U.-H. Yoon ・ G.-S. Lee ・ C.-K. Kim ・ J.-S. Lee ・ J.-H. Hahn ・ D.-W. Yun ・ H.-S. Ji ・ J.-H. Lee ・ S.-H. Park ・ G.-W. Kim ・ M.-S. Seo ・ Y.-H Kim ( )
국립농업과학원
(Genomics Division, National Academy of Agricultural Science, RDA, Suwon, Korea)
e-mail: [email protected] T.-H. Lee
명지대학교
(Bioscience and Bioinformatics Division, MyongJi University, Yongin, Korea)
벼 발아종자 발현유전자의 발현특성분석
윤웅한 ・ 이강섭 ・ 김창국 ・ 이정숙 ・ 한장호 ・ 윤도원 ・ 지현소 ・ 이태호 ・ 이정화 ・ 박성한 ・ 김건욱 ・ 서미숙 ・ 김용환
Analysis of germinating seed stage expressed sequence tags in Oryza sativa L.
Ung-Han Yoon ・ Gang-Seob Lee ・ Chang-Kug Kim ・ Jung-Sook Lee ・ Jang-Ho Hahn ・ Doh-Won Yun ・ Hyeon-So Ji ・ Tae-Ho Lee ・ Jeong-Hwa Lee ・ Sung-Han Park ・ Gun-Wook Kim ・ Mi-Suk Seo ・
Yong-Hwan Kim
Received: 4 September 2009 / Accepted: 18 September 2009
ⓒKorean Society for Plant Biotechnology
Abstract Seed germination is the important stage to ex- press many genes for regulation of energy metabolism, starch degradation and cell division from seed dormancy state. For the functional analysis of seed germination mech- anisms, we were analyzed the rice cDNA clones (Oryza sativa cultivar Ilpum) obtained from seed imbibition during 48 hours. Total number of 18,101 Expressed Sequence Tags (ESTs) were clustered using SeqMan program. Among them, 8,836 clones were identified as unique clones. We identified the chitinase gene specifically expressed in seed germination and amylase gene involved to starch degradation from the full length cDNA analysis, and several genes were registered to NCBI GeneBank. To analyzed the commonly expressed genes between inmature seed and germinated seed, 25,668 inmature ESTs and 18,101 germinated ESTs were clustered using SeqMan program and identified 2,514 clones as com- monly expressed unigene. Among them, alpha-glubulin and alcohol dehydrogenase I were supposed to LEA genes only expressed in the immature and germinated seed stages.
For the clustering of orthologous group genes, we further analyzed the 8,836 EST clones from germinating seeds using NCBI clusters of orthologous groups database. Among the clones, 5,076 clones were categorized into information storage and processing, cellular processes and signaling, metabolism and poorly characterized genes, proportioning 783 (14.29%), 1,484 (27%), 1,363 (24.8%) and 1,869 (34%) clones to the previous four categories, respectively.
서 론
벼는 세계인의 주곡작물이며 게놈연구를 위한 화본과 식물의 모델작물로 이용되고 있다. 최근 한국 등 10개국 이 참여한 국제 벼 염색체 완전해독 연구 (IRGSP 2005, http://rgp.dna.affrc.go.jp/IRGSP/)가 완료되고 이들 염기서열 정보가 공공부분에 공개됨에 따라 이를 활용할 수 있는 시스템 구축과 기능분석 연구들이 많이 이루어지고 있다 (Jung et al. 2008, Kim et al. 2009, Park et al. 2009).
벼의 주요 농업형질관련 유전자의 기능해석을 위하여 발현유전자 분리 및 확보가 필요하며 다양한 품종에서 대량발현유전자분석 연구가 수행되고 있다. 현재 우리나 라의 벼 종자발현유전자 구조분석은 농촌진흥청을 중심 으로 진행되고 있으며 Yoon 등 (2009)은 종자발달과정에 관여하는 유전자의 기능분석을 위하여 25,668개 일품벼 미숙종자 EST의 구조분석 및 정보해석을 행하였다. 일본 의 경우 Kikuchi 등 (2003)은 닛본바레 품종에서 32,127개 완전장 발현유전자의 구조분석을 행한 후 KOME 데이터 베이스 (http://cdna01.dna.affrc.go.jp/cDNA/)를 통하여 정보 Research Article
를 공개하고 있다. 현재 (2009년 8월) NCBI에 등록된 각 종 벼 EST는 1,248,955개이며 unigene 으로는 40,973개를 나타내고 있다. 벼 유전체 정보종합 DB로는 RAP-DB (http://rapdb.dna.affrc.go.jp/)와 TIGR Pseudomolecules version6 을 기반으로 구축된 벼 유전체정보 DB (http://rice.plantbiology.
msu.edu/)가 공개되고 있다 (Ouyang et al. 2007).
이러한 벼 발현유전자 및 유전자정보를 이용하여 세 계 각국은 농업형질유전자 대량기능분석과 유전자분리 에 적극 활용하고 있다 (Nakamura et al. 2007).
특히 식량원으로서 중요한 벼 종자 발달단계의 발현 유전자 분석 및 발현특성 연구는 종자형질 개선을 위한 연구자들에게는 매우 중요한 자료를 제공한다.
또한 LEA (Late embryogenesis abundant) 유전자들은 종 자성숙기와 발아기에 주로 발현되며 종자발아에 영향을 미친다는 점에서 매우 중요시 되고 있다 (Galau et al.
