토마토 marker-assisted background selection을 위한 SNP 마커 선발 데이터베이스
김지은 ・ 이봉우 ・ 김상미 ・ 이보미 ・ 이정희 ・ 조성환*
㈜씨더스
Genome-wide SNP Database for Marker-assisted Background Selection in Tomato
Ji-Eun Kim, Bong-Woo Lee, Sang-Mi Kim, Bo-Mi Lee, Jeong-Hee Lee, and Sung-Hwan Jo*
SEEDERS Inc., Daejeon, 305-509, Korea
Abstract : Backcrossing is a plant breeding method most commonly used to incorporate one or a few genes into an adapted or elite variety. To facilitate MAB (marker-assisted backcrossing) in a practice breeding program, we developed a SNP database and a program for providing selected markers for background selection from genome-wide SNPs of seven tomato accessions downloaded from NCBI-SRA. We identified 410,074 SNPs among 21 parental combinations with data from seven transcriptomes and developed a SNP database. To select the optimized number of markers for background selection, we divided 12 chromosomes according to physical length and genetic length. Initially, each chromosome was equally divided into five blocks according to physical length, and three SNPs were positioned per block. Additionally, we applied the genetic distance from tomato-EXPEN 2000 map because the frequency of recombination can vary greatly among chromosomal regions. When considering genetic distance, each chromosome was divided into fifteen blocks unequally and one SNP was positioned per block. The program for background selection was designed to be simple and easy to use, and it is available at http://tgsol.seeders.co.kr/index.php/tg/mab.
When the user selects the parental combination, the program provides selected markers with primer information. The value of this program for tomato breeding will further increase if more accession numbers are added to the database.
Keywords : MAB, Background selection, Genome-wide SNPs, NGS, Tomato
*Corresponding author (E-mail: [email protected], Tel: +82-42- 710-4035, Fax: +82-42-710-4036)
(Received on June 10, 2013. Revised on August 11, 2013.
Accepted on August 16, 2013.)
Copyright ⓒ 2013 by the Korean Society of Breeding Science
232This is an Open-Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
서 언
표준유전체 정보가 밝혀지고 NGS (Next generation sequ- encing)를 이용한 대용량 시퀀스 생산이 가속화되면서 유전 체 기반의 분자육종은 종자시장 및 육종산업에 새로운 동력 으로 각광받고 있다. 분자육종은 원하는 형질의 유무를 형질 에서 유래한 표현형의 관찰 없이 DNA 염기서열의 차이를 보 이는 분자마커(molecular marker)를 이용해 판별하는 기법이 다(Edwards & Batley 2010). 분자육종 과정은 크게 개량하 고자 하는 새로운 형질을 도입하는 과정과 회복친(recurrent)
의 우수형질을 잘 유지시키는 과정으로 나눌 수 있다. 이 중 MAB (Marker-assisted Backcrossing)는 후자에 속하는 기술 로 3개의 하위단계를 포함하는데, 먼저는 목표형질 혹은 QTL 로의 대체를 위한 마커를 사용하는 ‘foreground selection’이 있고, 두 번째로 목표유전자와 연관된 마커 사이의 재조합이 일어나는 여교잡 자손을 선발하는 ‘recombination selection’
이 있다. 마지막이자 본 연구의 목적인 ‘background selection’
은 목표유전자와 연관되어 있지 않은 마커를 사용하여 상당 부분 회복친의 유전체 구성을 갖는 여교잡 자손을 선발하는 것이다(Collard & Mackill 2008). 다시 말하면, MAB는 분 자마커를 사용하여 염색체 전체를 확인함으로써 여교잡으로 얻어진 자손이 우수친으로 회복하는데 걸리는 시간을 단축시 키는 중요 육종기법이다(Edwards & Batley 2010, Jena &
Mackill 2008).
