저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
The role of RSF1 in DNA damage signaling pathway
and DSB-induced transcriptional regulation
by
Sunwoo Min
Major in Cancer Biology
Department of Biomedical Sciences
The Graduate School, Ajou University
The role of RSF1 in DNA damage signaling pathway
and DSB-induced transcriptional regulation
by
Sunwoo Min
A dissertation submitted to The Graduate School of Ajou
University in Partial Fulfillment of the Requirements for
the Degree of Ph.D. in Biomedical Sciences
Supervised by
Hyeseong Cho, Ph.D.
Major in Cancer Biology
Department of Biomedical Sciences
The Graduate School, Ajou University
i -ABSTRACT-
The role of RSF1 in DNA damage signaling pathway and
DSB-induced transcriptional regulation
Chromatin remodeling factors are known as a key determinant of chromatin modification in DNA replication, transcription, and double strand break (DSB) repair. As a member of imitation switch (ISWI) family in ATP-dependent chromatin remodeling factors, the remodeling and spacing factor (RSF) complex consists of two subunits, SNF2h ATPase and RSF1. Recent studies suggest that the function of chromatin remodeling factor is to temporally regulate the chromatin modification in the crosstalk between DSB signaling pathway and transcriptional regulation for the efficient DSB repair. Although it has been reported that SNF2h ATPase is recruited to DSB sites in poly(ADP-ribosyl) polymerase 1 (PARP1)-dependent manner, the function of RSF1 is still elusive.
Here various cellular analyses confirmed that RSF1 is recruited and accumulated at DSBs in ATM-dependent manner, and the putative pSQ motifs of RSF1 by ATM are required for its accumulation at DSBs. In addition, depletion of RSF1 attenuates the activation of DNA damage checkpoint signals upon DNA damage. This defect is rescued by Trichostatin (TSA) treatment via chromatin relaxation. Thus, chromatin relaxation by RSF1 as chromatin
remodeling factor is required for the propagation of ɣH2AX signaling pathway. As a result, RSF1 promotes homologous recombination repair (HRR) by recruiting HR factors.
Although RSF1 propagates ɣH2AX signal pathway for the efficient repair as one of chromatin remodeling factors, the function of RSF1 in crosstalk between transcription and
ii
DDR is still elusive. Here inducible transcription system at DSB sites showed that RSF1 promotes DSB-induced transcriptional silencing, while SNF2h is dispensable for
transcriptional silencing at DSB sites. The major determinant of DSB-induced transcriptional silencing, ATM signaling, is also impaired in RSF1 depleted cells. To determine the
molecular mechanism of DSB-induced transcriptional silencing regulated by RSF1, the proteins in RSF1 mass spectrometry were screened by microirradiation. On the basis of the screening results from mass spectrometry analysis, the recruitment of transcriptional repressors at DSB sites in RSF1-dependent manner promotes DSB-induced transcriptional silencing. In addition, RSF1 interacts with polycomb repressive complex (PRC) at
transcriptionally active site, and Swi3, Ada2, N-Cor, and TFIIIB (SANT) domain of enhancer of zeste homolog 2 (EZH2) is required for its recruitment and its interaction with RSF1. The impaired recruitment of EZH2 at DSB sites also leads to the defects in recruitment of
RING1B and its substrate, H2AK119 ubiquitination at DSB sites. In addition, the defect in deacetylation of H2AK118 at DSB sites by HDAC1 in RSF1 depleted cells reduces the level of H2AK119 ubiquitination at DSB sites and eventually leads to the failure in DSB-induced transcriptional silencing. Finally, transcriptome analysis by RNA-sequencing reveals that transcriptome in cells upon DNA damage is changed in RSF1 depleted cells, and as a result, in RSF1 depleted cells, cell death is remarkably reduced upon the continuous DNA damage.
Altogether, these data reveal that RSF1 is recruited at DSB sites and regulates ATM-dependent checkpoint signaling pathway by chromatin relaxation. In parallel, RSF1 also regulates DSB-induced transcription silencing through PRC complex and HDAC1 and the crosstalk between the histone modifications by these histone modifiers.
