AMINO ACIDS METABOLISM
2019 년 1 학기
Text : Harper’s Biochemistry Refs: Devlin’s, Lippincott’s
Byung Kwan Jin, Ph.D.
Professor
Dept. of Biochemistry & Molecular Biology
School of Medicine Kyung Hee University
Ch. 27.
Biosynthesis
of the Nutritionally Nonessential Amino Acids
Ch. 28. Catabolism of Proteins & of Amino Acid
Nitrogen
Ch. 29. Catabolism of the
Carbon Skeletons
of Amino Acids
Ch. 30.
Conversion
of Amino Acids to Specialized Products
AMINO ACIDS METABOLISM
Car-bons
AMINO ACIDS METABOLISM
Car-bons
NH
3Urea
Chapter 27.
Biosynthesis of the Nutritionally Nonessential
Amino Acids
Biomedical Importance
* Medical implications related to the Amino acid (AA) deficiency:
If nutritionally essential AAs are absent from the diet,
or are present in inadequate amounts
* Amino acid deficiency:
kwashiorkor
(단백열량부족증 )marasmus
(소모증 )scurvy
(괴혈병 )Nutritionally essential & Nutritionally nonessential AAs
* All 20 AAs are essential to ensure health.
•Nutritionally essential AAs: (8) must be present in the human diet.
•Nutritionally nonessential AAs: (12) need not be present in the diet
(Table 27-1)
• Nutritionally nonessential AAs have small number of enzymes required to
synthesize them
