저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
The role of ATM responding to DNA damage induced by
Xrcc1 deficiency during oligodendrocyte genesis
by
Inseo Choi
Major in Cancer Biology
Department of Biomedical Sciences
The Graduate School, Ajou University
The role of ATM responding to DNA damage induced by
Xrcc1 deficiency during oligodendrocyte genesis
by
Inseo Choi
A Thesis Submitted to The Graduate School of Ajou
University in Partial Fulfillment of the Requirements for
the Degree of Master of Biomedical Sciences
Supervised by
Youngsoo Lee, Ph.D
Major in Cancer Biology
Department of Biomedical Sciences
The Graduate School, Ajou University
This certifies that the dissertation of
Inseo Choi is approved.
SUPERVISORY COMMITTEE
Hyeseong Cho
Jae-Ho Lee
Youngsoo Lee
The Graduate School, Ajou University
i -ABSTRACT-
The role of ATM responding to DNA damage induced by Xrcc1
deficiency during oligodendrocyte genesis
Inseo Choi
Department of Biomedical Sciences, Major in Cancer Biology The Graduate School, Ajou University
(Supervised by Assistant Professor Youngsoo Lee)
Ataxia telangiectasia (A-T) is a prime example of hereditary disease about DNA damage signaling and repair protein deficiency. A-T is an autosomal recessive neurodegenerative disorder associated the defective ATM (Ataxia telangiectasia mutated) gene that is estimated to affect 1 in 40,000-300,000 people. The clinical features manifested in A-T patients are multisystem symptoms, including cerebellar atrophy, ataxia, abnormal eyes movement, telangiectatic blood vessel, immunodeficiency and hyper-radiosensitivity. Currently there is no cure and no way to slow neuropathological progression of the A-T patients. Because, different from other animal models to mimic diseaseswith DNA repair protein deficiency, ATM-/- mice do not recapitulate the phenotypes of A-T patients, making it difficult to study
the mechanism of neurological phenotypes of this disease in mice.
To make theproper mice model for A-T, we generated Xrcc1 and Atmdouble knockout mice (hereafter referred to as Xrcc1nes-cre; Atm-/-) to determine the function of DNA damage sensor
kinase Atm upon persistentDNA damage condition by Xrcc1 inactivation during brain development.Xrcc1 is a scaffolding protein that is recruited to DNA single strand break site
ii
with several interacting proteins such as PARP1, DNA polymeraseβ and Ligase 3. Loss of Xrcc1 led to the persistence of endogenous DNA strand breaks. Xrcc1nes-cre; Atm-/- mouse
recapitulates the neurological phenotypes of A-T patients including cerebellar ataxia, cerebellar atrophy, defect of Purkinje cell and dysmyelination in the brain. In the case of dysmyelination, this is a subset of white matter disorders characterized by differentiation defect of oligodendrocytes, also known as leukodystrophy.
We found that neurons and astrocytes in the Xrcc1nes-cre; Atm-/- brain underwent normal
differentiation and maturation in vivo and in vitro. But, fully mature oligodendrocyte markers, MBP, PLP and CNPase were downregulated in the Xrcc1nes-cre; Atm-/-mouse cortex
and cerebellum. But, the preceding state of fully mature oligodendrocytes, oligodendrocyte progenitor cells (OPC) were intact without any defect such as cell cycle progression and cell death in vivo and in vitro. This outcome was affected by the low protein levels of the oligodendrocyte transcription factor, Olig1.Taken together, our data demonstrate Atm is critical for the proper development of cerebellum and neural cell differentiation, especially oligodendrocytes.
iii
TABLE OF CONTENTS
-ABSTRACT-………...i TABLE OF CONTENTS………..ii LIST OF FIGURES………..v LIST OF TABLES………...………vi I. INTRODUCTION……….1II.MATERIAL AND METHODS………7
A. Generation of Xrcc1 and Atm knockout animal model………..7
B. Isolation and differentiation of primary mouse neural stem cell………..10
C. Determination of cell number in proliferation………..10
D. Antibodies for histology and western blot………11
E. Immunohistochemistry………..11
F. TUNEL assay……….12
G. Protein extraction (nuclear/cytosol) and western blot analysis……….12
H. RNA extraction and quantitative real time PCR……….……..13
iv
1. Double inactivation of Xrcc1 and Atm in the central nervous system………..15
2. Loss of Xrcc1 and Atm results in DNA repair deficiency in the brain………..18
3. Cerebellar interneurons are not rescued in the Xrcc1nes-cre; Atm-/- brain…………...….19
4. Purkinje cells are susceptible to Xrcc1 and Atm loss………23
5. Xrcc1 and Atm required for oligodendrocyte differentiation………26
6. Oligodendrocyte progenitor cell formation in the Xrcc1nes-cre; Atm-/- brain…………...28
7.Oligodendrocyte specific transcription factor Olig1 is affected by Atm signaling…...37
IV. DISCUSSION………...………41
V. REFERENCES………...47 -국문요약-………..54
v
LIST OF FIGURES
Figure 1. Strategy of generating Xrcc1 and Atm knockout mouse model
………16
Figure 2. Generation of an Xrcc1 and Atm double knockout mouse
……….17
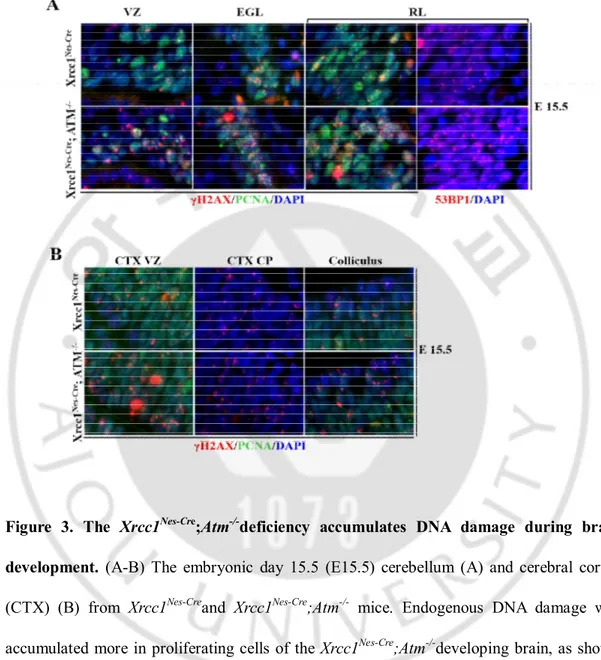
Figure 3. The Xrcc1nes-cre; Atm-/- deficiency accumulates DNA damage during brain development...……….20
Figure 4. Atm inactivation does not rescue cerebellar interneuron loss in the Xrcc1nes-cre
cerebellum………..……….22
Figure 5. Cerebellar atrophy and astrogliosis in the Xrcc1nes-cre; Atm-/- mice brain
...………..24
Figure 6. Fully mature Oligodendrocytes are missing in the Xrcc1nes-cre; Atm-/- brain
………...29-31
Figure 7. Oligodendrocyte progenitor cells exist in the Xrcc1nes-cre; Atm-/-
………..34-36
Figure 8.Oligodendorcye specific transcription factor might be affected by Atm signaling ………..39-40
vi
LIST OF TABLES
Table 1. Human neurological diseases linked to mutation in DNA repair gene and its mouse model ...……….3
1
I. INTRODUCTION
Thegenome is the genetic material for all living organisms. It is the entire set of hereditary information for building, running, and maintaining an organism and transmitting its informationtonext generation. Transmission of genetic information is constantly in a selective balance between the maintenance of genetic stability and elimination of mutational change. The genome is made of chemical DNA (Deoxyribonucleic acid). The DNA molecule is being continuously exposed to various types of damage caused by exogenous source (e.g. ionizing radiation, ultraviolet light from sunlight, chemicals, and pollutants) and endogenous sources (e.g. stalled forks during DNA replication and reactive oxygen species during respiration) during our lifetime. From the structural point of DNA damage lesions, it can be divided into two types of DNA damage such asDNA single strand break and DNA double strand breaks under large perspective (De Bont and van Larebeke, 2004; Lopez-Contreras and Fernandez-Capetillo, 2012). The quantity of lesions that every cell faces is up to 105
spontaneous or induced DNA lesion per day (Ames et al., 1993; Nakamura et al., 1998; Vilenchik and Knudson, 2003; De Bont and van Larebeke, 2004). It estimates that each individual could deal withmore than 105spontaneous or induced DNA lesions per day. To
counteract DNA damage, eukaryotic cells have developed a complex signaling network of repair processes known as DNA damage response to recruit and activate the right factors in the right place at the right time (Ciccia and Elledge, 2010).