기존의 여교잡 육종이 6-7세대 이상의 시간이 소요되는 것 에 비하여 MAB 육종은 여교잡 2세대에서부터 선발이 가능 하다고 보고되어(Collard & Mackill 2008) 신품종 육성 기간 을 단축하고, 작물개량 시 소요되는 노력과 비용을 절감해 육 종의 규모와 효율성을 증대시켜 경쟁우위를 확보할 수 있는 이점을 제공한다(Ibitoye & Akin-Idowu 2010, Xu & Crouch 2008). 벼를 비롯한 다양한 작물에서 MAB 육종기술을 성공 적으로 적용한 사례들이 보고되고 있는데(Neeraja et al. 2007), 이러한 MAB 기술이 육종현장에 적용되는데 필요한 조건에 는 첫째, 충분한 수의 genome-wide한 MAB용 마커를 확보 하여야 하며, 둘째로 교배조합이 바뀔 때마다 적용 가능한 마 커를 쉽게 확인할 수 있어야 한다. 또한 genotyping에 필요한 분자마커를 최소화하여 비용을 최소화하여야 한다. 하지만 육 종에 실질적인 도움이 되는 이러한 MAB 기술들은 아직 미흡 한 상황이다.
가장 대표적인 분자마커 중의 하나인 SNPs (Single Nucleotide Polymorphisms)는 DNA 서열에서 일어나는 단일 염기의 변 이로 유전체 전체적으로 가장 빈번하게 나타나며, 안정적으로 이용할 수 있는 장점이 있다(Gupta et al. 2001). 이에 따라, 최근 여러 작물에서 NGS를 통해 해독된 유전체 정보를 기반 으로 한 genome-wide SNPs 발굴로 대량의 분자마커를 빠르 게 확보하고 있다(Hyten et al. 2010, Trebbi et al. 2011, Chagné et al. 2012). 또한 SNP는 in silico 분석을 통해 농업 적으로 중요한 유전자를 확인할 수 있는 유용한 MAB 마커로 사용되는 등 응용 범위가 확대되고 있다(An et al. 2010, Cuesta-Marcos et al. 2010, Xu et al. 2012).
토마토는 전세계적으로 경제적 가치가 높은 채소일 뿐 아 니라 가지과 작물의 육종모델로 이용되고 있다(Foolad 2007).
토마토에 관한 다양한 유전체 정보(The Tomato Genome Consortium 2012), 주요 육종 형질 관련 분자마커 및 유전자 지도(Fulton et al. 2002, Shirasawa et al. 2010a) 등이 대표 적으로 SGN 데이터베이스(Sol Genomics Network; http://
solgenomics.net/)에 잘 정리되어 있다(Bombarely et al. 2011).
하지만 현재 대부분의 공개된 데이터베이스는 기존 대용량의 유전체 정보를 정리하여 기초연구를 지원하는데 초점을 두고 있어, 육종현장에 필요한 정보, 즉 실제 이용할 수 있는 MAB 분자마커를 제공하는 기능적 데이터베이스 구축이 필요하다.
따라서 본 연구는 토마토 MAB 분자마커의 활용성을 높이 기 위하여 공개된 토마토 7 품종의 NGS 데이터를 이용하여 genome-wide SNPs를 확인하고, 교배조합별 MAB용 분자마
커 및 primer를 선발하는 프로그램 개발 및 데이터베이스화 를 수행하였다.
재료 및 방법
토마토 NGS 데이터의 genome-wide SNPs 분석 차세대 염기서열분석을 통해 생산된 토마토의 sequencing data는 공개 데이터베이스 NCBI-SRA (Short Read Archive database; http://www.ncbi.nlm.nih.gov/sra)에서 수집하였다.
이들은 토마토 7개 계통의 전사체(transcriptome) 데이터 10 개로 구성되며(Hamilton et al. 2012), 이를 이용하여 토마토 표준유전체(S. lycopersicum Heinz 1706, ITAG 2.3) (SGN.
2011) 대비 발생한 genome-wide SNPs를 추출하였다.
서열의 품질(sequence quality)을 측정하고, 기준 이상 품 질의 서열로 filtering하는 SolexaQA package 프로그램을 이 용하여 데이터 전처리 과정을 진행하였다(Cox et al. 2010).