• Nutritionally nonessential AAs have short biosynthetic pathways
* Reactions and intermediates involved in the Biosynthesis (by human tissues) of
the 12 nutritionally nonessential 12 AAs
* Selected nutritional and metabolic disorders associated with metabolism
Chapter 27.
Biosynthesis of the Nutritionally Nonessential
Amino Acids
Carbons
Glutamate dehydrogenase
Glutamine synthetase
Aminotransferase
Carbons
NH
3AA biosynthesis :
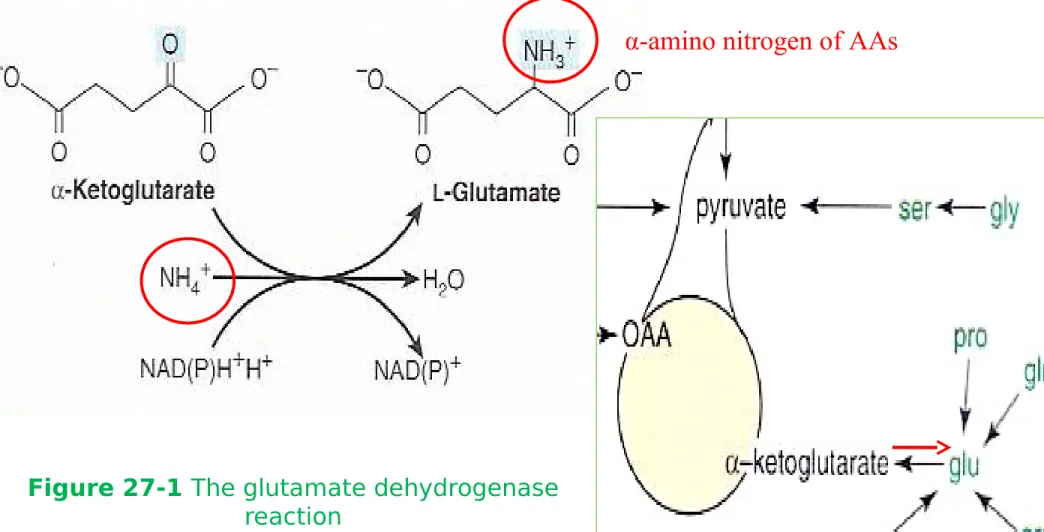
Figure 27-1 The glutamate dehydrogenase
reaction
Glutamate
- Reductive amidation of α-ketoglutarate by glutamate dehydrogenase
- 1
ststep in the biosynthesis of the ‘glutamate family’
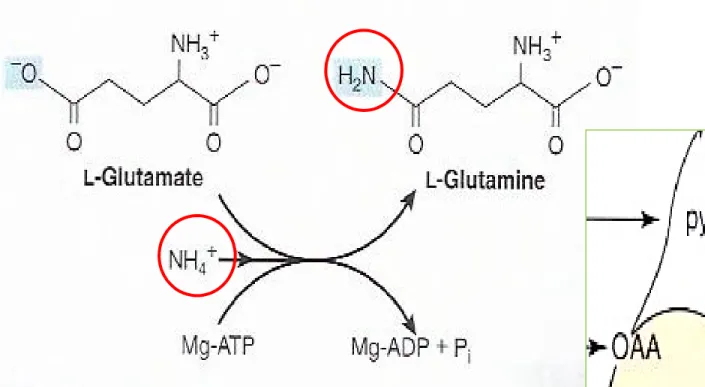
Figure 27-2 The
glutamine synthetasere-action
Glutamine
Alanine
:
Transamination
of pyruvate
Aspartate
:
Transamination
of oxaloacetate
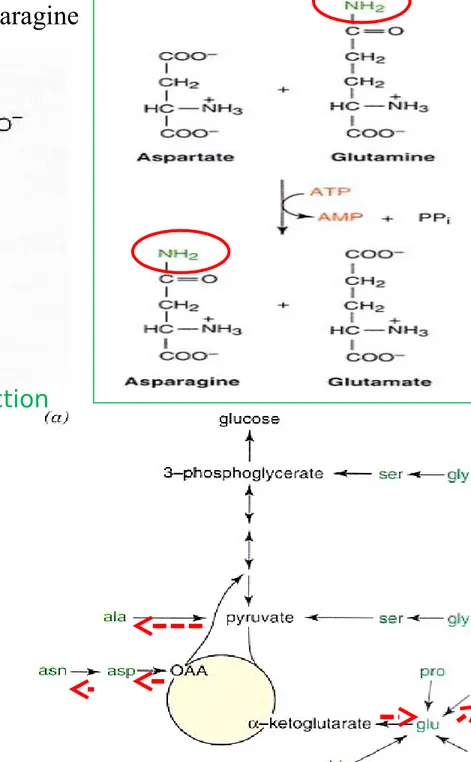
Amino donor Oxaloac-etate Aspartate Glutamate -Ketoglu-tarate Figure 27-3 Formation of alanineFigure 27-4 The asparagine synthetase reaction
Asparagine
: Conversion of Aspartate to Asparagine
Amin o
donor
Figure 27-2 The glutamine synthetase reaction
Amin o
Figure 27-5 Serine biosynthesis (-AA, -amino acids; -KA, -keto
acids)
Serine