1987, Ishibashi et al. 1990). 벼 단백질체 2D 분석을 통하여 발아종자의 단백질변화를 분석한 결과 148종의 단백질 변화가 나타났으며 전분분해 및 대사관련 유전자들의 발 현이 증대되고 저장단백질 등이 감소됨을 보고 하였다 (Yang et al. 2007). Xiao 등 (2008)은 GC-MS 분석을 통하여 발아종자의 대사체 변화를 분석 한 결과 174개의 변화를 검출하였다. Xiao 등 (2007)은 LEA 유전자를 벼에 과발현 시킨 결과 건조저항성이 증대되었다고 보고하였다. 이와 같은 결과는 발아특이 발현유전자가 발아시기에 중요한 역할을 하고 있음을 나타내며 이들 유전자의 특성을 파 악하고 활용함으로서 발아조절 등 농업형질 개선연구에 이용할 수 있을 것으로 생각한다.
발현유전자의 기능분류를 위하여 일반적으로 GO분류 와 NCBI의 COG DB를 이용한 COG 분류를 이용하고 있 다. COGs (Cluster of orthologous group)는 유전체 염기서열 로부터 얻어진 단백질정보를 이용하여 계통발생학적 분 류를 하는데 중요한 정보를 제공 한다 (Tatusov et al.
1997). 현재 원핵생물의 기능분류는 66종 게놈정보에서 192,987개 단백질정보를 분석한 후 4,872개 COG로 기능 분류를 하고 있다. 진핵생물 기능분류의 경우는 Caenorhabditis elegans, Drosophila melanogaster, Homo sapiens, Arabidopsis thaliana, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Encephalitozoon cuniculi등 7종의 유전체정보를 이용하여 110,655개 유전자정보로부터 59,838개의 단백질을 분석한 후 4,852개 KOGs (Eukaryotic cluster of orthologous group)로 기능을 분류하였다 (Tatusov et al. 2003). KOG의 기능분류 는 4개의 대분류군 (Information storage and processing, cellular processes and signaling, metabolism, Poorly characterized)내 에 15개의 기능별로 분류하고 있으며 이러한 COGs 및 KOGs 분류 결과는 NCBI의 DB에 수록하여 공개하고 있 다 (http://www.ncbi.nlm.nih.gov/COG/).
본 연구는 발아종자 발달단계의 cDNA 합성과 발아종
자에서 발현하는 발현유전자의 대량 염기서열 분석 및 발현유전자 정보종합화를 목적으로 수행 하였다. 이러한 발아종자 발현유전자정보의 종합화는 벼 및 식물전반의 구조 및 기능유전체를 연구하는 연구자들에게 보다 유익 한 정보를 제공할 수 있을 것이며 발아종자 유전자의 발 현특성을 분석함으로써 종자 발아에 관련된 발아율 향 상, 수발아 방지 및 농업형질개선 등의 실용화 연구에 기 여할 수 있을 것으로 기대되어 진다.
재료 및 방법
벼 발아종자 유전자은행 제작 및 발현유전자 염기서열결정
본 연구에 사용된 일품 벼 발아종자는 종자의 껍질을 벗긴 후 1% 과산화수소용액에 5분 소독한 후 멸균 증류 수로 세척을 하였으며 물에 침지하여 24시간 흡습시킨 종자를 30℃ 항온기에서 48시간 발아시킨 발아종자를 사 용하였다. 일품벼 발아종자 발현유전자은행 (cDNA) 제작 을 위한 RNA 분리는 Trizol 시약 (Invitrogen Life Technologies) 을 이용하여 total RNA를 분리한 후 PolyA Tract mRNA kit (Promega)를 사용하여 Poly(A) mRNA를 분리하였다. 발아 종자 cDNA 은행제작은 ZAP-cDNA Gigapack III 골드클로 닝 kit (Clontech)를 사용하였다. 발아종자 cDNA 100 ng을 1 ug의 Uni-ZAP vector에 삽입한 후 Gigapack III Gold Pack- aging Extract에 packaging하여 cDNA 은행제작을 하였다.
개별 유전자 클론의 플라스미드 DNA 분리는 96 well 밀 리포어 멀티스크린 (Millipore MANANLY50, MAFBNOB50) 을 이용하여 대량으로 플라스미드 DNA를 분리하였다.
발아종자 삽입단편의 크기 확인은 플라스미드 DNA를 EcoRI, XhoI 제한효소로 처리한 후 1.2% Agarose gel 전기 영동으로 하여 삽입 단편을 확인 하였다.
발아종자 발현유전자 염기서열 결정은 각 클론의 5‘말 단을 BigDye terminator ver 3.1 (Applied Biosystems)과 SK 프라이머 (5’-cgctctagaactagtggatc-3’)를 이용하여 염기서열 분석반응을 수행한 후 ABI3100 및 ABI3730 유전자 염기 서열분석기 (Applied Biosystems)를 사용하여 벼 발아종자 발현유전자에 대한 염기서열을 결정하였다.
벼 발아종자 발현 단일유전자 선발
발아종자 EST 염기서열 정보분석을 위하여 각 클론의 발현유전자 염기서열을 결정한 후 SeqMan 프로그램 (DNASTAR Inc)을 사용하여 벡터부위를 포함하는 염기서 열부분과 불명확한 염기서열을 제거하였다. 발아종자내 발현하는 unigene 선발은 SeqMan 프로그램을 이용하여 100 bp씩 95%의 상동성으로 유전자 염기서열들을 연결
하여 얻어진 singleton 유전자와 두개 이상의 유전자로 cluster된 contig 수를 합하여 얻어진 수를 unigene 수로 결 정하였다 (Kikuchi et al. 2003, Yoon et al. 2009).
