Received: May 31, 2012 / Accepted: June 10, 2012 Address for correspondence: Jin-Hong Shin, MD, PhD
Department of Neurology, Pusan National University Yangsan Hospital,
#20 Geumo-ro, Mulgeum-eup, Yangsan 626-770, Korea
Tel: +82-55-360-2122, Fax: +82-55-360-2152, E-mail: shinzh@gmail.com
근디스트로피의 유전자 치료
양산부산대학교병원 신경과
신 진 홍
Gene Therapy of Muscular Dystrophy
Jin-Hong Shin, MD, PhD
Department of Neurology, Pusan National University Yangsan Hospital, Yangsan, Korea
KEYWORDS Gene therapy, Muscular dystrophy, Read-through, Exon skipping, Gene replacement, Adeno-associated virus
Gene therapy has been the research of few success for a long time. Finally, encouraging results are emerging in recent clinical trials. Here we review most promising approaches in gene therapy of muscular dystrophy. Read-through of nonsense mutation may mitigate 15% of Duchenne muscular dystrophy patients. Expression of dystrophin was noted on clinical trial but 6-minute walk test did not improve. Exon skipping by antisense oligo is the one closest to the approval. Phase 2 clinical trials on Duchenne muscular dystrophy are currently underway. However, not all mutations can be benefited by this method. Adeno-associated virus (AAV) for gene replacement is the most versatile tool in gene therapy. Packaging capacity is the limiting factor, that dystrophin gene is miniaturized to fit into AAV.
Most other genes of autosomal recessive limb-girdle muscular dystrophy can be transferred by AAV without modification. Control of immune reaction will be the key to its success.
서 론
질병의 원인이 되는 유전자의 이상을 근본적인 유전자 수준에서 직접 개입하여 치료하는 것은 유전 물질의 발견 이후 인류의 오랜 숙원이었다. 수차례에 걸친 비극적인 결 과를 통해 요원하게만 느껴졌던 유전자 치료는,1 오랜 연 구 결과의 축적에 힘입어 마침내 일부 질환에서 성공적인 임상 시험례가 보고되고 있다.2
근디스트로피는 유전자의 이상으로 발생하며 스테로이 드 및 대증 요법 이상의 치료 방법이 없다는 점에서 유전 자 치료 연구의 일차적 대상이 된다.3 한편 근디스트로피
는 체중의 40%나 차지하며 600개 이상의 근육으로 나뉘어 져 있는 전신의 골격근을 침범하며, 심근까지 침범하는 경 우도 있으므로 치료 전략상 쉬운 대상은 아니다.
근육질환에서의 유전자 치료 연구는 Duchenne 근디스트 로피(Duchenne muscular dystrophy, DMD)에 집중되어 왔다.
DMD는 X염색체 단완에 위치한 디스트로핀(dystrophin) 유 전자의 이상에 의해 발생하는 질환으로 남아 3500명 중 한 명 꼴로 발생하여 진행성 근디스트로피 중 발생 빈도가 가 장 높다. 따라서 DMD를 중심으로 유전자 치료의 방법들 을 소개하고 다른 근디스트로피에의 적용 가능성을 덧붙 이고자 한다.
Ribosome mRNA
Translation
Protein
Nonsense mutation
Nonsense- mediated decay
Premature
termination Read-through Full-length
protein Figure 1. Read-through of nonsense mutation.
50
49 51 52
Pre-mRNA, exon 49-52
50
49 51 52
mRNA, wild type
49 51 52
mRNA, Δ50 Æ Reading frame disrupted
49 52
mRNA, Δ50 + skipping of 51
Reading frame preserved
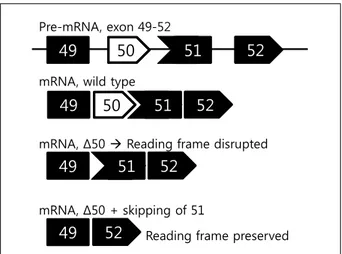
Figure 2. Skipping of dystrophin exon 51 to restore reading frame.