2
DNA repair is extremely important in the nervous system because DNA damage occurs spontaneously during brain development and in the adult brain, (Lu et al., 2004; Shull et al., 2009; Suberbielle et al., 2013). In the nervous system unrepaired DNA lesions and mutations can have an enormous effect on the formation of a functional nervous system (McKinnon, 2013). Defects in cellular DNA damage signaling and repair processes can lead to many human hereditary syndromes(Table 1). Representatively, Ataxia oculomotor apraxia (AOA1) and Spinocerebellar ataxia with axonal neuropathy (SCAN1) are related to disfunctional DNA single strand break repair proteins. AOA1 and SCAN1 result from mutations in the Aprataxin (APTX) and Tyrosyl-DNA phosphodiesterase 1 (TDP1) genes respectively. APTX acts for repairing 5’-end resulting from abortive DNA ligation reactions. TDP1 is an enzyme required for the 3’-end tyrosyl DNA procession after abortive Topoisomerase 1 (Top1) mediated torsional stress induced DNA single strand break. Theconsequence of defects in APTX orTDP1 ischaracterized by cerebellarataxia and sensorimotor neuropathy (McKinnon and Caldecott, 2007; Rass et al., 2007). An interesting feature is that the pathology in these disorders is almost exclusively restricted to the nervous system implying the importance of maintaining genomic stability during brain development.
3
Table 1. Human neurological diseases linked to mutation in DNA repair gene and its mouse model
The reports of mouse model are listed below. 1)(El-Khamisy et al., 2009),
2) (Katyal et al., 2007), 3) (Barlow et al., 1996), 4) (Buis et al., 2008), 5) (de Klein et al., 2000), 6) (Murga et al., 2009), 7) (Zhu et al., 2001),
4
Ataxia telangiectasia (A-T) is another prime example of the relationship between DNA damage responsedefect and neurodegenerative disorder. A-T is caused by mutation in the ATM gene (Ataxia Telangiectasia mutated). ATM is a serine/threonine (Ser/The) protein kinase and belongs to the phosphoinositide 3-kinase (PI3K) family, which is also includes ATR (Ataxia telangiectasia and Rad3-related protein) and DNA-PKcs (catalytic subunit of DNA-dependent protein kinase) (Savitsky et al., 1995). These two proteins are also involved in DNA damage responses. ATM is rapidly recruited to DNA double strand break (DSB) sites and phosphorylates many downstream substrates to activate the cellular DSB responses including DNA damage repair, cell cycle checkpoint and apoptosis (Shiloh and Ziv, 2013). The well-known tumor suppressor; p53 was the first substrate of ATM identified in vitro and
in vivo(Banin et al., 1998; Canman et al., 1998; Khanna et al., 1998). In A-T cells (ATM
deficient human cells), the stabilization and activation of p53 was defected, and these cells were characterized by defective G1-S checkpoint, in which p53 has a central role (Kastan et al., 1992; Khanna and Lavin, 1993).
A-T is an autosomal recessive disorder that is estimated to affected 1 in 40,000-100,000 people (Crawford, 1998). It has been reported that the 432 different mutations in the ATM gene without any hotspots(Lavin et al., 2007). This is an early childhood onset disease at aroundage 12 months-2 years, when ataxia of gait and posture is an important sign of the clinical features as the child begins to walk (Boder and Sedgwick, 1958). The clinical features of A-T patients are sterility, chromosomal instability, immunodeficiency and neurologic impairments including cerebellar ataxia and abnormal eye movement (oculomotor apraxia). Affected persons also manifest both sensitivity to ionizing radiation
5
and cerebellar atrophy (Taylor et al., 1975; Cabana et al., 1998; Crawford et al., 2000). In many clinical reports, A-T patients showed dysmyelinated white matter in the brain called leukodystrophy (Chung et al., 1994; Sardanelli et al., 1995; Opeskin et al., 1998; Habek et al., 2008). Up to 30% of A-T patients develop lymphoid tumors since ATM plays a critical role in the differentiation of T and B cells (Lumsden et al., 2004). At present there is no cure and no way to slow the neuropathological progression of the A-T patients. It is possible to alleviate some of the symptoms associated with immunodeficiency (Lavin et al., 2007).
A slow progress has been made in the research of neuropathologicalphenotypes of human A-T patients. A great deal of knowledge to understand neurological defects in human diseases comes from analyzing proper animal models that mimic neurological phenotypes in human. However, unfortunately most of animal models for A-T did not show any sign of neuropathology such as ataxia and neurodegeneration. One of possibilities is that the kind of endogenous DNA damage to induce ATM signaling is different from what has been reported.
To investigate the ATM function for maintaining genomic stability during brain development, we generated anXrcc1 and Atmdouble inactivatedmouse model using a tissue specific gene deletion system. We hypothesized that chronic DNA damage by Xrcc1 inactivation to block both base excision repair (BER) and DNA single strand break repair (SSBR) triggers Atm dependent signaling for homeostasis during brain development. Xrcc1 interacts with various DNA repair factors, including PARP1, APTX, DNA polymerase β, DNA ligase 3 and indirectly TDP1(McKinnon and Caldecott, 2007). Inactivation of Xrcc1 couldinducechronic genotoxic stress due to unrepaired endogenous DNA damage during brain development (Lee et al., 2009). Bysimultaneous inactivation of Xrcc1 and Atm in the
6
mouse, we examined the function of Atmto maintain genomic stability during brain development. We found that Atmis required for proper formation of the cerebellar structure and full differentiation of oligodendrocytes. We also found that in cerebellar interneuron progenitor cells, p53, one of the ATM substrates, could be regulatedby another protein kinase rather than ATM upon DNA damage induced by XRCC1 deficiency. The more important point we want to make clear is that in contrast with existing A-T mouse models, our Xrcc1 and Atmdouble knockout mouse showed many similar neurological phenotypesto those observed inhuman A-T patients. Our new A-T mouse model is quite valuable to reveal the molecular mechanisms and etiology of A-T neural phenotypes, and to establish brain specific cellular function of ATM and its signaling pathway.
7
II.MATERIALS AND METHODS
A. Generation of Atmand Xrcc1 knockout animal model
Atm and Xrcc1 mouse lines have been previously reported (Herzog et al., 1998; Lee et al., 2009). To inactivate simultaneously the Atmand Xrcc1 genes in the nervous system, we bred
Xrcc1loxP/loxP; ATM+/- with Nes-cre mice. Xrcc1Nes-cre; Atm-/- mice were obtained by crossing
male Xrcc1loxP/loxP; Atm+/-;orXrcc1loxP/+; Atm+/- mice and femaleXrcc1loxP/loxP; Atm+/- ;Nestin-cre orXrcc1loxP/+; Atm+/-; Nestin-cremice. We maintained Nes-creexpression only in females
to avoid ectopic expression of Cre recombinase.Littermates were used as controls including
Xrcc1+/+; Atm+/+(wildtype: WT), Xrcc1loxP/+; Atm+/+, Xrcc1loxP/loxP; Atm+/+, Xrcc1+/+; Atm+/-, and Xrcc1loxP/+; Atm+/-.Atmheterozygotes (Atm+/-)and Xrcc1loxP+; Nes-cre animals did not
show any difference from WT. The presence of a virginal plug was considered embryonic day 0.5 (E0.5) and the day of birth as postnatal day 0 (P0).
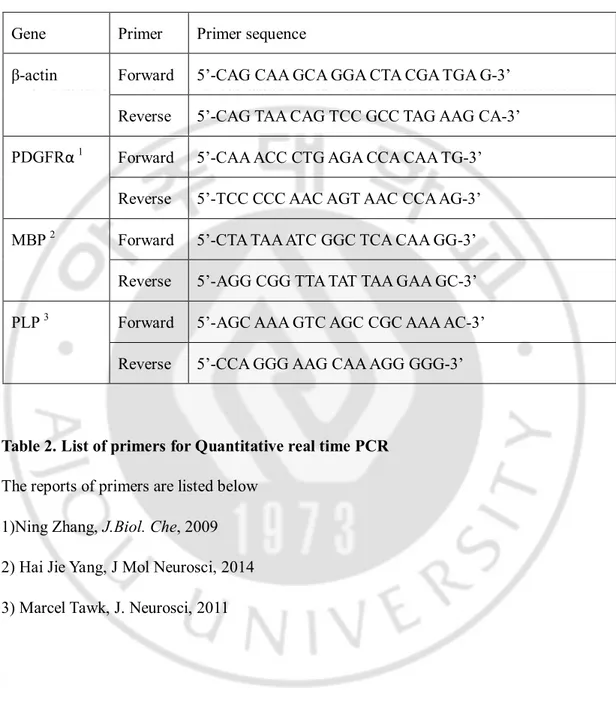
The genotypes of the mice were determined by PCR (polymerase chain reaction). Primer sequences for the Xrcc1, Cre, andAtmgenes are listed in below (Table 2).
l PCR for the Xrcc1 gene
WT (Xrcc1+/+) and Floxed (Xrcc1loxP/loxP) alleles
A combination of primers is PC1 and PC2:WT (245bp) and the Xrcc1loxP/loxPgene (279bp)
PCR products were amplified for 35cycles of 94℃ for 30 sec, 58℃ for 45 sec and 72℃ for 45 sec.
8
l PCR for the Cre recombinase gene Cre recombinaseallele
A combination of primers isCre3 and Cre4
PCRproduct were amplified for 36 cycles of 94℃ for 30 sec, 60℃ for 45 sec and 72℃ for 45 sec.
l PCR for the Atm gene
WT allele of the Atm gene
A combination of primers Atm WT1 and WT2
PCRproduct were amplified for 36 cycles of 94℃ for 1 min, 60℃ for 1 min and 72℃ for 1 min.