발아종자 및 미숙종자 공통발현유전자 분석
일품벼 발아종자와 미숙종자에 공통으로 발현하는 발 현유전자분석을 위하여 SeqMan 프로그램을 이용 하였다.
미숙종자와 발아종자의 공통발현 유전자수 산정은 각각 의 미숙종자와 발아종자의 unigene을 합한 수에서 발아종 자와 미숙종자 발현유전자를 같이 assembly하여 얻어진 unigene의 개수를 제외하고 얻어진 수를 공통발현 유전자 수로서 산정하였다 (Yoon et al. 2009).
발아종자 발현유전자단편 정보분석
발아종자 발현유전자단편의 정보분석은 국립농업과 학원 농업생명공학정보센터 (http://www.niab.go.kr)에 설치 된 webblast 및 NCBI의 BLAST (http:/www.ncbi.nlm.nih.gov) 을 이용하여 상동성 검사를 수행하였다.
발아종자 발현유전자의 벼 염색체내의 위치결정을 위 하여 일본의 벼 명명 프로젝터에서 구축한 데이터베이스 (RAP DB, http:/rapdb.dna.affrc.go.jp)와 미국 미시간대학교 벼 게놈 명명프로젝터 데이터베이스 (RGAP DB, http:/rice.
plantbiology.msu.edu)의 TIGR Pseudomolecule ver. 6 정보를 이용하였다. 각각의 발아종자 발현유전자들의 염색체내 위치결정은 BLASTN 분석을 통해 얻어진 발현유전자 염 기서열의 유사성이 99% 이상을 나타내는 염색체 내 지역 을 판별하여 각 유전자의 염색체내 위치로 나타내었다.
NCBI KOG DB 기반 유전자 기능분류
발아종자로부터 얻은 unigene들에 대한 기능분류는 NCBI 에서 애기장대 식물 등 7개 주요 진핵생물 단백질 서열로부 터 구축된 KOG (Eukaryotic Clusters of Orthologous Groups) DB 인 KOG DB를 기반으로 하여 수행하였다. 먼저, BLASTX 분석을 통해 KOG DB 내 서열 중, unigene 서열 각각에 대해 가장 높은 유사성을 가지는 단백질 서열을 검색하였다. 이 때 검색 조건으로 e-value가 1.0e-20 이하인 것들만을 추출하 여 유사서열이 아닌 것이 나올 확률을 최소화하였다. 다음 으로 이렇게 얻어진 KOG DB내 단백질 유사서열의 분류정 보를 바탕으로 unigene 서열의 기능 분류를 수행하였다.
결과 및 고찰
벼 발아종자 유전자은행 제작 및 발현유전자 염기서열결정
일품벼 발아종자 cDNA 유전자은행제작 결과 2.0 X 106 pfu/ml의 phagemid를 얻었다. 발아종자 발현유전자 삽 입단편 크기는 1.2% 아가로즈 전기영동으로 확인한 결과 0.5 - 3.0 kb의 삽입단편을 포함하고 있는 것으로 나타났 다. 본 연구에서 cDNA 합성을 위하여 사용한 poly(A) RNA 분리 및 oligo(dT) primer와 reverse transcriptase 효소를 이 용한 방법으로는 3 kb 이상 길이를 가진 cDNA 합성에 어 려움이 있다. 향후 효율적인 완전장유전자의 분리를 위 하여서는 CAP trapper 방법을 이용한 완전장 cDNA 은행 제작이 요구되어 진다 (Carnini P, 1996, Kikuchi S, 2003).
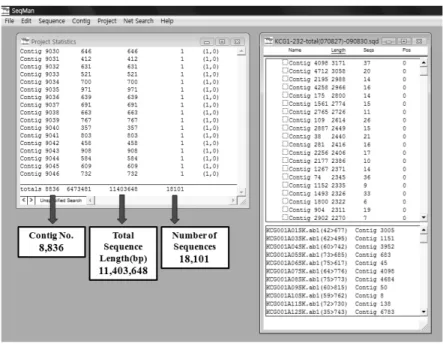
대량 염기서열분석을 위하여 밀리포어 96 well 멀티스 크린 시스템을 사용하여 150 ul TB 배지에서 5 - 8 ug의 플라스미드 DNA를 얻을 수 있었으며 무엇보다 균일한 DNA양을 얻을 수 있었다. 각각의 발아종자 발현유전자 클론의 염기서열 결정은 96well PCR plate를 이용하여 각 well당 200 ng의 DNA, 5 pmol의 SK primer, 2 ul의 Bigdye terminater sequence reaction, 1 ul의 5 X buffer를 넣은 후 염 기서열분석 반응을 시키고 ABI3100과 ABI3730 염기서열 분석기를 이용하여 염기서열을 결정하였다. 발아종자 염 기서열분석은 총 22,272 (96well 232plate) 개의 발현유전 자를 분석하였으며 이들 클론 중에서 150 bp 이하 길이의 염기서열정보를 가진 클론과 불량 염기서열부분의 제거 를 행하여 최종적으로 18,101개 클론의 염기서열정보를 얻을 수 있었다. 발아종자 발현유전자 염기서열분석 성 공률은 81.2%로 나타났다. 발아종자 18,101개 클론으로부 터 얻어진 전체 염기서열 길이는 11,403,648 bp였으며 발 아종자 발현유전자 1개 클론의 평균 길이는 630 bp로 확 인되었다. 앞서 발표한 미숙종자 발현유전자의 염기서열 분석 성공률 76%와 1개 클론 평균 염기서열 길이 550bp (Yoon et al. 2009) 보다 높은 경향을 나타내고 있다 (표 1).