서열 품질의 기준 값은 phred score 20으로 주어, 서열을 이 루는 염기(base) 하나의 품질이 20 보다 낮은 것은 잘라버린 후, 남은 서열의 길이가 25 bp 이상인 서열만 얻는 작업을 수 행하였다(Ness et al. 2011, Garg et al. 2011).
수집한 데이터는 전사체이므로 gene splicing junction을 고려한 TopHat (v1.4.1; Trapnell et al. 2009)을 이용하여 표 준유전체로의 alignment를 수행하였고, 적용한 옵션은 다음 과 같다. Min-intron-size는 40, max-intron-size는 23,000, mismatches는 1로 설정하였다. 전사체 데이터 중 2개의 PI212816 데이터와 3개의 M82 데이터는 표준유전체와의 alignment 후에, 각각 1개의 파일로 합친 후, SNP 분석을 진 행하였다. SAMtools (v0.1.16; Li et al. 2009)를 이용하여 각 샘플의 alignment 결과에서 genome-wide SNPs를 추출하 였다. SAMtools 사용 옵션은 기본값으로 수행하였고, 추출된 SNP 결과에서 정확도 높은 SNP를 선발하기 위해 mapping quality 값을 기본값 25보다 높은 30으로 적용하고, 적어도 3 개 이상의 raw reads를 갖는 SNP로 필터링하였다.
교배조합별 이용 가능한 SNP 분자마커 데이터베이스 개발 분석에 이용한 7 품종의 토마토 계통을 이용하여 교배 가 능한 21개 조합을 도출하고, 조합별로 이용 가능한 SNP를 선 발하여 데이터베이스화 하였다. 또한 MAB genotyping을 위 하여 표준유전체 정보를 이용해 SNP 검출용 프라이머(primer) 를 디자인하였고, 프라이머 제작에는 Primer3 (v2.3.5; http://
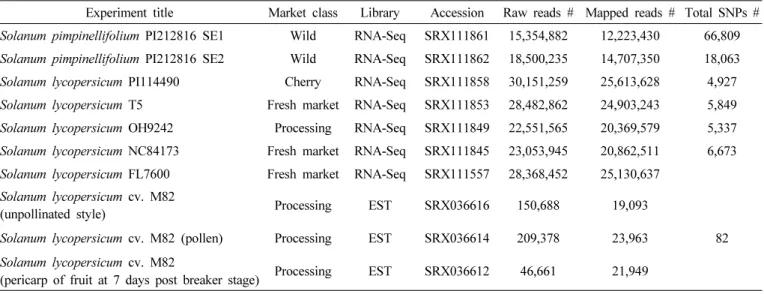
Table 1. Summary of tomato sequencing data. Each row represents dataset information used in this study.
Experiment title Market class Library Accession Raw reads # Mapped reads # Total SNPs # Solanum pimpinellifolium PI212816 SE1 Wild RNA-Seq SRX111861 15,354,882 12,223,430 66,809 Solanum pimpinellifolium PI212816 SE2 Wild RNA-Seq SRX111862 18,500,235 14,707,350 18,063 Solanum lycopersicum PI114490 Cherry RNA-Seq SRX111858 30,151,259 25,613,628 4,927 Solanum lycopersicum T5 Fresh market RNA-Seq SRX111853 28,482,862 24,903,243 5,849 Solanum lycopersicum OH9242 Processing RNA-Seq SRX111849 22,551,565 20,369,579 5,337 Solanum lycopersicum NC84173 Fresh market RNA-Seq SRX111845 23,053,945 20,862,511 6,673 Solanum lycopersicum FL7600 Fresh market RNA-Seq SRX111557 28,368,452 25,130,637
Solanum lycopersicum cv. M82
(unpollinated style) Processing EST SRX036616 150,688 19,093
Solanum lycopersicum cv. M82 (pollen) Processing EST SRX036614 209,378 23,963 82 Solanum lycopersicum cv. M82
(pericarp of fruit at 7 days post breaker stage) Processing EST SRX036612 46,661 21,949
primer3.sourceforge.net/releases.php) 프로그램을 이용하였다(You et al. 2008). 프라이머 제작 조건은 다음과 같다. 프 라이머 크기는 18~22 bp, 증폭산물의 크기는 180~220 bp, annealing 온도는 55~65℃ 로 설정하였다. 디자인된 프라이 머 서열을 모두 토마토 표준 유전체에 mapping 하여, SNP 지점 외에 다른 영역에 결합되지 않는지 검정하였다. Mapping 조건 중, mismatch 값을 2로 주었을 때 SNP 지점에만 결합 되는 프라이머 만을 선발하여 DB화 하였다.