본 론
1. 정지돌연변이(nonsense mutation)의 번역초과(read-through) (Fig. 1)
정지돌연변이란 특정 아미노산 코돈(codon)을 가지고 있 어야 할 유전자 서열이 돌연변이로 인해 정지 코돈 중 한 가지로 바뀌어 단백질 합성 시에 중도 종료되어 버리는 것 을 말한다. 이 경우 단순히 정상보다 짧은 단백질이 발현 되는 것이 아니라 nonsense-mediated decay라는 기전에 의 해 전체 mRNA가 소멸되어 버려 해당 유전자의 단백질이 총체적으로 결손되는 결과를 가져온다.4 DMD의 15%는 정 지돌연변이에 기인한다.5
단백질 합성은 세포질 내 리보솜에서 성숙 mRNA의 정 보를 번역하면서 이루어지는데, 정지 코돈을 무시하고 번 역을 계속하는 번역초과 오류는 정상 세포에서도 드물게 일어난다. 아미노글리코사이드계 항생제를 투여하면 이 현상이 항진되는 것이 알려져 왔으며, gentamicin을 사용한 임상시험에서 정지돌연변이를 가진 DMD 환자의 디스트 로핀 발현을 증가시키는 것이 확인되었다.6 하지만 이미 알려진 gentamicin의 신독성과 정맥투여 필요성으로 인해 실용화 되지는 못하였다.
이후의 연구는 번역초과를 효율적으로 일으키면서도 독 성이 없고 경구 복용이 가능한 소분자 화합물을 찾는 것으 로 전환되었으며 마침내 찾아진 약물 PTC124는 Atarulen이 라고 명명되었다.7 전임상 시험을 거쳐 제1상 임상시험에 서 안전성이 확보되고,8 Duchenne 근디스트로피에서 디스 트로핀 발현을 증가시키는 것이 확인되었다. 그러나 제2상 임상시험 단계에서 6-minute walk test의 결과를 호전시키 지 못하자 약물의 실효성에 의문이 제기되었고, 현재 임상 시험 중단 후 전반적인 재평가 과정 중에 있다.3
2. 엑손 스키핑(exon skipping) (Fig. 2)
디스트로핀은 79개의 엑손으로 이루어진 거대한 유전자 로, DMD의 반수는 한 개 또는 수 개 엑손의 결실로 인해 일어나게 된다. 한편 개별 엑손은 꼭 3의 배수 개의 핵산으 로 구성되어 있는 것이 아니어서 특정 엑손의 결실 이후 코돈의 해독틀(reading frame)은 보존되지 않을 수도 있다 는 것이 주목해야 할 부분이다. 대규모의 유전형-표현형 조사의 결과 해독틀이 보존되는 경우는 임상 양상이 경한 Becker 근디스트로피(Becker muscular dystrophy, BMD), 그 렇지 못한 경우에는 DMD로 될 가능성이 크다는 결론을 얻게 되었다.9
그렇다면 디스트로핀의 일부분에 결실이 있더라도 해독 틀을 회복시켜 주면 DMD의 임상 양상을 BMD 정도로 호 전시킬 수 있을 것으로 추론할 수 있으며, 이를 구현한 것 이 antisense mediated exon skipping이다. 이 방법의 골자는, 생물학적으로 분해되지 않는 핵산 유사체를 투여하여 pre-mRNA의 특정 부분에 상보 결합시킴으로써 특정 엑손 을 이어맞추기(splicing)로부터 차폐시키는 것이다. 원래 결 실이 있는 엑손 이외에도 차폐된 엑손의 추가적인 결실이 발생하게 되지만 해독틀이 회복됨으로써 디스트로핀의 발 현은 오히려 증가된다.
마우스 모델을 거쳐 개 모델에서 매우 성공적인 결과가 확인되었으며,10,11 이를 바탕으로 제2상 임상시험이 진행
중이다.12,13 안전성의 문제는 발견되지 않았으며 추적 근육
생검에서 디스트로핀 단백질 발현의 증가가 확인되었다. 실제 기능적 호전에 있어서는 아직 만족할 만한 성과가 확 인되지 않았으나 계속되는 임상 시험의 추이를 관찰해 볼 필요가 있다.
Full-length dystrophin
R16-17/∆C
micro-dystrophin ABD H1 R1 R16 R17 H3 R24 H4 CR
ABD H1 R1 R2 R3 H2 R4 R19 H3 R20 R21 R22 R23 R24 H4 CCRR CT
ITR
ITR CMV R16-17/∆C micro-dystrophin pA
rAAV genome
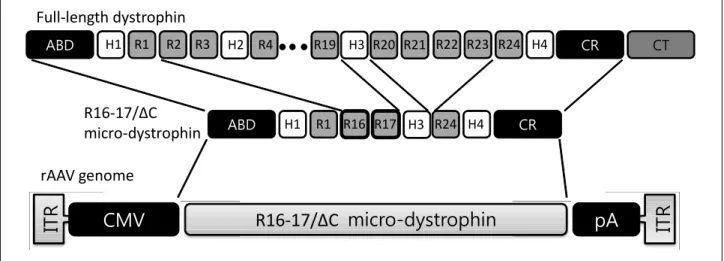
Figure 3. Structures of dystrophin, micro-dystrophin, and micro-dystrophin in recombinant AAV genome.