Mutant allele of the Atm gene
A combination of primers Atm Atm4380 and Neo5
PCRproduct were amplified for 36 cycles of 94℃ for 1 min, 55℃ for 1 min and 72℃ for 1 min.
9 Gene Primer Primer sequence
XRCC1 PC1 5’-TAT GCT TGC TGT ACA GGG ATT GGG-3’ PC2 5’-TGG ACC ATG AAA AAG CTG TGT GC-3’ Cre Cre3 5’-CTG CCA CGA CCA AT GAC AGC-3’
Cre4 5’-ACC TGC GGT GCT AAC CAG CG-3’
Atm WT WT1 5’-GCC TGT TAT CTT CTA TGT GCA CCG TCT TCG C-3’ WT2 5’-GGT GCG GTG TGG ATG GGA CTG GAG G-3’
Atm Mutant Atm4380 5’-GTG ATG GAC CTG AGA CAA GAT GCT GTC-3’ Neo 5 5’-GGG AAG ACA ATA GCA GGC ATG C -3’
10
B. Isolation and differentiation of primary mouse neural stem cell.
Primary neural stem cells (NSC) were purified from embryonic brains at embryonic day 14 (E14.5).The embryonic forebrain was dissected after removingthe scalp, skull and meninges. The isolated forebrain was triturated mechanically into a single cell-suspension using 1ml micropipette set at approximately 950μl, 200μl micropipette set at approximately 190μl and polished Pasteur pipettes gently by piptetting up and down 20 times each. Single NSCs were cultured in neural stem cell basal medium (STEM CELL technology) supplemented with proliferation supplements (STEM CELL technology) and 20ng/mlEpidermal growth factor (EGF, STEM CELL technology) in T-25 tissue culture flasks (SPL) until neurospheres were established. Neurospheres were split every 6~7 days. For in vitroNSC differentiation assay, E14.5 neural stem cells were cultured at 1× L-ornithine (Sigma) coated plate and in neural stem cell basal medium (STEM CELL technology) supplemented with differentiation supplements (STEM CELL technology). Differentiation medium was partially replaced every 2~3 days. The status of differentiation was checked on DIV (days in vitro) 14 or DIV 21. Both proliferation and differentiation culture were established at 37 ℃ in a humidified, CO2-regulated (5%) incubator.
C. Determination of cell number in proliferation
Neural stem cells were plated at 5x104cells/well in neural stem cell basal medium
11
STEM CELL technology). Neurospheres were collected at Day 7 after plating and then dissociated to the single cell level, and total single neural stem cell numbers were counted using a hemocytometer.
D. Antibodies for histology and western blot
For immunohistochemistry and immunocytochemistry, we used antibodies to calbindin D28K (mouse, 1:2000, Sigma), GABA receptor α6 (rabbit, 1:500, Chemicon), GFAP (mouse 1:1000, sSigma), Pax2 (rabbit, 1:500, Zymed), gH2AX (rabbit, 1:500, Abcam), 53BP1 (rabbit, 1:500, Bethyl Laboratory), PCNA (mouse, 1:500, Santa Cruz), Olig2 (rabbit, 1:1000, Millipore), PDGFRα (rabbit, 1: 1000, Santa Cruz), MBP (rabbit, 1:500, Abcam). The antibodies listed above were used with a citrate buffer based antigen retrieval treatment.
This antibody was used without antigen retrieval: Olig1 (rabbit, 1:1000, LS Bio).
For western blot, we used antibodies to NeuN clone A60 (mouse 1:1000, Millipore), Actin (mouse, 1:5000, Abcam), CNPase (mouse, 1:1000, Sigma), Olig1 (rabbit, 1:1000, LS Bio), Olig2 (rabbit, 1:1000, Millipore).
E. Immunohistochemistry
Embryos and brain tissues were removed at indicated ages after cardiac perfusion with 4% (wt/vol) buffered paraformaldehyde, cryoprotected in buffered 25% sucrose (wt/vol) solution and sectioned at 10 μm using an MEV cryostat (SLEE).
12
Immunohistochemical analysis of tissue and cells, were perform using the antibodies listed above. Sections were incubated with antibodies overnight after quenching endogenous peroxidase using 0.6% (vol/vol) H2O2 in methanol. After washing slides with
Phosphate-buffered saline (PBS) two times, the tissues were treated with biotinylated secondary antibody (Jackson ImmunoResearch) and avidin-biotin complex (Vector Labs). Immunoreactivity was amplified with VIP substrate kit (Vector labs). Sections were counterstained with 0.5% methyl green, dehydrated and mounted in DPX (Sigma). For fluorescence immunoreactions of sections, FITC- or Cy3 conjugated Secondary antibodies (Jackson ImmunoResearch) were used and counterstained with DAPI (Vector Labs). All stained slides were examined and imaged using an OPTIKA B-600TiFL equipped with a Leica DFC310 FX digital camera.
F. TUNEL assay
Embryos and brain tissues were labeled with TdT enzyme and Anti-digoxigenin conjugate antibody to detection of cell death using ApopTag®Fluorescine In situ Apoptosis Detection
Kit S7110 (Millipore) following manufacturer’s instructions.
G. Protein extraction (nuclear/cytosol) and western blot analysis
Tris-13
HCl, 150mM NaCl, 50mM NaF, 1% Tween-20 (vol/vol), 0.2% NP-40, 1mM DTT,phosphatase inhibitor cocktail 2 (Sigma), Protease inhibitor tablet (Sigma)). AlsoNucleus and Cytosol protein were extracted from P14 mouse brains using Nuclear/Cytosol Fractionation Kit (Biovision). All protein samples were quantified by Bradford assay (Bio-Rad).Proteins (30μg per lane or 50μg per lane) were separated through a Bis-Tris 10% (wt/vol) polyacrylamide gel and transferred in to nitrocellulose membrane (GE Healthcare).
Primary antibodies were listed above. Secondary antibodies were used Goat- anti mouse, anti- rabbit HRP-conjugated antibody (Bio-Rad) and detected using ECL (Bio-Rad). Actin antibody (1:2500, Santa Cruz Biotech) served as a protein-loading control. For the nuclear extracts, H3 antibody was used for fractionation efficiency and loading control.
H. RNA extraction and quantitative real time PCR
Total RNA was extracted from the brain tissue or Neural stem cells using an TRIzol reagent (Invitrogen). To generate cDNA, 4 μg of total RNA was used in 20 μl of revers transcription reaction, using Super-ScriptⅢ RT-PCR Kit (Invitrogen). Quantitative real time PCR was performed using Rotor-Gene SYBR Green RT PCR Kit (Qiagen). Data were normalized to the internal control b-actin and analyzed DDCt method. Primer list used in quantitative PCR for mouse are given in below table (table 3).