일품벼 발아종자 unigene 선발
일품벼 발아종자 EST의 염기서열 정보분석 및 기능분 류를 위하여 발아종자의 unigene을 분리하였다. 발아종자 의 unigene 분리는 DNASTAR사의 SeqMan 프로그램을 이 용하였으며 발아종자 18,101개 EST의 basecalling과 clustering 을 행한 결과 2개 이상의 클론이 중복된 2,735개의 contig 와 6,101개의 singleton 등 8,836개의 unigene을 얻었다 (그 림 1, 표 1). 발아종자에서 많이 발현하는 유전자들의 분 포도 분석은 앞서 SeqMan으로 분석된 clustering 결과를 이용 하였다. 우선 50개 이상의 클론으로 구성된 contig를 분석한 결과 2,735개 contig 중에서 14개의 contig가 50개 이상의 클론으로 구성되어있었다 (그림 2). 이러한 경향 은 콩 유래 68,661개의 EST 염기서열로부터 cluster 분석 을 한 결과 60개 이상의 클론을 포함하는 contig는 10개 미만인 것으로 보고 한 것과 유사한 결과를 나타내고 있
Fig. 1 Identification of unigenes using SeqMan program. To estimate the level of redundancy, the Ilpumbyeo germinating seed ESTs were clustered with each other using the SeqManII program (DNASTAR), which considers two sequences being originated from the same transcript when they have 95% nucleotide identity over a minimum of 100 bp. Poor quality sequence (<100 bp) and vector sequence were eliminated
Table 1 Summary of assembly and clustering of cDNA clones from germinating seed of rice cv. Illpumbyeo
Groups Records Number of initial clones
Number of sequenced clones Total sequence length (bp) Average length (bp) Singletons
Clustering A total Unigene
22,272 18,101 11,403,648 630 6,101 2,735 8,836
6100 1359
536 242143 117
57 50 30 29
110 29
12
5 3 4 3
1 1 2 1
10 100 1000 10000
1 2 3 4 5 6 7 8 9 10 A1 A2 A3 A4 A5 A6 A7 A8 A9 A10 Number of clones
Number of scaffolds
Fig. 2 Cluster distribution of cDNA clones from germinating seed of rice.
A1 : 11-20, A2 : 21-30, A3 : 31-40, A4 : 41-50, A5 : 51-60, A6 : 61-70, A7 : 71-80, A8 : 81-90, A9 : 91-100, A10 : 101-150
A total of 18,101 EST clones were clustered using SeqMan program. Among 8,836 clones were identified as unique clones 다 (Umezawa et al. 2008). 이와 달리 앞서 발표한 벼 미숙
종자의 경우 50개 이상의 클론을 가진 contig는 32개였으 며 그중 3개의 contig는 1000개 이상의 glutelin 유전자로 구성되어 있었다 (Yoon et al. 2009). 이와 같은 결과는 종 자발아 시기에는 종자내부의 전분분해와 일반적인 대사 관련 유전자의 발현이 다양하게 이루어지는 것으로 생각 되어지며 상대적으로 glutelin 등 특정 단백질 합성 유전 자 발현이 많이 나타나는 미숙종자와는 다른 경향을 나 타내고 있었다.
벼 발아종자 및 미숙종자 공통발현유전자 분석
종자성숙 및 발아 시기의 일련의 과정에서 이루어지 는 유전자들의 발현 특성분석을 위하여 미숙종자와 발아 종자 공통적으로 발현하는 다양한 유전자를 동정하고자 한다.
우선 앞서 분석된 벼 미숙종자 발현유전자 25,643개와
발아종자 발현유전자 18,101개 등 43,744개의 발현유전자 를 SeqMan 프로그램으로 cluster 분석을 행한 결과 13,989 개의 unigene을 얻었다. 발아종자와 미숙종자에서 공통적 으로 발현하는 유전자를 분석하기위하여 미숙종자의 7,694개 unigene (Yoon et al. 2009)과 발아종자의 8,836개 unigene을 합한 16,530개에서 미숙종자·발아종자 unigene 13,989개를 제외시킨 결과 2,541개의 공통 발현 unigene을 얻었다 (그림 3). 발아종자와 미숙종자에 공통으로 발현 하는 2,541개 unigene 중에는 LEA 유전자들이 많이 포함 되어 있으며 식물조직별 RT-PCR 및 microarray실험 등을 통하여 보다 자세한 LEA 유전자 분석연구를 진행하고 있 다 (data not shown). Yang (2007) 등은 단백질체 분석을 통
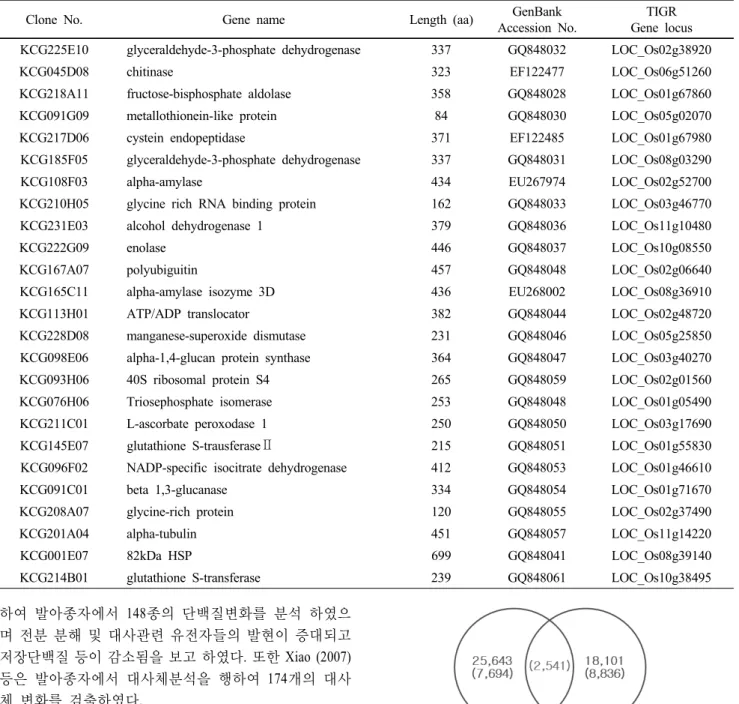
Table 2 The list of abundantly expressed genes from germinating seed of rice cv. Illpumbyeo
Clone No. Gene name Length (aa) GenBank
Accession No.