엑손 스키핑에 사용되는 핵산 유사체는 정맥 또는 피하 투여가 필요하며 근육 내에서 반감기가 짧아 자주 반복 투 여해야 하는 불편이 있다. 또한 심근 세포에 들어가지 않 는 것이 큰 단점으로 이를 극복하기 위해 peptide-con- jugated morpholino 등이 연구되고 있다.14 DMD의 정지돌연 변이에의 적용 가능성도 연구 중이며,15 팔다리이음 근디 스트로피(limb-girdle muscular dystrophy, LGMD) 2B 또는 미요시 근병증의 원인이 되는 dysferlin을 비롯하여 다른 유전자의 이상에도 같은 원리를 적용할 수 있을 것으로 예 상된다.16 하지만 어느 경우에나 환자 각각의 돌연변이에 따른 맞춤 설계가 필요하며, 모든 종류의 돌연변이에 적용 할 수는 없다는 제한이 있다.
3. 유전자 대체(gene replacement)
결손된 유전자를 대체해 넣는 것은 기능 결손(loss of function)에 의한 유전 질환을 치료하는 가장 단순 명쾌한 발상이며 유전자 치료의 원형이라 할 수 있다. 진핵세포에 유전 물질을 전달하는 방법으로는 바이러스 매개체를 이 용하는 것이 가장 효율적이며, 이 중 주목 받고 있는 것은 parvovirus의 일종인 아데노 유반 바이러스(adeno-associated virus, AAV)이다.17
아데노 바이러스의 오염체로서 발견된 AAV는 단독 증 식이 불가능하여 증식을 위해 아데노 바이러스나 헤르페 스 바이러스의 공감염이 요구된다. 또한 유전자 재조합을 통해 양단의 역위 반복 서열(inverted terminal repeat, ITR) 을 제외한 모든 바이러스 유전체를 제거하고 목적 유전자 로 치환해 넣을 수 있어, 유전자 치료 시 바이러스 유래 단 백질의 발현이 전혀 없다. 알려진 병원성이 없고 면역 반
응 유발이 경미하며 대부분 숙주 염색체에 삽입 없이 핵내 episome의 형태로 장기간 발현한다. 또한 비분열 세포를 잘 감염시키고 혈청형에 따라 골격근 또는 심근에 친화력 이 높아 근육 질환의 유전자 치료에 이상적인 매개체로 자 리매김 하고 있다.
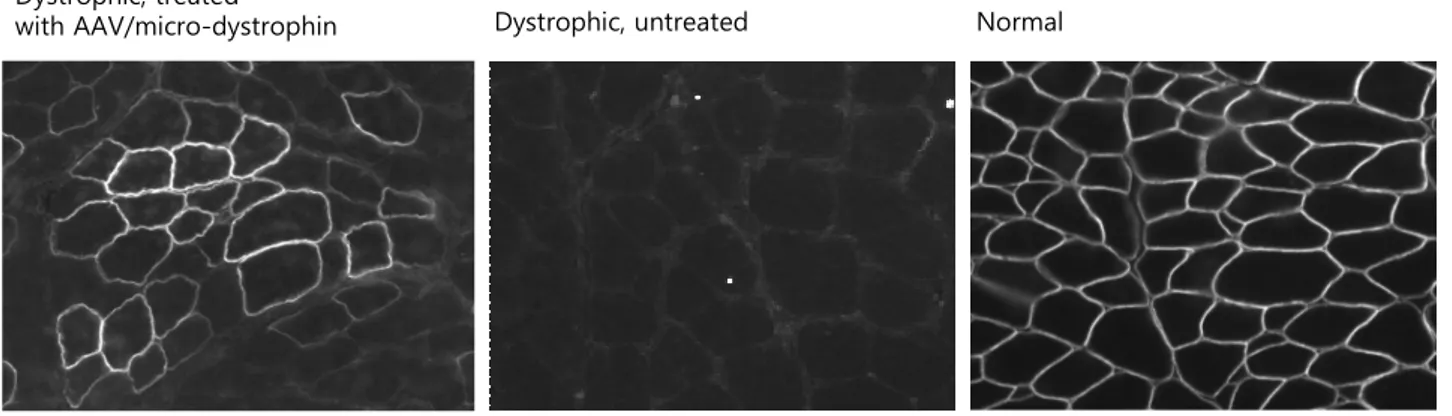
전달할 수 있는 유전자의 크기가 매우 제한되어 있는 것 은 AAV의 중요한 단점이다. 전체 4.9 kb 중 유전자 발현에 필수적인 부분을 넣고 나면 목표 유전자를 위해 할애할 수 있는 용량은 4.0 kb도 되지 않는데, 디스트로핀 유전자에서 단백질 합성에 직접 사용되는 서열만도 11kb로 이를 훌쩍 초과한다. 한편 디스트로핀 유전자는 대량 결실이 있어도 임상형이 매우 경미할 수도 있다는 것이 오래 전부터 알려 져 왔다. 이에 착안하여 디스트로핀에서 기능적으로 필수 적인 부분만 추출하여 AAV의 크기에 맞게 소형화하는 연 구가 시행되어 마이크로-디스트로핀(micro-dystrophin)이 개발되었다(Fig. 3).18 마이크로-디스트로핀은 마우스 모델 에서 성공적인 기능 회복을 이루었으며,19 DMD의 골격근 이상 뿐 아니라 심근병증에도 치료 효과를 나타내어 매우 고무적이라 할 수 있다(Fig. 4).20,21