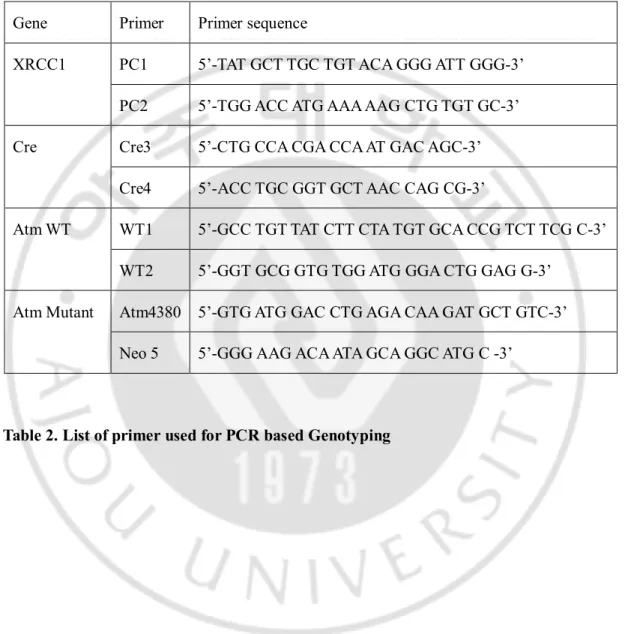
14 Gene Primer Primer sequence
β-actin Forward 5’-CAG CAA GCA GGA CTA CGA TGA G-3’ Reverse 5’-CAG TAA CAG TCC GCC TAG AAG CA-3’ PDGFRα 1 Forward 5’-CAA ACC CTG AGA CCA CAA TG-3’
Reverse 5’-TCC CCC AAC AGT AAC CCA AG-3’ MBP 2 Forward 5’-CTA TAA ATC GGC TCA CAA GG-3’
Reverse 5’-AGG CGG TTA TAT TAA GAA GC-3’ PLP 3 Forward 5’-AGC AAA GTC AGC CGC AAA AC-3’
Reverse 5’-CCA GGG AAG CAA AGG GGG-3’
Table 2. List of primers for Quantitative real time PCR
The reports of primers are listed below 1)Ning Zhang, J.Biol. Che, 2009 2) Hai Jie Yang, J Mol Neurosci, 2014 3) Marcel Tawk, J. Neurosci, 2011
15
III.RESULTS
1.Double inactivation of Xrcc1 and Atm in the central nervous system
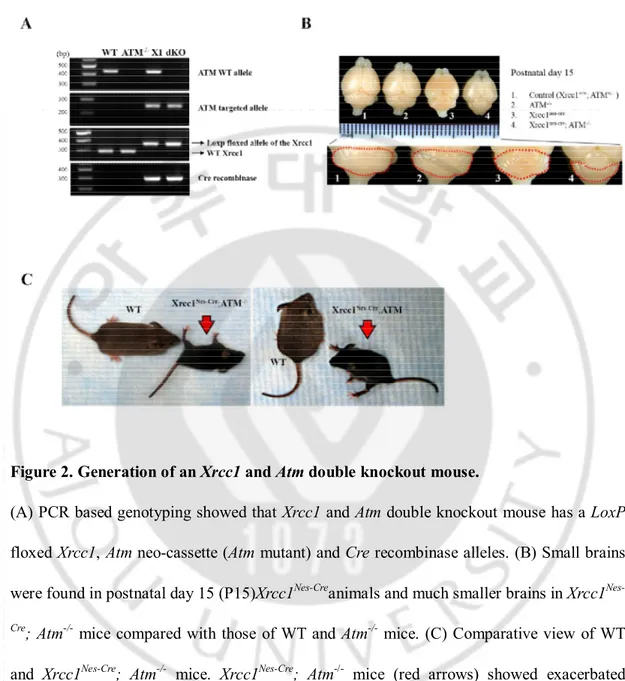
Deletion of theXrcc1 gene increased the number of endogenous DNA Single strand breaks(Khoronenkova and Dianov, 2015). To determine the role of ATM upon DNA damage during brain development, we generatedXrcc1 and Atm double knockout mice.Xrcc1 germline knockout mice showed early embryonic lethality before brain development (Tebbs et al., 1999). To resolve this, we used a Nestin-Cre system to generate Xrcc1conditional knockout mice which induce tissue specific DNA damage, particularlyin the central nervous system during development.This Xrcc1conditional knockout mouse modelhasbeen previously described (Lee et al., 2009). Xrcc1conditionally targeted mice (hereafter referred to as Xrcc1nes-cre) were crossbred with Atm germline knockout mice (hereafter referred to as Atm-/-) (Herzog et al., 1998)(Figure 1). Generated Xrcc1 and Atm double knockout mice
(hereafter referred to as Xrcc1nes-cre; Atm-/-) died beforepostnatal week 3. The genotype of
mouse in these experimentswas confirmed at the DNA level (Figure 2A). Xrcc1nes-cre; Atm
-/-mice had smaller brain than those of littermates of WT and Atm-/- especially the cerebellum
(Figure 2B). Gross behavior observation indicated that unlike WT and Atm-/- mice, Xrcc1 nes-creanimals showed mild ataxia with episodic spasms as previously reported(Lee et al., 2009).
StrikinglyXrcc1nes-cre; Atm-/- animals showed severe ataxia and exacerbated neurological
16
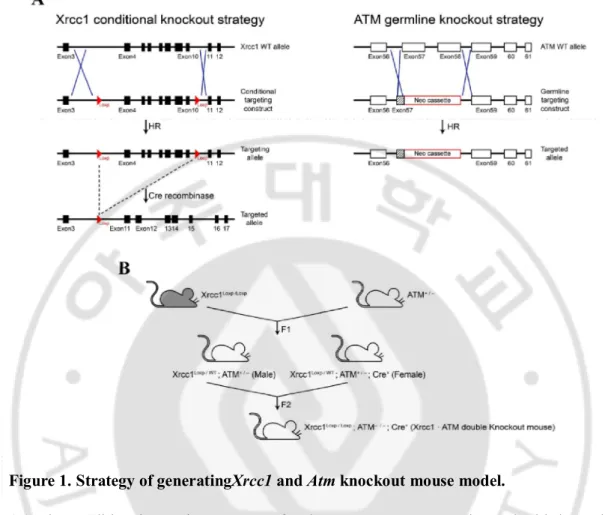
Figure 1. Strategy of generatingXrcc1 and Atm knockout mouse model.
(A) The conditional targeting construct for the Xrcc1 gene was engineered with loxP sites flanking exons 4-10. The Atm germline targeting construct was engineered with Neo gene to interrupt exon 57 and replace a part of exon 58 of the Atm gene. The targeted region is the phosphoinositol 3-kinase domain (PI3K domain) which is catalytic activity domain of Atm. (B) To inactivate the Xrcc1 and Atmgene in the nervous system, first we gained the
Xrcc1loxP/+;Atm+/- mice. And then Xrcc1loxP/loxP;Atm-/-;Nestin-Cre+mice were obtained
bybreeding male Xrcc1loxP/+;Atm+/-with femaleXrcc1loxP/+;Atm+/-;Nestin-Cre+mice.We
17
Figure 2. Generation of an Xrcc1 and Atm double knockout mouse.
(A) PCR based genotyping showed that Xrcc1 and Atm double knockout mouse has a LoxP floxed Xrcc1, Atm neo-cassette (Atm mutant) and Cre recombinase alleles. (B) Small brains were found in postnatal day 15 (P15)Xrcc1Nes-Creanimals and much smaller brains in Xrcc1 Nes-Cre; Atm-/- mice compared with those of WT and Atm-/- mice. (C) Comparative view of WT
and Xrcc1Nes-Cre; Atm-/- mice. Xrcc1Nes-Cre; Atm-/- mice (red arrows) showed exacerbated
18
2. Loss of Xrcc1 and Atm results in DNA repair deficiency in the brain
Xrcc1 and ATM are critical for DNA single strand break repair and double strand break repair respectively. To estimate the role of Xrcc1 and Atmin repairing DNA damage in the nervous system, we analyzed the brains of Xrcc1nes-cre; Atm-/-mice for the accumulation of
DNA strand breaks. We used gH2AXas a marker for DNA damage that could be detected as foci formation in the nucleus. gH2AXis the phosphorylatedform of histone H2AXthatis accumulated at the sites of DNA strand breaks to act as a sensor for DNA damage and to initiate DNA damage repair responses(Rogakou et al., 1998).Although gH2AX typically involves in DNA double strand breaks, DNA single strand breaks by Xrcc1 deficiency can be converted to DNA double strand break that arise during replication or random damage accumulation leading to adjacent breaks.
During early neural development,there was widespread accumulation of DNA damage throughout theXrcc1nes-cre; Atm-/-mouse brain more than that of the Xrcc1nes-cre
brain.gH2AXfoci formation occurred in proliferating cells in the various areas of both
Xrcc1nes-creand Xrcc1nes-cre; Atm-/- mice brain.WhenXrcc1nes-creand Xrcc1nes-cre; Atm-/- animals
were compared,the Xrcc1nes-cre; Atm-/-brain showedmore accumulation of gH2AX signal than
the Xrcc1nes-crebrain, suggesting that Xrcc1nes-cre; Atm-/- animals faced increased challenge to
DNA damage and by inactivating Atm, the load of DNA damage was affected (Figure 3A,B). Moreover, these DNA strand breakswas not repaired until proliferating cells becoming post-mitotic differentiated cells.WT andAtm-/-mice brain did not show any noticeable gH2AX
19
shown).This sign of DNA damage accumulation was also confirmed with 53BP1 foci; another DNA damage marker (Figure 3A). 53BP1 is one of many early responding proteins to DNA damage which is phosphorylated upon DNA damage, and could be detected as foci formation in the nucleus (Dimitrova et al., 2008).
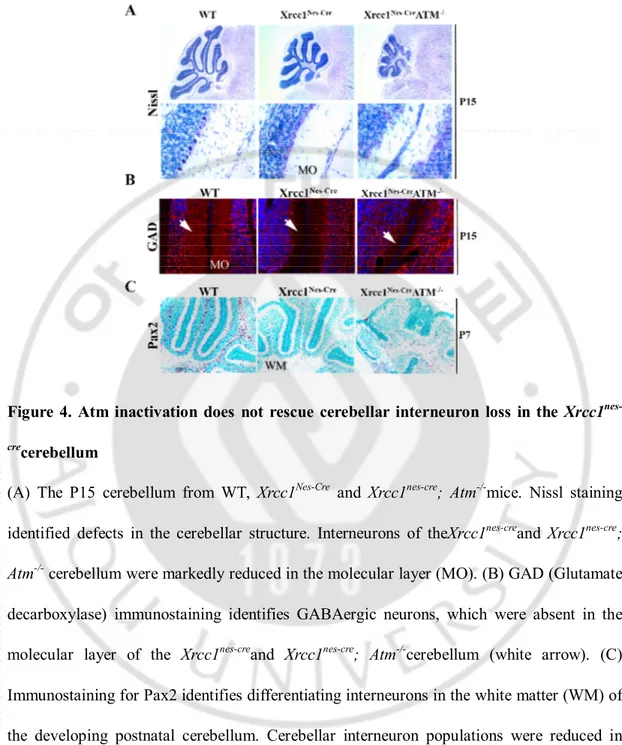
3. Cerebellar interneurons are not rescued inthe Xrcc1nes-cre; Atm-/-brain
To analyze the function of ATMformaintenance of genomic stability during brain development,we had surveyed the Xrcc1nes-cre; Atm-/- mouse brain using various markers of
differentiated neural cells. In the previous study, Xrcc1nes-crebrains showed profound and
widespread loss of cerebellar interneurons in the white matter (Lee et al., 2009). Interneuron progenitors of the Xrcc1nes-crecerebellumunderwent DNA damage induced p53 dependent cell
cycle arrest rather than apoptosis resulting in absence of interneurons. To confirm this, theXrcc1nes-cre; p53-/-mouse had been generated and rescue of interneuron loss in the
cerebellum was observed. p53 wasidentified as the first substrate of ATM in vitro and in
vivo(Banin et al., 1998; Canman et al., 1998; Khanna et al., 1998).DNA damage triggers
several signal transduction pathways that lead either to damage repair coupled with attenuation of cell cycle arrest, or to programmed cell death (apoptosis) (Choi et al., 2012; Sullivan et al., 2012). Upon DNA damage, p53 is stabilized and activated through various posttranslational modifications including phosphorylation by ATM(Banin et al., 1998).