염색체 부위별 교차율 조사 및 염색체 분획
MAB 마커를 선발하기에 앞서 토마토 염색체의 분획은 단 순 물리적인 거리(physical distance) 혹은 유전적 거리(genetic distance; cM)를 반영한 2가지 경우를 모두 적용하였다. 먼저 각 염색체의 물리적 길이를 통해 5개의 동일한 크기의 구획으 로 나누었고, 각 구획 안에서 이용 가능한 마커를 선발하였다.
이 때 사용한 마커는 토마토 7 품종의 교배조합별로 프라이머 가 이용 가능한 SNP를 이용하였다.
두번째로는 물리적 거리와 유전적 거리의 상관관계를 분석 하여 염색체를 분획하였다. 유전자 재조합 비율을 측정하기 위해 이용한 마커는 S. lycopersicum LA925 와 S. pennellii LA716 type F2.2000 간 tomato-EXPEN 2000 map의 분자 마커 2,604개(Fulton et al. 2002)를 SGN (ftp://ftp.solgenomics.
net/maps_and_markers/Markers/curr/Tomato-EXPEN-2000.
fasta)에서 수집하였다(Shirasawa et al. 2010a, Shirasawa et al. 2010b). 수집된 마커를 BLAST를 통해 토마토 genome
sequence 에 mapping 한 결과, 실제로 mapping 되는 1,924 개 마커를 이용하여 재조합 비율을 분석하였다(Bowers et al.
2012). 분석한 재조합 비율을 적용하여 각 염색체를 15개로 구획하였고, 각 구획 안에서 이용 가능한 마커를 선발하였다.
15개의 각 영역에서 선발되는 마커는 토마토 7 품종의 교배 조합별로 프라이머가 이용 가능한 SNP를 이용하였다.
MAB 분자마커 선발 프로그램 개발
MAB 분자마커 선발 프로그램은 웹 기반으로 작성되었고, 이용하고자 하는 교배조합을 선택하는 선택창을 제공하여 선 택된 교배조합에 따라 자동으로 적용 가능한 SNP의 염색체 별 분포를 그래픽화 하였다. 프로그램 하단의 표에는 교배조 합 간에 이용 가능한 SNP 마커와 마커 확인을 위해 제작된 프라이머 쌍에 대한 정보로 구성하여 제공하였다.
결과 및 고찰
토마토 NGS 데이터의 genome-wide SNPs 분석
NCBI-SRA로부터 수집된 토마토 7 품종의 transcriptome 데이터를 이용하여 genome-wide SNPs를 선발하였다(Table 1).