Oxidation of α-hydroxyl group of 3-phosphoglycerate to oxo acid transamination dephosphorylation Ser
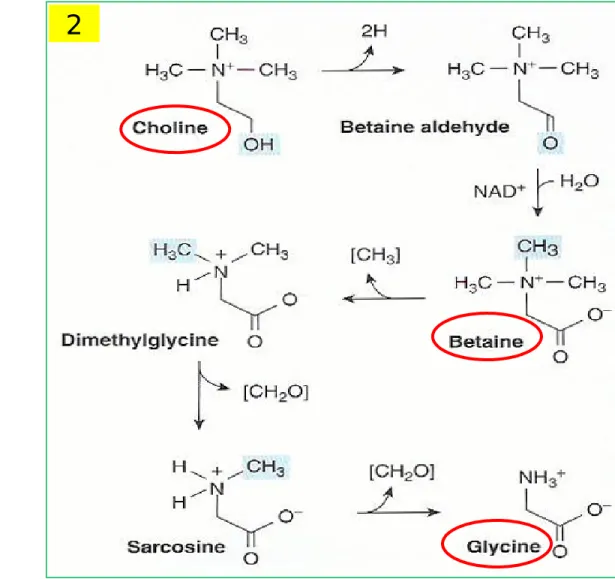
Figure 27-6 Formation of glycine from choline
Glycine
Glycine aminotransferase : Gly from glyoxylate and Glu or Ala
② From choline betaine Gly (Fig 27-6)
+ Glu or Ala
; Amino donor
Glycine aminotransferase
2
1
2
1
Glycine
① Glycine aminotransferase : Gly from glyoxylate and Glu or Ala ② From choline betaine Gly (Fig 27-6)
③ From Ser by serine hydroxymethyltransferase (Fig 27-7)
Figure 27-7
The serine hydroxymethyltransferase reac-tion
3
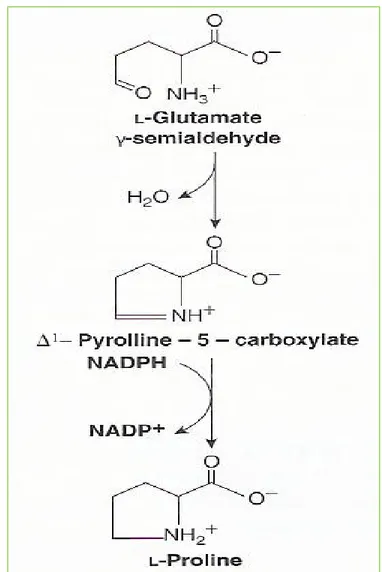
Figure 27-8 Biosynthesis of proline from glutamate
Proline
Proline
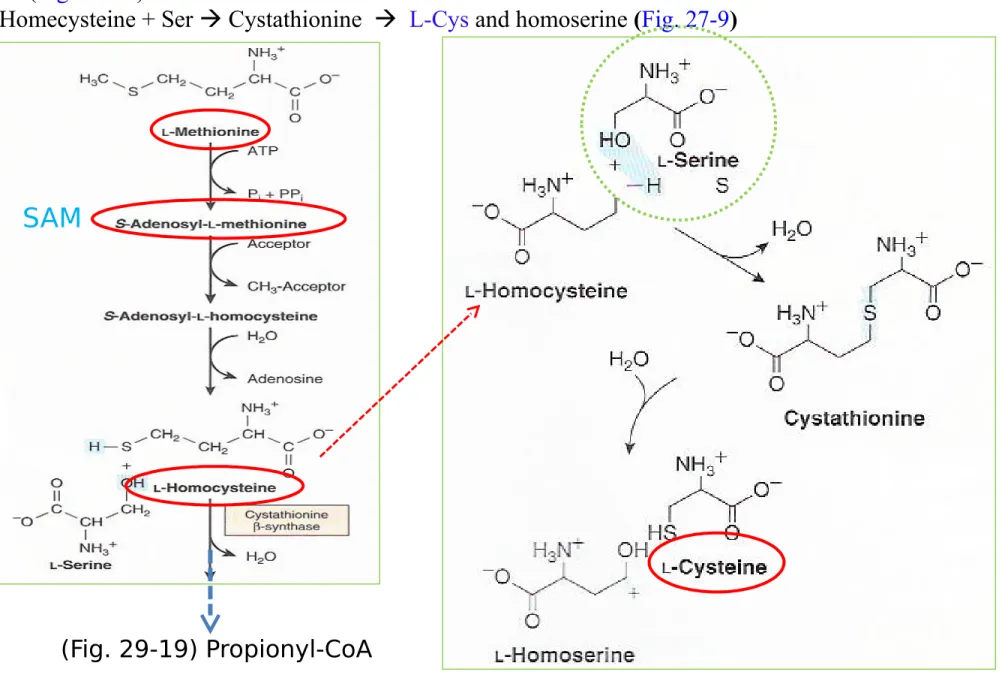
Cysteine
Cys (nutritionally nonessential AA) from Met (nutritionally essential AA) : Met SAM homocys-teine (Fig. 29-19)
L-Homecysteine + Ser Cystathionine L-Cys and homoserine (Fig. 27-9)
Figure 27-9 Conversion of homocysteine and serine to homoserine and cys-teine
SAM
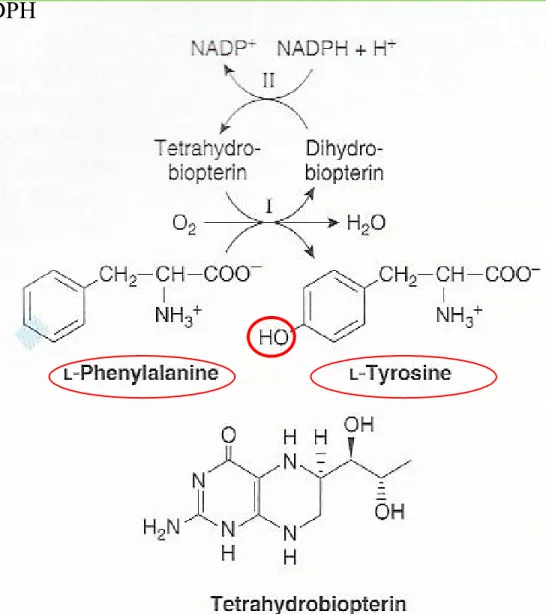
Figure 27-10 The phenylalanine hydroxylase re-action
Tyrosine
① Phenylalanine hydroxylase : Phe to Tyr (Fig. 27-10)
② Catalysis by mixed function oxygenase : O2 into Phe and water ③ Reducing power by tetrehydrobiopterin from NADPH