20
Figure 3. The Xrcc1Nes-Cre;Atm-/-deficiency accumulates DNA damage during brain
development. (A-B) The embryonic day 15.5 (E15.5) cerebellum (A) and cerebral cortex
(CTX) (B) from Xrcc1Nes-Creand Xrcc1Nes-Cre;Atm-/- mice. Endogenous DNA damage was
accumulated more in proliferating cells of the Xrcc1Nes-Cre;Atm-/-developing brain, as shown
by γH2AX and 53BP1 foci formation in the nucleus. Nuclei were stained with DAPI. VZ, Ventricular Zone; EGL. External granule cell layer; RL, rhombic lip; CP, cortical plate.
21
Thus, we had speculatedthatXrcc1 and Atm deficiency also rescued p53-dependent cell-cycle arrest in the cerebellar interneurons similar toXrcc1nes-cre; p53-/-cerebellar phenotype.
Histopathological analysis of both the Xrcc1nes-cre and Xrcc1nes-cre; Atm-/-cerebellum showed
the dramatic reduction in size and disruption of the cellular architecture of the cerebellum in the Xrcc1nes-cre; Atm-/-animals (Figure 4A). Furthermore, there was no sign of proper
interneuron genesis in the Xrcc1nes-cre; Atm-/-cerebellum.So in contrast tothe Xrcc1nes-cre; p53
-/-cerebellum, cerebellar interneurons were not rescued in the Xrcc1nes-cre; Atm-/-cerebellum.
These interneurons are critical for attenuating the output signal of granule and Purkinje cell thereby maintaining the balance of electrical activity between cerebellar neurons (Barmack and Yakhnitsa, 2008). The cerebellumcontains five different types of interneurons: the Stellate cell, Basket cell, Golgi cell, Lugaro cell and Unipolar brush cell(Harriman; John C. Eccles, 1967; Voogd and Glickstein, 1998; Ramnani, 2006).The Stellate and Basket cells are inhibitory GABAergic interneurons, found in the molecular layer of the cerebellum. Accordingly, we found thatthe expression level of Glutamic acid decarboxylase (GAD), the enzyme that catalyzes synthesis of g-aminobutyric acid (GABA) was almost lack in the
Xrcc1nes-cre and Xrcc1nes-cre; Atm-/-cerebellum (Figure4B). Cerebellar interneurons are
originated from progenitors in the white matter of the cerebellum and begin to differentiateafter birth(Yamanaka et al., 2004; Weisheit et al., 2006). To define the differentiation stage of interneurons, we measured cerebellar interneurons in differentiation using a interneuron early differentiation maker; Pax2 immunostaining.
22
Figure 4. Atm inactivation does not rescue cerebellar interneuron loss in the Xrcc1 nes-crecerebellum
(A) The P15 cerebellum from WT, Xrcc1Nes-Cre and Xrcc1nes-cre; Atm-/-mice. Nissl staining
identified defects in the cerebellar structure. Interneurons of theXrcc1nes-creand Xrcc1nes-cre; Atm-/- cerebellum were markedly reduced in the molecular layer (MO). (B) GAD (Glutamate
decarboxylase) immunostaining identifies GABAergic neurons, which were absent in the molecular layer of the Xrcc1nes-creand Xrcc1nes-cre; Atm-/-cerebellum (white arrow). (C)
Immunostaining for Pax2 identifies differentiating interneurons in the white matter (WM) of the developing postnatal cerebellum. Cerebellar interneuron populations were reduced in both Xrcc1nes-creand Xrcc1nes-cre; Atm-/-suggesting that ATM signaling is not involved in
23
Compared with wild-type and Atm-/-(data notshown) tissue, there was a marked reduction of
Pax2-positive interneuron progenitors in both theXrcc1nes-cre and Xrcc1nes-cre; Atm
-/-cerebellum(Figure 4C).These datasuggest two important outcomes that in the cerebellum Xrcc1 is necessary to maintain genomic stability for interneuron progenitors and to suppress DNA damage induced Atm independent p53-mediated cell cycle checkpoint activation.At least, ATM is not a sole p53 upstream kinase in cerebellar interneuron progenitors. This is distinct from the well-known for DNA damage signaling pathway.
4. Purkinje cells are susceptible to Xrcc1 and Atm loss
The most prominentclinical symptom of A-T patients is progressive neurodegeneration due to Purkinje cell loss (Boder, 1985; Gatti et al., 1991; Crawford, 1998).Purkinje cells are functionally important since they provide solo neuronal output signalsto the deep cerebellar nucleus. Their neuronal activity is controlled by the neuronal input from granule cells as well as interneurons. They are arranged as a monolayer and make up to 104dendritic connections.
Most Purkinje cell axons make inhibitory synaptic connections utilizing GABA as neurotransmitter to inhibit neurons in the deep cerebellar nuclei, which are located deep within the cerebellar white matter. As a result, the cerebellum regulate movement via Purkinje cell activity(Cerminara et al., 2015).
24
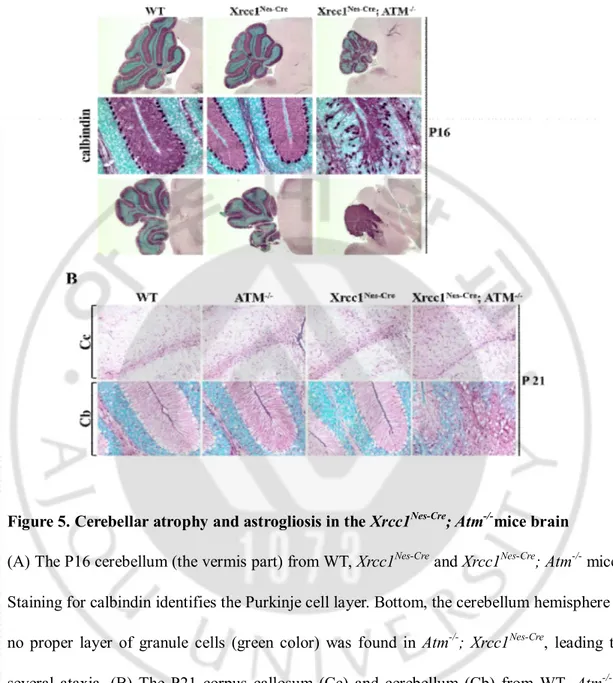
Figure 5. Cerebellar atrophy and astrogliosis in the Xrcc1Nes-Cre; Atm-/-mice brain
(A) The P16 cerebellum (the vermis part) from WT, Xrcc1Nes-Cre and Xrcc1Nes-Cre; Atm-/- mice.
Staining for calbindin identifies the Purkinje cell layer. Bottom, the cerebellum hemisphere – no proper layer of granule cells (green color) was found in Atm-/-; Xrcc1Nes-Cre, leading to
several ataxia. (B) The P21 corpus callosum (Cc) and cerebellum (Cb) from WT, Atm-/- , Xrcc1Nes-Cre and Xrcc1Nes-Cre; Atm-/- mice. Staining for GFAP identifies astrocytes. The Xrcc1Nes-Crebrain displayed increased GFAP (Glial fibrillary acidic protein) immunoreactivity,
25
The Purkinje cell layer in the experimental animals was visualized with Calbindin immuno-staining. In WT, Atm-/-andXrcc1nes-cre mouse, Purkinje cells are densely packed as a single
cell layer forming the proper Purkinje cell layer and exhibit dense dendritic arborizationwhich fill the molecular layer. Whereasin the Xrcc1nes-cre; Atm-/- mouse, Purkinje
cell density was markedly reduced and they appear heterotopically located in the molecular and granule cell layers. Also Purkinje cell bodies (nucleus) formed the irregular arrangement and their dendritic branches are lost in the Xrcc1nes-cre; Atm-/-cerebellum (Figure 5A). For
these reasonsXrcc1nes-cre;Atm-/-cerebellum showed unclear boundaries between the molecular
layer and granular layer. Overall the Xrcc1nes-cre;Atm-/-animalsdid not have proper cerebellum
structure which is most likely non-functional, resulting in severe ataxia. This Xrcc1 nes-cre;Atm-/-animal model is actually the first animal model displaying ataxia by deletion of the
Atm gene, mimicking ataxic phenotype of A-T patients.