TIGR Gene locus KCG225E10 glyceraldehyde-3-phosphate dehydrogenase 337 GQ848032 LOC_Os02g38920
KCG045D08 chitinase 323 EF122477 LOC_Os06g51260
KCG218A11 fructose-bisphosphate aldolase 358 GQ848028 LOC_Os01g67860
KCG091G09 metallothionein-like protein 84 GQ848030 LOC_Os05g02070
KCG217D06 cystein endopeptidase 371 EF122485 LOC_Os01g67980
KCG185F05 glyceraldehyde-3-phosphate dehydrogenase 337 GQ848031 LOC_Os08g03290
KCG108F03 alpha-amylase 434 EU267974 LOC_Os02g52700
KCG210H05 glycine rich RNA binding protein 162 GQ848033 LOC_Os03g46770
KCG231E03 alcohol dehydrogenase 1 379 GQ848036 LOC_Os11g10480
KCG222G09 enolase 446 GQ848037 LOC_Os10g08550
KCG167A07 polyubiguitin 457 GQ848048 LOC_Os02g06640
KCG165C11 alpha-amylase isozyme 3D 436 EU268002 LOC_Os08g36910
KCG113H01 ATP/ADP translocator 382 GQ848044 LOC_Os02g48720
KCG228D08 manganese-superoxide dismutase 231 GQ848046 LOC_Os05g25850
KCG098E06 alpha-1,4-glucan protein synthase 364 GQ848047 LOC_Os03g40270
KCG093H06 40S ribosomal protein S4 265 GQ848059 LOC_Os02g01560
KCG076H06 Triosephosphate isomerase 253 GQ848048 LOC_Os01g05490
KCG211C01 L-ascorbate peroxodase 1 250 GQ848050 LOC_Os03g17690
KCG145E07 glutathione S-trausferaseⅡ 215 GQ848051 LOC_Os01g55830
KCG096F02 NADP-specific isocitrate dehydrogenase 412 GQ848053 LOC_Os01g46610
KCG091C01 beta 1,3-glucanase 334 GQ848054 LOC_Os01g71670
KCG208A07 glycine-rich protein 120 GQ848055 LOC_Os02g37490
KCG201A04 alpha-tubulin 451 GQ848057 LOC_Os11g14220
KCG001E07 82kDa HSP 699 GQ848041 LOC_Os08g39140
KCG214B01 glutathione S-transferase 239 GQ848061 LOC_Os10g38495
Fig. 3 The patterns of differentially expressed genes detected by immature seed and germinating seed in rice.
We were sequenced 43,744 clones and selected 13,989 unique clones, also we were clustered genes with immature seed and germinating seed genes using SeqMan program
하여 발아종자에서 148종의 단백질변화를 분석 하였으 며 전분 분해 및 대사관련 유전자들의 발현이 증대되고 저장단백질 등이 감소됨을 보고 하였다. 또한 Xiao (2007) 등은 발아종자에서 대사체분석을 행하여 174개의 대사 체 변화를 검출하였다.
발아종자 발현유전자 발현특성분석
종자의 발아는 단백질수화작용, 심층세포의 구조변화, 호흡, 미세물질의 합성 그리고 신장 등의 복잡한 과정을 동반 한다 (Bewley et al. 1985).