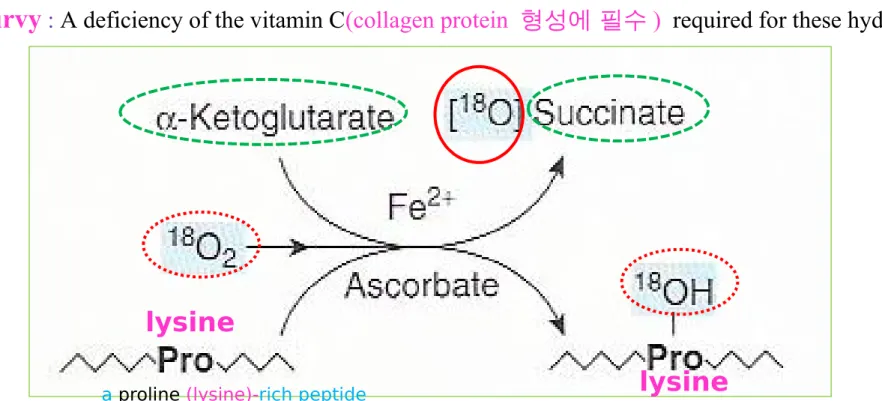
Figure 27-11 The prolyl (lysyl) hydroxylase re-action
Hydroxyproline and hydroxylysine
① Occur in collagen
② no tRNA for hydroxylated AA ㅡ > neither dietary hydroxyproline nor hydroxylysine is
incorporated during protein synthesis ㅡ > Post-translationally, hydroxylation by prolyl hydroxylase and lysyl hydroxylase of tissues (skin, skeletal muscle…) (Fig. 27-11)
③ Hydroxylases (mixed-function oxygenases)
substrate (proline <lysine> -rich peptide, O2, ascorbate, Fe2+ , and α-ketoglutarate succinate+ hydroxy-pro or hydroxy-lys
**
scurvy :
A deficiency of the vitamin C(collagen protein 형성에 필수 ) required for these hydroxylasesa proline (lysine)-rich peptide
lysine
Valine, Leucine, and Isoleucine:
nutritionally
essential AAs①
Tissue aminotransferases for three AAs and their corresponding α-keto acids② Thus, these α-keto acids can replace their AAs in the diet
Amino donor
Figure 27-12 Selenocysteine (top) and the reaction cat-alyzed
by selenophosphate systhetase (bottom)
Selenocysteine, the 21
stAmino acids
• SelenoCys in
active sites
of several enzymes: thioredoxin reductase, gluthathione
peroxi-dase..