Next, we analyzedastrocytes – one of glial cell populations in the nervous system – using glial fibrillary acid protein (GFAP)immunostaining. The Xrcc1nes-cre cortex(the Corpus
callosum is the white matter structure rich with astrocytes.) displayed increased immunoreactivity of GFAP suggestive of astrogliosis (Figure 5B). And the Xrcc1nes-cre;Atm
-/-brainshowed further increased immunoreactivity suggesting that the condition of the chronic DNA damage status was worsened by Atm inactivation. This astrogliosis has been implicated in both perturbation of brain homeostasis and neurodegenerative disease (Cirillo et al., 2011), Furthermore, astrogliosis occurs in many type of neurological disorders including spasms(Weidenheim et al., 2009).In case of the cerebellum (Cb), specialized glia
26
population called Bergmann glia is critical for migration of granule cells once granule cell precursor cells exit cell cycle to form granule cell layers (Sild and Ruthazer, 2011).The WT,
Atm-/- and Xrcc1nes-cre cerebellum showed a dense parallel array of GFAP immunoreactivity
in the molecular layer, typical Bergmann glia formation (Figure 5B). However, the Xrcc1 nes-cre;Atm-/-cerebellum displayed total disruption of Bergmann glia that indicates granule cell
migration is abnormal leading to malformation of the Purkinje cell layer.
5. Xrcc1 and Atmare required for oligodendrocyte differentiation
We approachedto another kind of differentiated glial cells in the central nervous system (CNS), oligodendrocyte. In the CNS, oligodendrocytes synthesize large quantities of myelin, a lipid-rich, multi-layered spiral-wrapped membrane structure which insulates nerve axons, so that neuronal electric impulse can be conducted with minimal loss. Myelination segments by oligodendrocytes refer to as the internode, which enables rapid and highly efficient neural conduction in neuronal networks.(Gieselmann et al., 1994; Miller and Mi, 2007; Haroutunian et al., 2014).
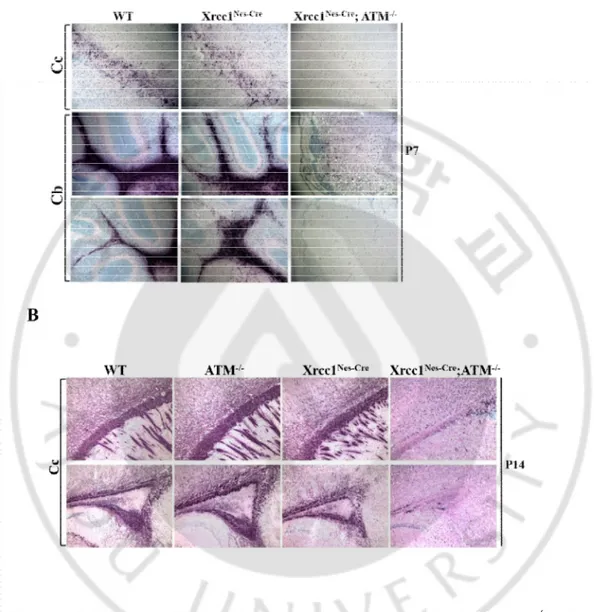
When we examined the brain tissues using Myelin basic protein (MBP) antibody, a marker for fully mature oligodendrocyte,surprisingly we found that the fully mature oligodendrocytes were absent in the cerebral cortex including the Corpus Callosum and to the minor extent in the cerebellum of the Xrcc1nes-cre; Atm-/-brain (Figure 6).Compared with
27
reduction of MBP-positive oligodendrocyte in the Xrcc1nes-cre; Atm-/-brain at postnatal
day7(P7) (Figure 6A). Even at postnatal day 14 (P14), MBP was not observed in the
Xrcc1nes-cre; Atm-/-brain (Figure 6B).Deficit of fully mature oligodendrocytes was further
confirmed by the expression of another oligodendrocyte marker CNPase at the protein level determined by western blotting.2',3'-Cyclic-nucleotide 3'-phosphodiesterase (CNPase) is a critical enzyme for generation of myelin. The expression level of CNPase was completely diminished in the Xrcc1nes-cre; Atm-/-cerebral cortexat P14.This failure of oligodendrocyte
differentiation persisted into postnatal 21 days that is the time point when the Xrcc1nes-cre; Atm-/-animals rarely survived.The Xrcc1nes-cre; Atm-/-mouse cerebellum showed minor
reduction of CNPase protein level. In contrast, the expression of neuronal marker, NeuN showed compatible expression pattern in theXrcc1nes-cre; Atm-/-cortex to that of the WT, Atm
-/-and Xrcc1nes-creanimals(Figure 6C). These data imply the strong possibility of involvement of
Atm and its signaling during oligodendrocyte genesis in the context of DNA damage particularly induced by Xrcc1 inactivation during brain development. This Atm involvement is quite exclusive to oligodendrocytes since other neural populations such as neurons and astrocytes were not affected in the Xrcc1nes-cre; Atm-/-brain.
Similar to the in vivofinding, we observed a complete absence of CNPase expression at the protein level in differentiated cells from neural stem cells (NSC) in vitro. NSC from WT,
Atm-/- andXrcc1nes-creembryonic forebrainexpressed CNPase 14 days (DIV14) after plating in
differentiation medium. However, we could not detect CNPase expression in cells differentiated from Xrcc1nes-cre; Atm-/-NSC (Figure 6D). Next, we checked the transcription
28
appliedquantitative real-time PCR to quantify the MBP and PLP mRNA levels in the P17 cortex and cerebellum. The expression level of target proteins was measured after synthesizing cDNA from total RNA. The MBP and PLP expression at the transcriptional level was low at P17 in the Xrcc1nes-cre; Atm-/- samples compared to the control groups
(Figure 6E,F). These data suggest that the very low protein level of MBP and PLP is not due to the matter of target protein stability, rather the diminished transcriptional level.
This phenomenon which is lack of mature oligodendrocytes in the brain, called leukodystrophy,was also found in several A-T patients(Chung et al., 1994; Sardanelli et al., 1995; Opeskin et al., 1998; Ciemins and Horowitz, 2000; Habek et al., 2008; Lin et al., 2014). So far, the underlying molecular mechanisms for leukodystrophy resulting from Atm mutation are not known. This is the first animal model with Atm deficiency to show defects in oligodendrocyte genesis. In future, it is warranty that the molecular and biochemical mechanisms for leukodystrophy will be revealed by analyzing further this animal model. Furthermore, a possible pharmaceutical application to relieve neurological symptoms will be explored using this animal model.
6. Oligodendrocyte progenitor cell formation in the Xrcc1nes-cre; Atm-/-brain.
Oligodendrogenesis begins with multipotent embryonic neural stem cells (NSC), which can be differentiated into neurons, astrocytes, and oligodendrocytes. The successive stages of oligodendrocyte differentiation include oligodendrocyte progenitor cells (OPC), pre-oligodendrocyte, and fully mature oligodendrocytes.
29
Figure 6. Fully mature Oligodendrocytes are missing in the Xrcc1nes-cre; Atm-/-brain.
(A-B) The Corpus callosum (Cc) and cerebellum (Cb) from WT, Atm-/-, Xrcc1Nes-Cre and Xrcc1nes-cre; Atm-/-mice at ages P7 and P14. Staining for MBP (myelin basic protein)
identified fully differentiated oligodendrocytes in the brain. Double inactivation of Atm and Xrcc1in the brain leads to complete absence of fully mature oligodendrocytes.
30
Figure 6. Fully mature Oligodendrocytes are missing in the Xrcc1nes-cre; Atm-/-brain
(Cont’d).
(C) Protein extracts from the P14 and P21 mouse cerebral cortex and cerebellum were subjected to western blot analysis for NeuN (neuron marker) and CNPase (3’-cyclic nucleotide 3’-phosphodiesterase, fully mature oligodendrocyte marker). β-actin was used as a loading control. (D) Neural stem cells from E14.5 WT, Atm-/-, Xrcc1Nes-Creand Xrcc1Nes-Cre; Atm-/-forebrain were cultured in NSC differentiation media. Proteins were extracted from in vitro differentiated cells after DIV (days in vitro) 14 and DIV21. β-actin was used as a
loading control.Arrow indicates NeuN positive western band (the upper band is none specific.)dKO, Xrcc1Nes-Cre; Atm-/-doubleknockout
31
Figure 6. Fully mature Oligodendrocytes are missing in the Xrcc1nes-cre; Atm-/-brain
(Cont’d).