벼 발아종자에 발현하는 유전자들의 발현특성을 분석 하기 위하여 우선 발아종자 발현유전자 cluster 분석을 통 하여 얻어진 contig 중에서 20개 이상의 클론들로 구성된 발현유전자들의 완전장 염기서열을 결정하였으며 25개 의 유전자를 GenBank에 등록하였다 (표 2). 이들 종자발아 발현유전자들의 염색체내 위치 결정은 RAP DB와 TIGR Pseudomolecule ver. 6 DB 에서 BLASTN 분석을 통해 유사
성이 99% 이상을 나타내는 염색체 부위를 판별하여 각 유전자의 염색체 위치를 결정하였다. 종자발아시기에 가 장 많이 발현하는 유전자는 당대사에 관여하는 glyceralde- hyde-3-phosphate dehydrogenase 유전자였다. 이 유전자는 TIGR DB에서는 벼 염색체내에 6종이 존재하는 것으로
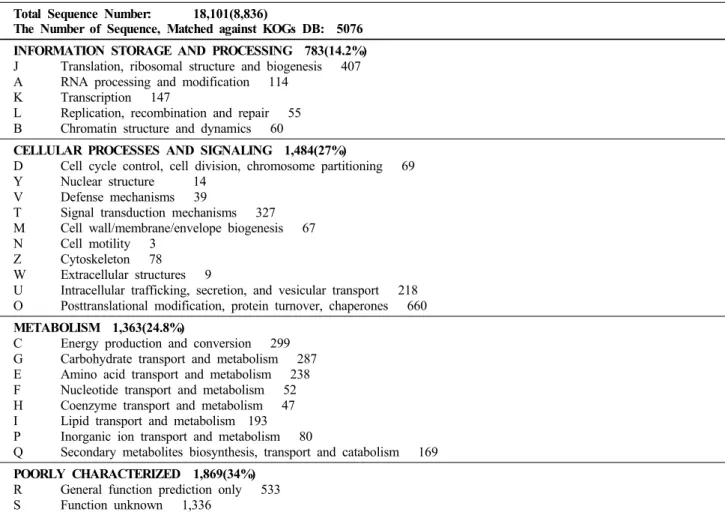
Table 3 Functional classification of unigenes by the KOG database Total Sequence Number: 18,101(8,836)
The Number of Sequence, Matched against KOGs DB: 5076 INFORMATION STORAGE AND PROCESSING 783(14.2%) J Translation, ribosomal structure and biogenesis 407 A RNA processing and modification 114
K Transcription 147
L Replication, recombination and repair 55 B Chromatin structure and dynamics 60
CELLULAR PROCESSES AND SIGNALING 1,484(27%)
D Cell cycle control, cell division, chromosome partitioning 69 Y Nuclear structure 14
V Defense mechanisms 39
T Signal transduction mechanisms 327 M Cell wall/membrane/envelope biogenesis 67 N Cell motility 3
Z Cytoskeleton 78 W Extracellular structures 9
U Intracellular trafficking, secretion, and vesicular transport 218 O Posttranslational modification, protein turnover, chaperones 660 METABOLISM 1,363(24.8%)
C Energy production and conversion 299 G Carbohydrate transport and metabolism 287 E Amino acid transport and metabolism 238 F Nucleotide transport and metabolism 52 H Coenzyme transport and metabolism 47 I Lipid transport and metabolism 193 P Inorganic ion transport and metabolism 80
Q Secondary metabolites biosynthesis, transport and catabolism 169 POORLY CHARACTERIZED 1,869(34%)
R General function prediction only 533 S Function unknown 1,336
The 8,836 unigenes of germinating seed were classified into functional groups by the KOG database. Data sets were obtained from public databases as NCBI KOGs
보고되어 있으며 본 연구 결과, 발아시기에 3종의 유전자 (KCG225E10 : GQ848032, KCG185F05 : GQ848031, KCG106H12 : EU267971)의 발현이 많이 나타났다. Mangneschi 등 (2009) 은 산소결핍 상태의 종자발아시기에 발현하는 유전자들 의 양상을 분석한 결과 3종의 glyceraldehyde-3-phosphate dehydrogenase 유전자의 발현 변화가 나타났으며 이들 유 전자중 2종은 높게 1종은 낮게 발현하는 것으로 보고하 고 있다.
전분분해효소인 alpha-amylase 유전자의 경우 벼에는 9 종이 존재하며 Yu 등 (1998)은 발아시기에 3종의 유전자 들이 발현하는 것으로 보고 하였으나 본 실험에서는 1종 (KCG108F03 : EU267974)이 많이 발현하였다. 단백질분해 효소로 chinase (KCG045D08 : EF122477)와 cystein endopeptidase (KCG217D06 : EF122485)등의 발현도 많이 나타났는데 저 장단백질의 분해에 관여 할 것으로 생각된다.
특히 미숙종자와 발아종자에 공통으로 발현하는 LEA 유전자들은 종자의 성숙, 생리대사 및 발아 등에 역할을 하는데 glyceraldehyde-3-phosphate dehydrogenase (KCG225E10 : GQ848032), alcohol dehydrogenase 1 (KCG231E03 : GQ848036), manganese-superoxide dismutase (KCG228D08 : GQ848046) 등
이 미숙종자와 발아종자에서 공통발현을 하고 있었다. 또 한 특이적으로 발아종자에서만 발현유전자들로는 Chitinase (EF122477), NADP-specific isocitrate dehydrogenase (GQ84053), glycine-rich protein (GQ848055), glutathione S-transferase (G 848061)등이 있었다. 그 외 enolase (KCG222G09 : EF122486), fructose-bisphosphate aldolase (KCG218A11 : GQ848028) 유 전자들이 많이 발현하였다.
본 연구를 통하여 얻어진 발아종자관련 정보들은 발 아종자의 생리연구 및 농업형질개선 연구에 기여할 수 있을 것으로 기대되어 진다.