• Tumorigenesis, atherosclerosis, selenium deficiency cardiomyopathy (Keshan disease)
Cys-teine
Chapter 28.
Catabolism of Proteins & of Amino Acid Nitrogen
* Biomedical Importance
①
How nitrogen of AAs is converted to
urea
and rare disorders that
accom-pany defects in urea biosynthesis
② Ammonia is highly toxic, tissues
convert ammonia to amide nitrogen
of
Gln (non-toxic)
Subsquent deamination of Gln in
liver releases ammonia
urea
(non-toxic)
③ Liver dysfunction (
간경화 , 간
염 ) elevated blood ammonia
levels clinical signs and
symp-toms
NH
3•Protein
turnover (degradation & synthesis)
occurs in all forms of life
① Humans turn over 1-2% of total body protein per day, principally muscle protein
② Liberated amino acids ~ 75% reutilized
③ Excess amino acids not stored but rapidly degraded nitrogen urea
*Protease & peptidases degrade proteins to AAs
① Half-life t1/2 (susceptibility) of protein:
30 min to >150 hrs (half-lives of liver protein) ② PEST sequences: rapid degradation
- regions rich in proline(p), glutamate(E), serine(S), thronine(T)
③ Peptide hydrolysis: intracellular proteases
Figure 28-1 Reactions involved in the attachment of ubiquitin (Ub) to pro-teins
⑤ Short half-lives, Abnormal and
Misfolded proteins : degraded in the cytosol
requires ATP and
ubiquitin (present in
all eukaryotic cells)
Ubs are attached by non-α-peptide bond
formed between carboxyl terminal of Ub and
α-amino groups of lysyl residues in target
protein.
E1 Ub activating Enz.
E2 Ub transferase (ligase)
E3 Ub ligase (transferase)
•Amino terminal affect ubiquitination:
amino terminal Met or Ser
retards,
whereas
Asp or Arg
accelerates
ubiquitination
* Degradation of ubiquitin-tagged proteins
takes place in
proteasome
④ Extracellular, membrane-associated, long-lived proteins degraded in lysosomes by ATP-inde-pendent processes Defects of Ubiquitina-tion; Angelman syndrome. Defect in E3 ligase; Hippel-Lindau syndrome
4
5
5
protea-someThe multiple ubiquitin-tagging hypothesis (1981)
: Novel prize for the discovery of ubiquitin-mediated protein degradation (2004)
Chemistry – Technion (Israel)
Avram Hershko
Aaron Ciechanover1
UC, Irvine,
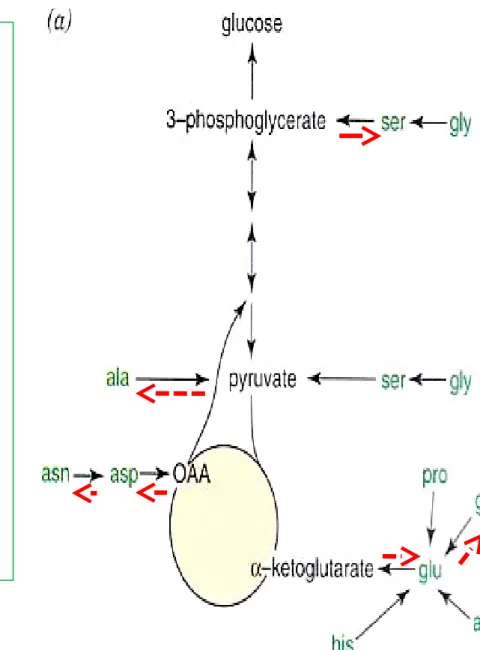
Figure 28-2 Interorgan amino acid exchange in normal post absorptive humans
*
Interorgan exchange maintains circulating levels of AAs
Muscle & liver : major roles in maintaining circulating AAs levels Muscle : generates over half of the total body pool of free AAs Liver: the site of urea cycle enzymes necessary for disposal of excess nitrogen
* Ala & Gln : released from muscle into circulation
* Ala : vehicle of nitrogen transport in the plasma
extracted primarily by the liver
Figure 28-3 The glucose-alanine cycle Figure 28-4 Summary of amino acid exchange between organs immediately after feeding
•Interorgan exchange maintains circulating levels of AAs
Ala: key gluconeogenic AA
Rate of hepatic gluconeogenesis from Ala >>>>> all other AAs
Branched-chain AAs (Leucine, Valine, Isoleucine) serve a special role in nitrogen metabolism, both in fasting state, when they provide brain with an energy source,
and after feeding, when they are extracted predominantly by muscles, having been spared by liver.
