1)의 발현을 통해 암세포의 전이활성을 증가시킨다는 사실을 보고하였고, 이를 통해 본 연구에서는 미토콘드리아 호흡기능 손상이 어떤 분자기전에 의해 Cln-1 의 발현을 조절시키는지 규명하고자 하였다. 미토콘드리아 호흡기능 손상 유도 모델로 미토콘드리아 전자전달계 복합체Ⅰ의 저해제인 rotenone 을 정상 간세포주인 Chang 세포에 처리하였다. Cln-1 의 mRNA 와 단백질 발현의 증가가 rotenone 에 의해 유도된 것을 확인하여, 전사수준에서 Cln-1 의 발현이 증가됨을 알 수 있었다. 같은 조건에서 미토콘드리아성 활성산소가 증가되어 있는 것을 활성산소에 의한 Cln-1 발현을40
조절하는 것을 확인하였다. 또한 rotenone 에 의해 증가된 Cln-1 의 발현이 항산화제인 N-acetylcysteine 의 전 처리에 의해 소실되는 것으로 보아, rotenone 에 의한 미토콘드리아 손상으로 유도되는 Cln-1 발현증가는 미토콘드리아에서 생성되는 활성산소에 의해 매개되는 것임을 알 수 있었다. 이러한 활성산소에 의해 어떠한 전사조절기전에 의해 Cln-1 의 발현이 조절되는 지를 규명하기 위해, Cln-1 의 -2700bp 에서 +300bp promoter 유전자 서열을 TFSEARCH program 을 이용하여 분석한 결과 Heat Shock Factor1 (HSF1) 전사인자의 결합부위가 여러 부분 존재하는 것을 확인하였다. 이를 확인하고자 Chang 세포주에서 rotenone 에 의해 HSF1 의 band shift 가 증가하는 것을 확인하였고, 해당 band 가 phosphorylation 에 의해서 activation 된 band 인지 확인하기 위해 lambda phosphatase 처리와 S326 잔기에 인산화된 HSF1 항체로 shift 된 band 가 HSF1 의 phosphorylation 에 의해서 activation 된 band 임을 확인 하였다. 그리고 NAC 처리로 band 의 shift 가 복구되는 것으로 보아 활성산소에 의해 HSF1 의 activitiy 가 조절됨을 알 수 있었다. HSF1 의 활성화에 의해서 Cln-1 이 조절됨을 확인하고자 HSF1 을 Knockdown 과 Overexpression 에 의해 Cln-1 발현이 조절됨을 알 수 있었다. 또한 Cln-1 의 promoter 를 이용한 chip assay 와 luciferase assay 를 통해 ROS 와 rotenone 에 의해 HSF1 의 binding activity 가 조절됨을 확인하여, 이로서 미토콘드리아의 호흡기능 억제에 의한 Cln-1 발현 증가가 활성산소를 매개로 한 HSF 에 activation 에 의해 조절된 것임을 알 수 있었다. 미토콘드리아의 기능손상이 저하되어 있고, Cln-1 과 HSF1 의 발현이 증가해 있는 간암세포주인 SNU 449 에서 HSF1 의 Knockdown 에 의해 Cln-1 의 발현이 저하되는 것과 동시에 침투활성이 감소하는 것을 확인하였다. 본 연구를 통해 암세포의 미토콘드리아 기능손상에 의한 활성산소의 증가로 HSF1 의 활성화가 유도되고, Cln-1 의 발현이 증가 함으로서 암세포의 침투활성이 증가됨을 알 수 있었다. 이 결과로 미토콘드리아 기능손상이 간암의 악성화에 기여하는 분자기전을 좀 더 구체적으로 이해할 수 있었다. 본 연구로41
미토콘드리아 기능 손상을 가지고 있는 간암치료에 대한 새로운 제어타겟을 제시하게 될 것으로 기대한다.