(E-F)Realtime PCR quantification of MBP and another fully mature oligodendrocyte marker, PLP mRNA expression in the cortex and cerebellum at postnatal day 17 (the mRNA level was normalized to the level of sample WT). These are representative graphs from two repeats that showed the same pattern of the expression difference.dKO, Xrcc1Nes-Cre; Atm
32
Accompanying this morphological change is the stage-specific expression of molecular markers; Platelet-derived growth factor receptor α (PDGFRα) in oligodendrocyte progenitors, Myelin basic protein (MBP), 2’,3’-cyclic nucleotide 3’-phosphodiesterase (CNPase) and Proteolipid protein (PLP) in mature myelinating OL. Also Oligodendrocyte transcription factor 1 and 2 (Olig1 and Olig2) are specific transcription factors to confine oligodendrocyte lineage(Eng et al., 1968; Hart et al., 1989; Karthigasan et al., 1996; Qi et al., 2001; Ligon et al., 2006).
To define the differentiation stage of oligodendrocyte susceptible to double inactivation of Xrcc1 and Atm, we immunostained OPC using PDGFRα antibody.In contrast with the lack of fully mature oligodendrocyte marker expression such as MBP and PLP(Figure 6A,B), we observeda normal level of PDGFRα expression in the Xrcc1nes-cre; Atm-/-Corpus Callosum
where oligodendrocytes are mainly generated at postnatal day 14 (Figure 7A). Consistently with this in vivo data, weconfirmed thatthrough NSC differentiationin vitro. TheXrcc1nes-cre; Atm-/-NSC differentiated into PDGFRα+OPC measured byimmunocytochemistry (Figure7
B,C).Also the transcription level of PDGFRα was not reduced in the Xrcc1nes-cre; Atm
-/-oligodendrocytesfrom the P7brain tissue measured by realtime PCR method (Figure7 D). Despite the normal OPC population, oligodendrocyteswere absent in the Xrcc1nes-cre; Atm
-/-brain.This lackof mature oligodendrocytes in the brain of Xrcc1nes-cre; Atm-/- mouse could
result from either DNA damage induced cell cycle arrest or apoptosis in PDGFRα positive oligodendrocyte precursor cells. To determine whether OPC undergo cell death or have defects in cell cycle, we performed dual staining of TUNEL (for apoptosis) or PCNA immunopositivity with PDGFRα immunoreactivity. We found thatthe Xrcc1nes-cre;Atm
-/-33
animals showed no discernible difference compared to control groups. Therewas no cell death signal in the cortex and cerebellum at P14 in matched brain parts of all examined genetic backgrounds. The rostral migratory stream (RMS) was only region that was positive forthe cell death signal.As expected, the RMS in theXrcc1nes-cre; Atm-/-brain had slightly more
TUNEL+ apoptosis signals. But none of PDGFRa positive cells were also positive for the TUNEL staining, suggesting that OPC did not selectively undergo apoptosis upon DNA damage in Xrcc1 and Atm null background (Figure7 E).Moreover,proliferation defect was not detected in theXrcc1nes-cre;Atm-/-OPC (Figure7 F).Collectively, our data indicate that OPC
can begenerated without any defects in the Xrcc1nes-cre; Atm-/-brain as demonstrated both in vivo and in vitro. Also malformation in differentiation of oligodendrocytes in the Xrcc1nes-cre; Atm-/-animals was not due to reduction of PDGFRa positive cells.
34
Figure 7. Oligodedrocyte progenitor cells exist in the Xrcc1nes-cre; Atm-/-brain
(A) The P14 Corpus callosum (cc) from WT, Atm-/-, and Xrcc1nes-creand Xrcc1nes-cre; Atm
-/-mice. PDGFRα staining for oligodendrocyte progenitor cells (OPCs) indicateda similar expression level of OPCs in the Xrcc1nes-cre; Atm-/-braincompared with controls groups. (B)
Neural stem cells from WT, Atm-/-, Xrcc1nes-cre and Xrcc1nes-cre; Atm-/-embryonic forebrain at
E14.5 were cultured in NSC differentiation media till day 14 (DIV14). Cells were immunostained with PDGFRα (red). Nuclei were stained with DAPI.
35
Figure 7. Oligodedrocyte progenitor cells exist in the Xrcc1nes-cre; Atm-/-brain (cont’d).
(C) Quantification of percentage of PDGFRα-positive cellsin vitro. (D) Realtime PCR quantification of PDGFRα mRNA expression in the cortex and cerebellum at postnatal day 7 (the mRNA level was normalized to the level of sample WT). These are representative graph from two repeats which showed a similar profile of expression pattern. dKO, Xrcc1Nes-Cre; Atm-/-doubleknockout
36
Figure 7. Oligodedrocyte progenitor cells exist in the Xrcc1nes-cre; Atm-/-brain (cont’d).
(E) Analysis of cell death signal in OPC, assessed by TUNEL and PDGFRα double immunostaining, throughout the Rostral migratory stream regions at P14 from WT, Atm-/- ,
and Xrcc1nes-creand Xrcc1nes-cre; Atm-/-mice.Apoptosis was not occurred to PDGFRα positive
OPCs in the all brain examined including the Xrcc1nes-cre; Atm-/-brain. (F) Analysis of the
proliferating signal in OPC, assessed by PCNA and PDGFRαdouble immunostaining, throughout the Rostral migratory stream regions at P14 from WT, Atm-/- , and Xrcc1nes-creand Xrcc1nes-cre; Atm-/-mice. Proliferation was not defected in thePDGFRα positive OPCs in the
37
7. Oligodendrocyte specific transcription factor Olig1 is affected by Atm signaling
Two transcription factors, Oligodendrocyte transcription factor 1 (Olig1) and Oligodendrocyte transcription factor 2 (Olig2) are closely related basic helix-loop-helix (bHLH) transcription factors that play critical roles in oligodendrocyte differentiation (Lu et al., 2002). Buttheir roles are quite distinct. Olig2 promotes the fate choice decision to form OPC and sustains the replication competent state so as to expand the OPC at earlier development stage.(Li et al., 2011). By contrast, Olig1 promotes the differentiation of OPC to fully mature oligodendrocyte by facilitating myelin gene expression to promote the maturation of OPCs (Li et al., 2007). Olig1-null mice showed reduced mRNA and protein levels for genes involved in oligodendrocyte maturation, including MBP, Myelin Oligodendrocyte Glycoprotein (MOG) and PLP(Xin et al., 2005; Guo et al., 2010). As previouslydescribed (Figure 6), the mRNA and protein expression levels of fully mature oligodendrocyte marker proteins, MBP, PLP and CNPase, were decreased in the Xrcc1nes-cre; Atm-/-brain. To test the possible transcription factor regulation by Atm signaling, we
examined the protein levels of Olig1 and Olig2 by western blot. The Xrcc1nes-cre; Atm-/-mouse
cerebellum showed compatible expression levels of Olig2. In contrast, Olig1 showed reduced expression pattern in the Xrcc1nes-cre; Atm-/-cortex and cerebellum (Figure 8A). We
confirmed that expression of Olig2 was not defected in the in the Xrcc1nes-cre; Atm-/- Corpus
Callosum and cerebellum at postnatal day 14 by tissue immunostaining (Figure 8B).In case of Olig1, during the brain development, Olig1 localizes to the nucleus at postnatal day 1, thentranslocates to the cytosol at postnatal day 7, by 14 most of oligodendrocyte lineage
38
cells display cytosolic Olig1 (Arnett et al., 2004). Also it was reported that cytosolic Olig1 is expressed in human oligodendrocyte for membrane expansion and full myelination to axon (Othman et al., 2011). We examined using immunofluorescence staining to check the expression level of Olig1 and localization in vivo. Olig1 is reduced in the Xrcc1nes-cre; Atm
-/-cortex and cerebellum as well as tendency to nuclear location (Figure 8C). This result is consistence with western blot analysis that as shown in (Figure8 A).
39
Figure 8. Low expression level of Olig1 in Xrcc1nes-cre; Atm
-/-(A)Protein extracts from the P14 mouse cerebral cortex and cerebellum were subjected to western blot analysis forOlig1 and Olig2 (Oligodendrocyte transcription factor) and CNPase (3’-cyclic nucleotide 3’-phosphodiesterase, fully mature oligodendrocyte marker). β-actin was used as a loading control.
40
Figure 8. Low expression level of Olig1 in Xrcc1nes-cre; Atm-/-(Cont’d)
(B) The P14 Corpus callosum (cc) and cerebellum from WT, Atm-/-, and Xrcc1nes-creand Xrcc1nes-cre; Atm-/-mice. Olig2 staining for oligodendrocyte transcription factor indicated a
similar expression level of OPCs in the Xrcc1nes-cre; Atm-/-braincompared with controls
groups.(C)Analysis of the localization of Olig1, stained by Olig1 and DAPI, throughout the cortex and cerebellum at P14 from WT, Atm-/- , and Xrcc1nes-creand Xrcc1nes-cre; Atm-/-mice.
41
IV. DISCUSSION
DNA repair mechanism is significantto maintaining the genomic stability. The nervous system is very sensitive to DNA damage, particularly in comparison with other tissues that contain non-replicating cell types. There are some unique properties of the nervous system that may account for this relative sensitivity. The brain metabolizes about 20% of consumed oxygen but has a lower capacity than other body parts to neutralize reactive oxygen species (ROS).Neurons are particularly susceptible to oxidative stress. Because of this, neural cells are continuously exposed to the physiological condition which induces DNA damage spontaneously (Lu et al., 2004; Shull et al., 2009; Suberbielle et al., 2013).