AAV에 의한 유전자 치료는 마우스 모델에서는 면역 반 응이 거의 일어나지 않고 마우스의 수명 한도까지 발현이 잘 유지되나, 개 모델의 경우에는 저명한 면역 반응을 일 으키는 것이 확인되었다. 근육내 투여로 시행된 임상시험 에서도 유의한 마이크로-디스트로핀 발현이 관찰되지 않 았으며 이는 면역 반응에 의한 것으로 추정된다.22 면역 반 응은 전달된 마이크로-디스트로핀 또는 AAV 자체에 대한 반응일 것이다. 따라서 AAV를 이용한 마이크로-디스트로 핀의 유전자 치료는 면역 반응을 어떻게 효율적으로 제어 하는가에 앞으로의 성패가 달려 있을 것으로 예측된다.23
Dystrophic, treated
with AAV/micro-dystrophin Dystrophic, untreated Normal
Figure 4. Skeletal muscle of dystrophic and normal dogs stained with anti-dystrophin antibody.
한편 면역 반응에 의한 문제점을 해결하고 AAV를 이용 한 DMD의 유전자 치료가 성공할 경우, 엑손 스키핑의 경 우와 달리 개별 환자의 돌연변이 종류에 관계없이 적용 가 능하다. 다른 근디스트로피에의 적용 또한 무척 용이할 것 으로 예측되며 일부 LGMD에는 이미 임상시험이 진행 중 이다.24 Dysferlin과 titin을 제외한 상염색체 열성 LGMD의 원인 유전자들은 모두 별도의 소형화 작업 없이도 AAV로 운반 가능하다. Dysferlin의 경우 두 개의 AAV를 이용하여 전장의 dysferlin을 전달하는 방법이 연구되고 있다.25
결 론
지금까지 근디스트로피의 유전자 치료 분야에서 임상 시험 단계까지 도달한 몇 가지 방법들을 살펴보았다. 이 외에도 주목할 만한 유전자 치료로는, 근긴장성 근디스트 로피에서 RNA의 분해 또는 차폐 유도,26 비결손 단백질의 과발현을 통한 기능 개선,27 AAV를 이용한 엑손 스키핑28 등이 있다.
단일유전질환 중에 임상 승인을 받은 유전자 치료는 아 직 전 세계 어디에도 존재하지 않으며, 수년 이내 실용화 가능성에 있어서도 낙관적 전망과 비관적 전망이 혼재해 있는 실정이다. 하지만 유전자 치료는 근디스트로피를 비 롯한 희귀 난치성 질환의 새로운 치료로 매력적인 접근 방 법이며 포기할 수 없는 희망이기에 꾸준히 연구되어야 할 분야임에 틀림이 없다.29
REFERENCES
1. Sheridan C. Gene therapy finds its niche. Nat Biotechnol 2011;29:121-128.
2. Simonelli F, Maguire AM, Testa F, Pierce EA, Mingozzi F,
Bennicelli JL, et al. Gene therapy for Leber’s congenital amau- rosis is safe and effective through 1.5 years after vector administration. Mol Ther 2010;18:643-650.