fasting state
•Animals convert α–amino nitrogen to varied end products
① Different animals excrete excess nitrogen as ammonia, uric acid, or urea
Fish : ammonotelic (excrete ammonia; highly toxic) Birds : uricotelic (excrete uric acid)
Humans : ureotelic (excrete nontoxic, water-soluble urea)
Biosynthesis of Urea
Four stages:
(1) Transamination,
(2) Oxidative deamination of Glu
(3) Ammonia transport
(4) Urea cycle
Figure 28-5 Overall flow of nitrogen
in amino acid catabolism
PLP in linkage to
aminotransferase
Biosynthesis of Urea
* Transamination transfers α-amino nitrogen to α-ketoglutarate, forming glutamate
① Transamination: α-amino acids and α-keto acids by aminotransferase (Fig. 28-6; Fig. 7-4; “ping-pong mechanism), except Lys, Thr, Pro, Hyp
② Coenzyme pyridoxal phosphate (PLP; A derivative of Vitamin B6, Fig. 44-12)
present at catalytic site of all aminotransferase
enzyme-bound PLP serve as a carrier of amino groups
** remaining carbon “skeleton” of an AA is degraded by pathways (Chap. 29)
Figure 28-6 Transamination
1
1
2
Figure 28-7 Alanine aminotransferase and
glutamate aminotransferase
Biosynthesis of Urea
•Transamination transfers α-amino nitrogen to α-ketoglutarate, forming glutamate
• ③ Alanine aminotransferase : (Alanine-pyuvate aminotransferase)
glutamate aminotransferase : (Glutamate-α-ketoglutarate aminotransferase) (Fig. 28-7)
glutamate aminotransferase
Alanine aminotransferase
3
Biosynthesis of Urea
•Transamination transfers α-amino nitrogen to α-ketoglutarate, forming glutamate
• L-Glu is the only AA that undergoes oxidative deamination by glutamate dehydrogenase (GDH) (Fig. 28-8; Fig. 27-1)
The formation of ammonia from α-amino groups thus occurs mainly via the α-amino nitrogen of L-Glu
⑤ Serum levels of aminotransferases are elevated in some disease (Table 7-2): ALT (SGPT), AST (SGOT)
Figure 28-8
The L-glutamate dehydrogenase reaction
(Figure 27-1)
4
Biosynthesis of Urea
Four stages:
(1) Transamination,
(2) Oxidative deamination of Glu,
(3) Ammonia transport
(4) Urea cycle
Figure 28-5 Overall flow of nitrogen
in amino acid catabolism
Figure 28-8 The L-glutamate dehydrogenase reac-tion
L-Glutamate dehydrogenase occupies a central position in nitrogen metabolism
① Release of ammonia by hepatic L-glutmate dehydrogenase (GDH) (Fig. 28-8)
>> Conversion of α-amino nitrogen to ammonia by concerted action of Glu aminotransferase and GDH transdeamination
>> Liver GDH is allosterically inhibited by ATP, GTP, and NADH and activated by ADP
GDH
Figure 28-8 The L-glutamate dehydrogenase reac-tion
α-amino nitrogen of AAs
Figure 27-1 The glutamate dehydrogenase reac-tion
α-amino nitrogen of AAs
Glutamate
- Reductive amidation of α-ketoglutarate by
glu-tamate dehydrogenase
- 1
ststep in the biosynthesis of the
‘glutamate family’
1
Figure 28-9 Oxidative deamination catalyzed by L-amino acid oxi-dase
* Amino acid oxidases also remove nitrogen as ammonia
① L-amino acid oxidases : amino acids to an α-imino acid
α-keto acid + ammonium ion (Fig. 28-9)
>>> Reduced flavin is reoxidized by oxygen H2O2
Ammonia intoxification is life-threatening