KOGs 분석을 이용한 유전자 기능분류
발아종자 18,101 개 EST의 8,836개 unigene에 대한 기능 분류를 위하여 KOG분석을 행한 결과 약 57.4%에 해당하 는 5,076개의 unigene이 KOG 분류에 속하였으며 4개의 대 분류군중 Information storage and processing 14.2%, Cellular processes and signaling 27%, Metabolism 24.8%, Poorly char- acterized 34%의 비율을 나타내었다 (표 3). 발아종자 발현 유전자의 기능분류 결과 번역, 발생, 리보솜 구조 등에
관한 유전자 발현이 많이 나타났으며 특히 전사 후 수정, 단백질 이동, 사페론의 유전자등이 가장 많이 발현하였 다. 앞서 발표한 미숙종자와의 발현분석 비교에서는 번 역에 관한 유전자의 발현이 현저히 감소하였으며 에너지 대사의 경우는 현저히 높게 발현하는 것으로 나타났다.
이러한 결과는 발아종자의 경우 종자휴면기에서 흡수과 정을 거쳐 발아를 위하여 에너지생산 및 아밀라제 등 전 분분해 대사관련 유전자의 발현이 많이 이루어지는 시기 로 이들 유전자들의 발현양이 미숙종자에 비해 상대적으 로 높게 나타나는 것으로 생각되어진다. Umezawa 등은 콩의 68,661개 EST 및 이들로부터 얻어진 4,712개 완전장 유전자를 이용하여 KOG 분석을 행한 결과 세포생리기 능관련 유전자 및 대사관련 유전자들의 발현은 동등한 비율로 발현을 나타내는 것으로 보고하였으며 발아종자 의 경우도 같은 비율을 나타내었다 그러나 벼 미숙종자 의 경우는 세포생리기능관련 유전자들의 발현이 대사관 련 유전자들 보다 7% 높은 비율로 높은 발현을 나타내었 다 (Yoon et al. 2009).
적 요
종자의 발아기는 휴면기에서 에너지대사, 전분분해 및 세포분열 등 일련의 많은 유전자 발현을 나타내는 시기 이다. 종자의 발아기에 관여하는 유전자의 기능을 분석 하기 위하여 일품 벼를 48시간 발아시킨 후 얻은 발아종 자 cDNA 클론의 염기서열을 결정하였다. 염기서열이 결 정되어진 18,101개의 EST를 SeqMan 프로그램을 이용하 여 cluster 분석을 행한 결과 8,836개의 unigene을 분리 하 였다. 발아종자에서 많이 발현하는 유전자의 완전장 구 조분석을 행한 결과 발아종자 특이적으로 발현하는 키티 나아제 및 전분분해에 관여하는 아밀라제 유전자등 몇 종류의 특이발현 유전자들을 분리 하였으며 이들 유전자 들은 GenBank에 등록하였다.
미숙종자와 발아종자 공통적으로 발현하는 유전자의 분석을 위하여 미숙종자 25,668개와 발아종자 18,101개 등 43,744개의 유전자를 SeqMan 프로그램으로 cluster 분 석을 행한 결과 2,541개의 공통발현 unigene을 얻을 수 있 었다. 이들 유전자중 alpha-glubulin 및 alcohol dehydrogenase 1 등은 미숙종자와 발아종자에서만 발현을 나타내어 LEA 유전자로 추정되어진다.
발아종자 8,836개의 유전자 기능분류를 NCBI KOG DB 를 이용하여 분석한 결과 정보축적 및 처리기능관련유전 자 783개 (14.2%), 세포생리기능관련 유전자 1,484개 (27%), 대사관련 유전자 1,363개 (24.8%) 그리고 기능미확인 유 전자 1,869개 (34%)의 비율을 나타내었다.
사 사
이 연구는 농촌진흥청 국립농업과학원 어젠더 2-7-11
“벼 변이집단 및 생물정보를 이용한 유용 농업형질 유전 자탐색” 과제, “벼·배추의 생장 및 대사관련 유전자발현 네트워크구축” 과제 및 “바이오그린21사업” 과제지원에 의해 수행되었다. 본 연구수행 중 발아종자 EST DNA 분 리와 염기서열 분석은 최은영, 양희은양이 수고해 주었다.