Solanum pimpinellifolium PI212816에서 가장 많은 66,809 개의 SNP가 추출되었는데, 이는 표준유전체와 근연 관계가 멀고, 또 분석에 사용된 서열의 양이 7개 데이터 중 가장 많 았기 때문인 것으로 판단하였다(Jiménez-Gómez & Maloof 2009). 표준유전체와 동일한 6개의 S. lycopersicum 전사체
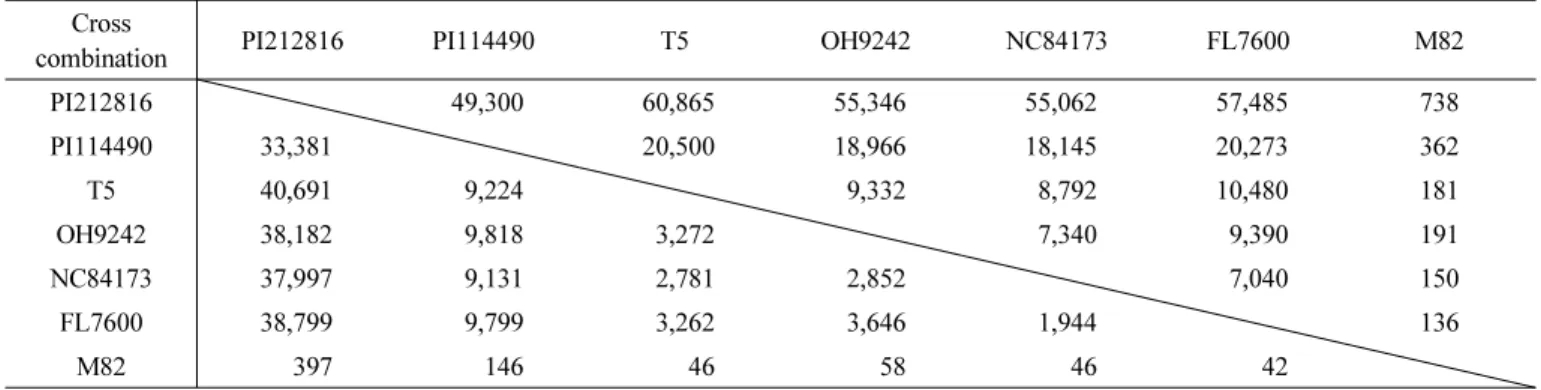
Table 2. SNP markers for tomato MAB depending on cross combination. The right top region indicates total SNP marker count (410,074) and left bottom region indicates SNP marker count with available primer (245,514).
Cross
combination PI212816 PI114490 T5 OH9242 NC84173 FL7600 M82
PI212816 49,300 60,865 55,346 55,062 57,485 738
PI114490 33,381 20,500 18,966 18,145 20,273 362
T5 40,691 9,224 9,332 8,792 10,480 181
OH9242 38,182 9,818 3,272 7,340 9,390 191
NC84173 37,997 9,131 2,781 2,852 7,040 150
FL7600 38,799 9,799 3,262 3,646 1,944 136
M82 397 146 46 58 46 42
데이터의 SNP는 82 개부터 18,063 개까지 추출되었다. 추출 된 SNP의 개수에 많은 차이를 보였는데, 이는 품종에 따른 표 준유전체와의 서열 유사 정도에 따라 나타난 polymorphism 의 차이로 보인다(Jiménez-Gómez & Maloof 2009, Jung et al. 2010). 가장 적은 82개 SNP가 추출된 M82 품종은 분석 에 사용된 서열 데이터 량이 약 105Mb로, 다른 데이터의 0.05~0.1%에 해당하는 매우 적은 양이었다. 이 요인 또한 가장 적은 SNP가 추출된 것에 영향을 준 것으로 예측되었다.
교배조합별 이용 가능한 SNP 분자마커 데이터베이스 구축 확보된 genome-wide SNPs는 표준유전체와 비교하여 얻 어졌으므로 비교대상이 바뀌면 SNP로 활용할 수 없는 경우 가 발생한다. 따라서 분석을 실시한 토마토 7 품종의 SNP를 이용하여 교배조합별 이용 가능한 SNP를 선발하였다(Table 2).