3. Mendell JR, Rodino-Klapac L, Sahenk Z, Malik V, Kaspar BK, Walker CM, et al. Gene therapy for muscular dystrophy: Lessons learned and path forward. Neurosc Lett 2012 May 17 [Epub].
doi:10.1016/j.neulet.2012.04.078
4. Amrani N, Sachs MS, Jacobson A. Early nonsense: mRNA decay solves a translational problem. Nature reviews. Mol Cell Biol 2006;7:415-425.
5. Mendell JR, Buzin CH, Feng J, Yan J, Serrano C, Sangani DS, et al. Diagnosis of Duchenne dystrophy by enhanced detection of small mutations. Neurology 2001;57:645-650 .
6. Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, Al-Dahhak R, et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol 2010;67:771-780.
7. Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007;447:87-91.
8. Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol 2007;47:430-444.
9. Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dys- trophy mutation database: an overview of mutation types and par- adoxical cases that confirm the reading-frame rule. Muscle Nerve 2006;34:135-144.
10. Alter J, Lou F, Rabinowitz A, Yin H, Rosenfeld J, Wilton SD, et al. Systemic delivery of morpholino oligonucleotide restores dys- trophin expression bodywide and improves dystrophic pathology.
Nature Med 2006;12:175-177.
11. Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S, et al. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol 2009;65:667-676.
12. Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon skipping and dystrophin restoration in pa- tients with Duchenne muscular dystrophy after systemic phos- phorodiamidate morpholino oligomer treatment: an open-label,
phase 2, dose-escalation study. Lancet 2011;378:595-605.
13. Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med 2011;364:1513-1522.
14. Jearawiriyapaisarn N, Moulton HM, Sazani P, Kole R, Willis MS.
Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc Res 2010;85:444-453.
15. Yokota T, Duddy W, Echigoya Y, Kolski H. Exon skipping for nonsense mutations in Duchenne muscular dystrophy: too many mutations, too few patients? Expert Opin Biol Ther 2012 Jun 1 [Epub]. doi:10.1517/14712598.2012.693469
16. Aartsma-Rus A, Singh KH, Fokkema IF, Ginjaar IB, van Ommen GJ, den Dunnen JT, et al. Therapeutic exon skipping for dysferli- nopathies? Eur J Hum Genet 2010;18:889-894.
17. Weitzman MD, Linden RM. Adeno-associated virus biology.
Methods Mol Biol 2011;807:1-23.
18. Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarco- lemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest 2009;119:624-635.
19. Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med 2006;12:787-789.
20. Bostick B, Shin JH, Yue Y, Duan D. AAV-microdystrophin ther- apy improves cardiac performance in aged female mdx mice. Mol Ther 2011;19:1826-1832.
21. Shin JH, Nitahara-Kasahara Y, Hayashita-Kinoh H, Ohshima-
Hosoyama S, Kinoshita K, Chiyo T, et al. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther 2011;18:910-919.
22. Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med 2010;363:1429-1437.
23. Wang Z, Tapscott SJ, Chamberlain JS, Storb R. Immunity and AAV-mediated gene therapy for muscular dystrophies in large an- imal models and human trials. Front Microbiol 2011;2:201.
24. Herson S, Hentati F, Rigolet A, Behin A, Romero NB, Leturcq F, et al. A phase I trial of adeno-associated virus serotype 1-γ-sarcoglycan gene therapy for limb girdle muscular dystrophy type 2C. Brain 2012;135(Pt 2):483-492.
25. Lostal W, Bartoli M, Bourg N, Roudaut C, Bentaï A, Miyake K, et al. Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum Mol Genet 2010;19:1897-1907.
26. Krzyzosiak WJ, Sobczak K, Wojciechowska M, Fiszer A, Mykowska A, Kozlowski P. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res 2012;
40:11-26.
27. Shin JH, Bostick B, Yue Y, Hajjar R, Duan D. SERCA2a gene transfer improves electrocardiographic performance in aged mdx mice. J Trans Med 2011;9:132.
28. Goyenvalle A, Babbs A, Wright J, Wilkins V, Powell D, Garcia L, et al. Rescue of severely affected dystrophin/utrophin-deficient mice through scAAV-U7snRNA-mediated exon skipping. Hum Mol Genet 2012;21:2559-2571.
29. Wilson JM. It’s time for gene therapy to get disruptive! Hum Gen Ther 2012;23:1-3.