In mammals, the major brain development step is almost completed during embryogenesis. At this period neuronal progenitor cells proliferate for self-renewal as well as to generate various types of neural cells at high rate. After birth, they escape the cell cycle and become post-mitotic cells to start migration and differentiation(Dehay and Kennedy, 2007). When DNA damage arises in eitherproliferating or post-mitotic neural cells, proliferating cells have more options to repair DNA damage, particularly strand breaks than post-mitotic cells(McKinnon, 2009). For example, DNA single strand breaks at stalled DNA replication sites can be converted into double strand breaks, and these can be accurately repaired through Homologous recombination repair (HRR), anerror-free repair mechanism, in the S/G2 phases of the cell cycle when a sister chromatid is available as a template for repair, whereas in post-mitotic cells, HRR is not functionalto repair DNA strand breaks since there is no sister chromatid available. Non-homologous end joining (NHEJ), an error-prone
42
repair mechanism, is the sole pathway available to prevent accumulation of DNA damage in the post-mitotic cells (Shull et al., 2009). During brain development, DNA damage naturally occurs, but its sources and types are unknown.For these reasons, understanding how DNA damage signaling pathway promotes neural development and preserves genomic integrity is essential to understand neuropathology found in several hereditary syndromes with genomic instability.
Defects in cellular DNA repair processes have been linked to genome instability and hereditary neurological syndrome. For example, defects in the DNA single strand break end processing enzymes APTX and TDP1 cause autosomal recessive disease AOA1 and SCAN1 respectively. The interesting feature is that the pathology in these disorders is almost exclusively restricted to the nervous system (McKinnon and Caldecott, 2007).
Another prime example is Ataxia telangiectasia (A-T), a well-characterized syndromeof the relationship that exists between DNA damage repair defects and multisystemhereditary disease. This early onset disease is characterized by immune system defects, hypersensitivity to ionizing radiation, cerebellar ataxia, and oculomotor apraxia (Boder and Sedgwick, 1958).Atmknockout mice (Atm-/-) exhibit many of the characteristics of A-T including
sterility, immunodeficiency and lymphoma, however they do not recapitulate the brain phenotypes of A-T patients, making it difficult to study the neurodegenerative aspects of this disease in mice. Although defects in ATM lead to neurodegeneration, its physiological role in the nervous system is unclear.
The neurological defects in A-T become obvious early in life, suggesting that they are originated during brain development. Thus,we generated a new A-T mouse model, Xrcc1and
43
Atmdouble inactivatedmouse (Xrcc1Nes-Cre;Atm-/-) to assess the role of Atm upon persistent
DNA damage during neural development.Xrcc1 isa scaffold protein involved in DNA single strand break repair/base excision repair (BER).Previous reports showed that inactivation of Xrcc1 during brain development led to persistence of DNA strand breaks throughout the brain and other cell lines.(Lee et al., 2009; Khoronenkova and Dianov, 2015).The Xrcc1 gene was selectively deleted in the nervous system during development using Cre recombinase whose expression is under control of Nestin promoter. Nestin is exclusively expressed in neuroprogenitor cells during development (Zimmerman et al., 1994). This persistent DNA damage could lead to genomic instability in the nervous system which is a good physiological situation to study the role of ATM during brain development and function.
The Xrcc1Nes-Cre;Atm-/- mouse had exacerbated neurological behavior phenotypes including
progressive severe ataxia.These Xrcc1Nes-Cre;Atm-/-micemarkedly affected the cerebellar
interneurons, Purkinje cells and mature oligodendrocytes. There was also a progressive accumulation of persistent DNA strand breaks in the affected brain.
In the previous reported paper, Xrcc1 is necessary for interneuron progenitors to suppress DNA damage. Inactivation of Xrcc1induced p53-mediated cell cycle checkpoint activation in interneuron progenitor cells. Researchers found a complete rescue of interneurons in the molecular layer of Xrcc1Nes-Cre;p53-/-mice. This rescue suggeststhe contribution of
p53-toward interneuron loss (Lee et al., 2009). In our research, we examined the cerebellar interneuron in the Xrcc1Nes-Cre;Atm-/-cerebellumto establish the ATM signaling pathway. We
44
p53. Xrcc1Nes-Cre;Atm-/- mouse, however,did not rescue p53-dependent cell cycle arrest of
cerebellar interneuron progenitor cells.Thesedata suggest that p53 could be activatedbyotherupstream kinases responding to DNA damage, not ATM. It may be possible that p53 is regulated by another member of PI3K family such as DNA-PK (DNA dependent protein kinase) and ATR (Ataxia telangiectasia and Rad3 related) after DNA damage in cerebellar interneuron progenitors.
We found another histological phenotype of the Xrcc1Nes-Cre;Atm-/-cerebellum. The Purkinje
cell layer was disrupted by irregular arrangement and its dendritic branches were lost. Purkinje cells are the primary output from the cerebellum. The stellate and basket cells, the interneurons of cerebellum, make inhibitory contacts with Purkinje cells to modulate neuronal signaling to the deep nuclei. Thus, defects of interneurons and Purkinje cells in the
Xrcc1Nes-Cre;Atm-/-cerebellum exacerbate the coordination of body motor functionleading to
severecerebellar ataxia.This histological and behavioral phenotype is consistent with neuro-clinical phenotypesof A-T patients(Boder, 1985).
In addition to the cerebellum, the oligodendrocyte was also affected by Xrcc1 and Atm inactivation. Oligodendrocytes inthe CNSare a kind of glial cells, and enable the myelination of axons for rapid and highly efficient nervous conduction in neuronal networks (Huang et al., 2015). Myelinated exons enable rapid transmission of action potentials through saltatory conduction, protectionfor axons, and allows for packing of greater axon densities during the evolution of brain complexity. Nerve conduction velocity in myelinated axons can reach 150 m/s, which is distinctly faster than 0.5 to 10 m/s measured in unmyelinated axons (Purves et
45 al., 2001).
We observed complete absence of mature oligodendrocytes when DNA damage persistedby Xrcc1 inactivation is not properly processed by inactivating Atm.We found that generation of OPC which is the stage before maturation ofoligodendrocytes is not affected in the Xrcc1Nes-Cre; Atm-/- cerebral cortex.Next we were wondering that whether reduction of
OPC contributes to lack of oligodendrocytes in the Xrcc1Nes-Cre; Atm-/- cerebral cortex. Dual
labeling analysis of apoptosis or cell cycle signals in OPC did not show difference in the experimental groups suggesting that Xrcc1 and Atmare dispensable in maintenance of OPC.
We speculated the oligodendrocyte defects happen during bona fide differentiation process which is dependent on function of Olig1, the transcription factor,whose expression levels were lower in the Xrcc1Nes-Cre; Atm-/- cerebral cortex and cerebellum. Our result clearly
demonstrate that ATM signaling is not only important to maintain global genomic stability in the nervous system during development, but also oligodendrocyte differentiation governed by transcription factor Olig1 upon a certain type of DNA damage, such as induced by Xrcc1 inactivation, during brain development. How Olig1 is under control of Atm at the protein or mRNA level upon DNA damage and its regulation will be the next step we will pursue to understand the underlying molecular mechanisms.
The similar phenotype was also foundin A-T patients. In many clinical reports,A-T patients showeddysmyelination in the brain; neuropathological feature ofleukodystrophy. Dysmylinationis characterized by abnormal white matter signals due to defective formation and function of the myelin sheath(Pouwels et al., 2014). Almost half of the brain is
46
comprised of tracts of myelinated axons in the white matter. A-T patients who had the sign of dysmyelination showed hyperintensity in the white matter on MR imaging (Chung et al., 1994; Sardanelli et al., 1995; Opeskin et al., 1998; Ciemins and Horowitz, 2000; Habek et al., 2008; Lin et al., 2014).In addition to A-T patients, more that 30% of Cockayne syndrome patients,another hereditary syndrome with the neurodegenerative phenotypesdue tothe CSA/CSBwhich are associated with nucleotide excisionrepair (NER), also show leukodystrophy(Weidenheim et al., 2009). Furthermore,DNA damagingagent-treated human shATM neural stem cell differentiated into normal number of neurons, but a reduced number of oligodendrocytes(Carlessi et al., 2009).These indicate directly or indirectly that DNA damage repair signaling, includingATM signaling, plays a selective role in differentiation of oligodendrocytes in the nervous system. What is the connection between DNA damage responses and oligodendrocyte differentiation at the molecular level is unknown.
Collectively, our data demonstrate for the first time that the Xrcc1Nes-Cre;Atm-/-mouse is the
new mouse model for A-T characterized by most similar neurological phenotypes such as cerebellar ataxia and leukodystrophy with human A-T. When aparticular type of endogenous DNA damage happens, ATM signaling pathway is critical for proper genesis of Purkinje cells and maturation of oligodendrocytes. These findings will contribute to further understanding of ATM role in DNA damage response and molecular mechanism for neurological features of A-T.