가능한 총 교배조합은 21개이고, 총 410,074 SNPs가 선발되 었다. 이 중 조건을 만족하는 primer를 디자인 할 수 있는 SNP는 245,514개이었다. 교배조합별 이용 가능한 SNP는 다 양하였는데 PI212816 × T5 조합은 가장 많은 60,865 SNPs 가 선발되었고, 그 중 primer를 이용할 수 있는 SNP는 40,691 개이었다. 반면 FL7600 × M82 조합은 가장 적은 136 SNPs 가 선발되었는데, 이용 가능한 primer를 가지는 SNPs 로는 42개가 존재하였다. M82의 경우 데이터 부족으로 인해 전체 적으로 적은 SNP가 추출되었고, M82와 교배조합을 이루는 경우 이용할 수 있는 SNP 개수가 제한될 수 있다. 적용 가능 한 21개 조합의 SNP를 선발 후, primer 제작 조건을 만족하 는 경우의 SNP 만을 데이터베이스화 하였다. 또한 이용 가능 한 모든 SNP는 표준유전체의 위치 정보를 데이터베이스에 포함시켜 위치에 따라 선발이 가능하도록 하였다.
염색체 부위별 교차율 조사 및 염색체 분획
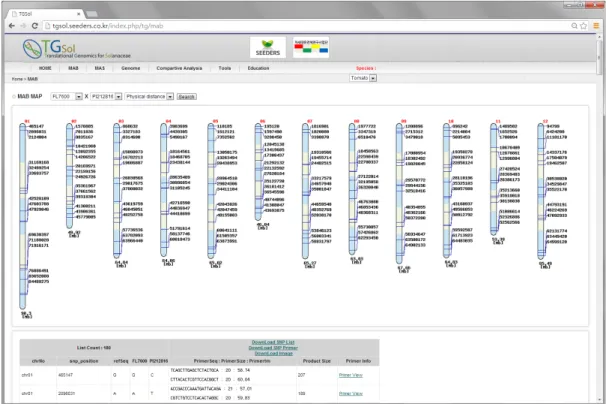
분자마커를 이용하여 회복친의 염색체 구성 정도를 효율적 으로 조사하기 위해서는 염색체를 적절히 구획하여 조사하여 야 한다. 따라서 토마토 염색체의 분획은 물리적인 거리를 반 영한 경우와 유전적 거리까지 고려한 경우, 2가지로 접근하였 다. 먼저 각 염색체의 물리적인 길이로 구획한 경우는 염색체 당 5개의 동일한 크기로 나누었다. 약 90Mb의 길이로 12개 염색체 중 가장 긴 1번 염색체는 5개의 18Mb 길이의 구획으 로 나뉘었고, 약 46Mb 길이의 가장 짧은 6번 염색체는 5개의 9Mb 길이로 분획되었다. 그리고 토마토 7 품종의 교배조합 별로 primer가 이용 가능한 SNP를 이용하여 한 구획 당 3개 의 SNP를 선발하여 나타내었다(Fig. 1).
염색체의 위치에 따라 유전자 재조합 빈도가 다르기 때문 에 재조합이 자주 일어나는 영역은 그렇지 않은 영역보다 더 많은 마커를 사용할 필요가 있으며, 동시에 MAB의 목적에 맞게 염색체 전체의 배경을 확인해야 한다. 따라서 사용하는 마커 수를 최소화하면서 효율적인 background selection을 위해 유전 거리를 고려하여 염색체를 차등적으로 나누었다 (King et al. 2002, Wu et al. 2003). 먼저 수집한 2,604개의 tomato-EXPEN 2000 map의 분자마커는 BLAST 분석을 통 해 토마토 표준유전체에서 위치를 확인하였고, 실제로 확인되 는 1,924개의 마커를 염색체에서 확보하였다. 이를 이용해 염 색체 내 물리적 거리와 유전적 거리를 비교하였다(Fig. 2). 12개 염색체 전반적으로, centromere를 포함하는 heterochromatin으 로 알려진 부위는 물리적 길이에 비해 유전적 길이가 매우 짧 게 나타나서 재조합이 매우 적은 것으로 나타나는 반면 염색 체의 양 말단은 물리적 길이의 변화와 유전적 길이 변화가 동 시에 증가하는 결과를 보여주었다. 그러나 토마토의 2번 염색 체의 경우 short arm에 NOR (Nucleolar Organizer Region)
Fig. 1. Snap shot of MAB marker selecting program using genome-wide SNPs divided by physical distance. FL7600×PI212816 combination was selected as an example. Partitioned regions in each chromosome were described with blue and yellow shade and blue lines indicate SNP positions in that region. At the foot of that page, info of each SNP and primer sets were listed.