인용문헌
Bewley DJ, Black M. (1985) Seeds, Physiology of development and germination, Plenum press, New York, pp135-168 Bove J, Jullien M, Grappin P. (2002) Functional genomics in the
study of seed germination. Genome Biol 3:1-5
Carninci P, Kvam C, Kitamura A, Ohsumi T, Okazaki Y, Itoh M, Kamiya M, Shibata K, Sasaki N, Izawa M, Muramatsu M, Hayashizaki Y, Schneider C. (1996) High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics 37:
327-36
Galau GA, Hughes DW. (1987) Coordinate accumulation of homeologous transcripts of seven cotton Lea genefamilies during embryogenesis and germination. Dev Biol 123:213-21 Huang J, Takano T, Akita S. (2000) Expression of alpha-expansin
genes in young seedlings of rice (Oryza sativa L.). Planta 211:467-473
International Rice Genome Sequencing Project. (2005) The map- based sequence of the rice genome. Nature 436:793-800 Ishibashi N, Yamauchi D, Minamikawa T. (1990) Stored mRNA in
cotyledons of Vigna unguiculata seeds: nucleotide sequence of cloned cDNA for a stored mRNA and induction of its synthesis by precocious germination. Plant Mol Biol 15:59-64 Jung KH, An G, Ronald PC. (2008) Towards a better bowl of rice:
assigning function to tens of thousands of rice genes. Nat Rev Genet 9:91-101
Kikuchi S, Satoh K, Nagata T, Kawagashira N, Doi K, Kishimoto N, Yazaki J, Ishikawa M, Yamada H, Ooka H, Hotta I, Kojima K, Namiki T, Ohneda E, Yahagi W, Suzuki K, Li CJ, Ohtsuki K, Shishiki T; Foundation of Advancement of International Science Genome Sequencing & Analysis Group, Otomo Y, Murakami K, Iida Y, Sugano S, Fujimura T, Suzuki Y, Tsunoda Y, Kurosaki T, Kodama T, Masuda H, Kobayashi M, Xie Q, Lu M, Narikawa R, Sugiyama A, Mizuno K, Yokomizo S, Niikura J, Ikeda R, Ishibiki J, Kawamata M, Yoshimura A, Miura J, Kusumegi T, Oka M, Ryu R, Ueda M, Matsubara K;
RIKEN, Kawai J, Carninci P, Adachi J, Aizawa K, Arakawa T, Fukuda S, Hara A, Hashizume W, Hayatsu N, Imotani K, Ishii Y, Itoh M, Kagawa I, Kondo S, Konno H, Miyazaki A, Osato N, Ota Y, Saito R, Sasaki D, Sato K, Shibata K, Shinagawa A, Shiraki T, Yoshino M, Hayashizaki Y, Yasunishi A. (2003) Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science 301:376-9
Kim CK, Kikuchi S, Satoh K, Kim JA, Kim DH, Kim YH, Park SH, Lee JH, Yoon UH. (2009) Genetic analysis os seed-specific gene expression for pigmentation in colored rice. Biochip Journal 3:125-129
Lu T, Huang X, Zhu C, Huang T, Zhao Q, Xie K, Xiong L, Zhang Q, Han B. (2008) RICD: a rice indica cDNA database resource for rice functional genomics. BMC Plant Biol 8:118 Magneschi L, Perata P. (2009) Rice germination and seedling growth
in the absence of oxygen. Annals of Botany 103:181-196 Nakamura H, Hakata M, Amano K, Miyao A, Toki N, Kajikawa M,
Pang J, Higashi N, Ando S, Toki S, Fujita M, Enju A, Seki M, Nakazawa M, Ichikawa T, Shinozaki K, Matsui M, Nagamura Y, Hirochika H, Ichikawa H. (2007) A genome-wide gain-of function analysis of rice genes using the FOX-hunting system.
Plant Mol Biol 65:357-71
Ouyang S, Zhu W, Hamilton J, Lin H, Campbell M, Childs K, Thibaud-Nissen F, Malek RL, Lee Y, Zheng L, Orvis J, Haas B, Wortman J, Buell CR. (2007) The TIGR Rice Genome An- notation Resource: improvements and new features. Nucleic Acids Res. 35(Database issue):D883-7
Park DS, Park SK, Han AI, Wang HJ, Jun NS, Manigbas NL, Woo YM, Ahn BO, Yun DW, Yoon UH, Kim YW, Lee MC, Kim DH, Nam MH, Han CD, Kang HW, Yi G. (2009) Genetic variation through Dissociation(Ds) insertional mutagenesis system for rice in Korea : progress and current ststus. Mo- lecular Breeding 24:1-15
Rice Annotation Project. (2008) The Rice Annotation Project Database (RAP-DB). Nucleic Acids Res. 36(Database issue):
D1028-33
Smith AM, Zeeman SC, Smith SM. (2005) Starch degradation.
Annu Rev Plant Biol 56:73-98
Song XJ, Huang W, Shi M, Zhu MZ, Lin HX. (2007) A QTL for rice grain width and weight encodes a previously unknown
RING-type E3 ubiquitin ligase. Nat Genet 39:623-30 Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B,
Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41
Tatusov RL, Koonin EV, Lipman DJ. (1997) A genomic perspec- tive on protein families. Science 278:631-637
Umezawa T, Sakurai T, Totoki Y, Toyoda A, Seki M, Ishiwata A, Akiyama K, Kurotani A, Yoshida T, Mochida K, Kasuga M, Todaka D, Maruyama K, Nakashima K, Enju A, Mizukado S, Ahmed S, Yoshiwara K, Harada K, Tsubokura Y, Hayashi M, Sato S, Anai T, Ishimoto M, Funatsuki H, Teraishi M, Osaki M, Shinano T, Akashi R, Sakaki Y, Yamaguchi-Shinozaki K, Shinozaki K. (2008) Sequencing and analysis of approxi- mately 40,000 soybean cDNA clones from a full-length- enriched cDNA library. DNA Res 15:333-46
Xiao B, Huang Y, Tang N, Xiong L. (2007) Over-expression of a LEA gene in rice improves drought resistance under the field conditions. Theor Appl Genet 115:35-46
Xiao LS, Frank T, Shu QY, Engel KH. (2008) Metabolite profiling of germinating rice seeds. J. Agric. Food Chem 56:11612-11620 Yang P, Li X, Wang X, Chen H, Chen F, Shen S. (2007) Proteomic analysis of rice (Oryza sativa) seeds during germination.
Proteomics 7:3358-68
Yoon UH, Lee GS, Lee JS, Hahn JH, Kim CK, Kikuchi S, Satoh K, Kim JA, Lee JH, Lee TH, Kim YH. (2009) Structural Analysis of Seed Developmental Stage ESTs in Oryza sativa L. Korean J of Plant Biotechnology 36:130-136
Yu SM. (1998) Molecular biology of rice, “Regulation of alpha- amylase gene expression”, Springer-Verlag press, pp 161-178