>> Ammonia produced by enteric bacteria and absorbed into portal venous blood and produced by tissues removed from circulation by liver and converted to urea
>> Ammonia is toxic to CNS; 10-20 μg/dL in peripheral blood >> Portal blood bypass the liver
Ammonia intoxification: tremor, slurred speech, coma, death
Ammonia in brain+ α-ketoglutarate Glu
: depletion of α-ketoglutarate impairs TCA cycle in neurons
GDH
1
Glutamine synthetase fixes ammonia as glutamine
- Formation of glutamine by mito. Gln synthetase (Fig. 28-10): Glu + ammonium ion Gln>> Key function of Gln : Sequester ammonia in a nontoxic form
Figure 28-10 The glutamate synthetase reaction strongly fa-vors
glutamine synthesis
1
Glutaminase & asparginase deamidate glutamine & aspargine
① Hydrolytic release of ammonia from amide nitrogen of Gln by glutaminase (Fig. 28-11)
Asparagine
Asparagi-nase
Aspar-tate
Figure 28-11 The glutaminase reaction proceeds essentially
irreversibly
in the direction of glutamate and NH4+ formationGlutaminase & asparginase deamidate glutamine & aspargine
>>> Hydrolytic release of ammonia from amide nitrogen of Gln by glutaminase (Fig. 28-11)
② Concerted action of Gln synthetase and glutaminase
interconversion of free ammonium ion and Gln
Figure 28-11
Figure 28-10
Concerted reaction of aminotransferase and GDH
Formation & secretion of ammonia maintains acid-base balance
① Excretion into urine of ammonia
cation conservation and regulation of acid-base balance ② Ammonia production from renal amino acid (esp. Gln):
* Urea is the major end product of nitrogen catabolism in humans
① Synthesis of 1 mol urea : 3 mol ATP + 1 mol ammonium ion + 1 mol of amino nitrogen of Aspartate ② Five enzymes (Fig. 28-12).
- Of the six participating AAs, N-acetylglutamate solely functions as enzyme activator
Mitochon-dria
Figure 28-12 Reactions and intermediates of urea biosyn-thesis
(1) Carbamoyl phosphate synthetase I initiates urea biosynthesis
① CO2 + ammonia + ATP carbamoyl phosphateby mito. carbamoyl phosphate synthase I (CPS I)
(cf. CPS II, cytosolic form of CPS1, use Gln as a source of nitrogen donor in pyrimidine biosynthesis (see Chap. 33)
② CPSI is active only in the presence of N-acetylglutamate (enzyme activator)
③ Concerted action of GDH and CPSI: nitrogen carbamoyl phosphate
Mitochon-dria
(2) Carbamoyl phosphate plus ornithine forms citrulline
① L-Ornithine transcarbamoylase (mitochondria) citrulline ② Entry of ornithine into mito. and exodus of citrulline from mito.: inner membrane transport system
Mitochon-dria
(3) Citrulline plus aspartate forms arginosuccinate
① Arginosuccinate synthetase links Asp and citrulline via the amino group of Asp and provides 2nd nitrogen of urea
Arginosuccinate
Mitochon-dria
(4) Cleavage of argininosuccinate forms arginine & fumarate
① Cleavage of argininosuccinate by argininosuccinase arginine + fumarate ( malate oxaloaetate) ② Transamination of oxaloacetate Asp
Mitochon-dria
Cytosol
Aspartate Fumarate Aspartate Fumarate(5) Cleavage of arginine releases
urea
& re-forms ornithine
① Hydrolysis of Arg by arginase release urea② Ornithine and lysine are inhibitors of arginase
③ Arginine as a precursor of NO by NO synthase (see Fig. 48-12)