Fig. 2. Recombination rate using physical distance (x axis) and genetic distance (y axis) from tomato-EXPEN 2000 map (S. lycopersicum
LA925, S. pennellii LA716 type F2.2000).
Fig. 3. Snap shot of MAB marker selecting program divided by genetic distance. Like as program in figure 1, partitioned regions in each chromosome were described with blue and yellow shade. Each blue or purple line is SNP position in that region.
을 가지고 있어 heterochromatin과 동일한 경향을 나타냈다.
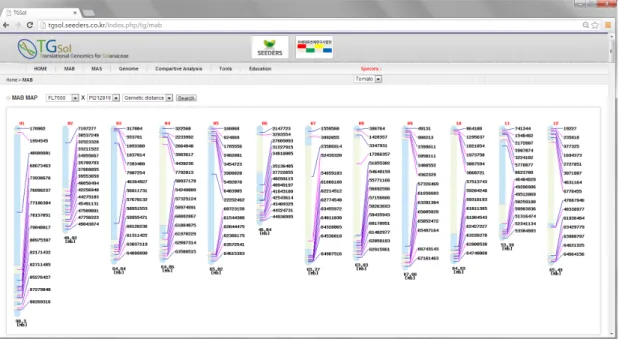
본 결과를 통해 염색체의 위치에 따라 유전자 재조합 빈도 가 상당히 다르게 발생하는 것을 확인하였고, 이를 반영하여 염색체 구획을 나누었다. 즉 재조합이 적게 일어나는 영역으 로 염색체 상에서 주로 중앙 부분은 구획을 넓게 나누었고, 재조합이 비교적 자주 일어나는 영역인 염색체의 양끝 말단 부분은 마커 선발 구획 또한 세분화하여 나누어 자세히 조사 하도록 하였다. 결과적으로 각 염색체를 15등분하되 교차 비 율을 적용하여 염색체의 구획의 크기를 차등화하였고, 토마토 7 품종의 교배조합별로 primer가 이용 가능한 SNP를 이용하 여 한 구획 당 1개의 SNP 마커를 선발하여 나타내었다(Fig. 3).
교배조합별 MAB 마커 선발 프로그램 개발
토마토 MAB 분자마커의 활용성을 높이기 위하여 토마토 교배조합별로 이용할 수 있는 마커를 12개 염색체 상에 그래 픽적으로 표현하여 웹 기반의 MAB용 마커 선발 프로그램을 개발하였다. 이 프로그램은 적용하고자 하는 교배조합을 선택 하면 저장된 데이터베이스에서 이용 가능한 SNP를 선발하여 12개의 염색체에 표현한다. 예로 FL7600 과 PI212816 간에 프라이머 제작이 가능한 MAB 마커는 38,799개이며, 프로그 램을 새로고침 할 때마다 38,799개의 SNP 마커 데이터베이 스에서 자동으로 새로운 마커 세트들이 선발되어 표현된다 (Fig. 1). 각 SNP를 확인할 수 있는 프라이머에 대한 자세한
정보를 추가로 제공하도록 프로그램 하단의 테이블을 구성하 였고, 선발된 마커 정보는 테이블 첫 행의 링크를 통해 텍스 트 파일 및 그림 파일로 다운로드 받을 수 있으며, 프라이머 서열 파일 또한 받을 수 있도록 하였다. 프로그램에서 표현되 는 마커 개수는 한 구획 당 3개의 SNP이므로 12개 염색체에 는 총 180개의 마커가 표현된다. 단, 데이터 량이 적어 추출 된 SNP 개수도 적은 M82 품종과의 교배조합을 이룰 때에, 교배조합 간 이용 가능 한 SNP 수가 제한될 경우 180개보다 적은 수의 SNP 마커가 선발될 수 있다. 또한 SNP 수가 비교 적 많은 교배조합 간에도 차이를 보이는 SNP가 적어 이용할 수 있는 SNP가 제한되거나 혹은 SNP 수가 많을지라도 SNP 가 일부 영역에 밀집되어 존재하는 경우, 프로그램 상에서 보 여질 때 염색체 전체적으로 골고루 분포하지 않는 것처럼 보 일 수 있다.
염색체 부위에 따라 교차비율이 다르게 나타나는 점을 고 려하여 유전적 거리를 반영한 MAB 마커 선발 프로그램 역시 토마토 12개 염색체 상에 마커를 그래픽적으로 표현하여 웹 기반의 프로그램으로 개발하였다(Fig. 3). 이 프로그램 또한 적용하고자 하는 교배조합을 선택하면 저장된 데이터베이스 에서 이용 가능한 SNP 마커를 선발하여 12개의 염색체에 표 현한다. 한 구획당 1개의 마커가 선발되므로 한 염색체당 15 개의 마커가 표현되며, 따라서 12개 염색체에는 총 180개의 마커를 표현했다. 이 프로그램도 새로고침을 실시하면 적용
가능한 새로운 마커 세트를 확인할 수 있다.
이러한 토마토 MAB용 분자마커를 제공하는 프로그램은 유용 유전자원의 발굴 및 작물개선 등 실제적인 육종현장으 로 적용되어 분자마커의 활용을 높이고, 육종효율을 증진시킬 것이다.
적 요
본 연구에서는 토마토 MAB에 활용하고자 토마토 7 품종 의 genome-wide SNPs 데이터베이스를 구축하고, MAB를 위한 분자마커 선발 프로그램을 개발하였다. 토마토 전사체 데이터를 NCBI-SRA에서 다운로드 하여 in silico 분석으로 SNP를 추출하였다. 전사체 데이터에서 추출된 SNP를 재료 로 7 품종의 토마토 계통을 이용해 총 21개 교배조합별 SNP 분자마커를 선발하였고, primer가 이용 가능한 마커를 이용하 여 데이터베이스를 구축하였다. 마커를 선발하기에 앞서 염색 체의 분획으로 두 가지 방법을 사용하였는데, 물리적 거리에 따른 분획과 유전거리에 따른 분획 방법이다. 물리적 거리를 이용한 분획은 각 염색체를 동일한 크기의 5개의 구획으로 나 누고, 한 구획 당 교배조합별 차이를 보이는 3개의 SNP를 선 발하였다. 교배조합이 바뀔 때마다 이용 가능한 SNP가 자동 으로 primer 정보와 함께 제공되도록 하였다. 유전거리를 반 영한 분획 방법은 각 염색체의 유전적 거리를 측정하여 물리 적 거리에 차등을 두어 염색체 구획을 설정하였다. 즉 재조합 이 자주 일어나는 염색체 양끝 말단 부분은 구획을 조밀하게 나누어 MAB 마커 또한 많이 할당하여 자세히 조사하도록 구 성하였다. 유전거리에 따른 마커 선발에는 1,924개의 tomato- EXPEN 2000 map 분자마커와 SNP 마커를 이용하였다. 교 배조합별로 이용할 수 있는 마커를 12개 염색체 상에 그래픽 적으로 제공함으로써 사용자가 쉽게 이해하고 이용할 수 있 는 MAB 위한 마커 선발 프로그램을 개발하였다. 이러한 토 마토 MAB용 분자마커를 제공하는 프로그램은 실제적인 여 교잡 선발 육종에 적용하여 분자마커의 활용을 높이고, 육종 효율을 증진시킬 것이다.
사 사
본 연구는 차세대바이오그린21사업(과제번호:PJ009063)의
“가지과 작물의 유전체 정보 실용화 연계 시스템 개발” 과제 의 지원에 의해 수행되었다.
인 용 문 헌