iii
Keywords: DNA damage response, DNA repair, Chromatin remodeling factor, DSB-induced transcriptional silencing
iv
TABLE OF CONTENTS
ABSTRACT... i
TABLE OF CONTENTS ... iv
LIST OF FIGURES ... vii
LIST OF TABLES ... ix
ABBREVIATION ... x
I. INTRODUCTION ... 1
A. DNA damage response ... 1
B. DSB repair pathways ... 2
C. Remodeling and spacing factor 1 in ISWI family ... 3
D. Chromatin remodeling factor in DNA damage response ... 3
E. DNA damage response in the context of chromatin ... 4
F. DSB-induced transcriptional silencing ... 6
G. Aim of this study ... 7
II. MATERIALS AND METHOD ... 10
1. Cell culture, reagents, and treatment ... 10
2. Plasmids and RNA interference ... 10
3. Mutagenesis ... 11
4. Laser micro-irradiation ... 11
5. Immunofluorescence microscopy ... 12
6. Antibodies ... 12
7. Purification of recombinant protein in vitro ... 13
8. Immunoblotting and protein membrane overlay assay ... 13
v
10. MNase assay ... 14
11. Homologous recombination (HR) and Non-homologous end joining (NHEJ) repair assay ... 15
12. Cell cycle analysis ... 15
13. Chromatin Immunoprecipitation (ChIP) ... 16
14. FokI assays ... 18
15. RNA isolation and RNA sequencing ... 18
16. Reverse transcription and quantitative RT-PCR ... 18
17. ATM retention assay ... 19
18. Nucleosome stability assay ... 20
19. Statistical analysis ... 20
III. RESULTS... 21
PART I. ATM-dependent chromatin remodeler RSF1 facilitates DNA damage checkpoints and homologous recombination repair ... 21
1. RSF1 is recruited at DNA double-strand break sites ... 21
2. ATM-dependent accumulation of RSF1 at DSBs ... 25
3. The putative motifs pSQ of RSF1 by ATM is required for its accumulation at DSBs . 29 4. Depletion of RSF1 attenuates DNA damage checkpoint signals ... 32
5. Depletion of RSF1 caused the failure in chromatin relaxation upon DNA damage ... 37
6. Depletion of RSF1 decreased histone H2A and H2B exchange at the sites of DSB ... 42
7. RSF1 facilitates homologous recombination repair by recruiting resection factors ... 45
PART II. RSF1 recruits EZH2 and HDAC1 for efficient ubiquitination of H2A to promote DSB-induced transcriptional silencing at DSB sites... 48
1. RSF1 leads to DSB-induced transcriptional silencing at DNA lesions ... 48
vi
3. RSF1 promotes DSB-induced transcriptional silencing by regulating ATM activity ... 55
4. Screening RSF1-interacting proteins identified that RSF1 recruits transcriptional repressors at DSB sites ... 58
5. Transcriptional repressors, recruited by RSF1, are involved in DSB-induced transcriptional silencing at DSB sites ... 61
6. RSF1 depletion impaired EZH2 recruitment at DSB sites ... 66
7. SANT domain of EZH2, interacting with RSF1, is important to its recruitment at DSB sites ... 69
8. RSF1 depletion induces DSB-induced transcriptional silencing by the reduction in H2A ubiquitination at transcriptionally active region upon DNA damage ... 73
9. RSF1 depletion impaired HDAC1 recruitment at DSB sites. ... 76
10. H2AK118 deacetylation by HDAC1 is required for the efficient monoubiquitination of H2A at K119 for transcriptional silencing at DSB sites ... 82
11. Failure of local transcriptional regulation resulted in inefficient repair of DNA double strand breaks ... 85
12. RSF1 regulates p53 signaling pathway, resulting in the reduction of cell death upon DNA damage ... 88
13. Acetylation of p53 at K392 is reduced in RSF1 depleted cells ... 93
IV. DISCUSSION ... 96
REFERENCE ... 103
vii
LIST OF FIGURES
Figure 1. RSF1 is recruited at DNA double-strand break site(s). ... 23 Figure 2. The accumulation of RSF1 at DSBs is dependent on ATM activity. ... 27 Figure 3. The putative pSQ motifs of RSF1 by ATM are required for its accumulation at DSBs. ... 30 Figure 4. Depletion of RSF1 attenuated DNA damage checkpoints. ... 33 Figure 5. Phosphorylation of RSF1 by ATM is important for DNA damage checkpoints ... 35 Figure 6. RSF1 relaxes the chromatin structure at DSB sites, and the forced chromatin
relaxation rescued the delayed propagation of γH2AX in response to DNA damage in RSF1 depleted cells. ... 40 Figure 7. histone H2A and H2B exchange was decreased in RSF1 depleted cells at the site of DSB. ... 43 Figure 8. RSF1 facilitates homologous recombination repair. ... 46 Figure 9. RSF1 leads to DSB-induced transcriptional silencing at DNA lesions. ... 50 Figure 10. SNF2h is not required for DSB-induced transcriptional silencing at sites of DSB. ... 53 Figure 11. RSF1 depletion reduced the level of pATM, resulting in the failure of
DSB-induced transcriptional silencing ... 56 Figure 12. Screening RSF1-interacting proteins identified that RSF1 recruits transcriptional repressors at DSB sites. ... 59 Figure 13-1. Transcriptional repressors, recruited by RSF1, are involved in DSB-induced transcription silencing at DSB sites. ... 62 Figure 13-2. RSF1 recruitment at DSB sites was dispensable to pre-existed transcriptional status at DSB sites. ... 64 Figure 14. RSF1 depletion impaired EZH2 (polycomb transcriptional repressor) recruitment at DSB sites. ... 67
viii
Figure 15. RSF1 directly interacts with SANT domain of EZH2, and its interaction is
required for the recruitment at DSB sites. ... 70,71 Figure 16. RSF1 depletion induces DSB-induced transcriptional silencing by the reduction in H2A ubiquitination at transcriptionally active region upon DNA damage ... 74 Figure 17. RSF1 depletion reduced the recruitment of HDAC1 at DSB sites. ... 78 Figure 18. The loss of H2AK118ac was reduced in RSF1 depleted cells, mediated by HDAC1. ... 80 Figure 19. H2AK118 deacetylation is required for the ubiqutination of H2AK119. ... 83 Figure 20. Failure of local transcriptional regulation resulted in inefficient repair of DNA double strand breaks. ... 86 Figure 21-1. RSF1 depletion showed the defects in p53 signaling pathway. ... 89 Figure 21-2. RSF1 regulates p53 signaling pathway, resulting in the reduction of cell death upon DNA damage. ... 91 Figure 22. Regulation of p53 enrichment on the promoters of apoptotic genes in RSF1 KO cells, resulted by reduced level of acetylation at p53 at K382. ... 94
ix
LIST OF TABLES
Table 1. siRNA sequences ... 11 Table 2. ChIP-qPCR primers ... 16 Table 3. qPCR primers ... 18
x
ABBREVIATION
ISWI: imitation switch
RSF: remodeling and spacing factor FACT: facilitates chromatin transcription TSA: Trichostatin
PRC: polycomb repressive complex SANT: Swi3, Ada2, N-Cor, and TFIIIB EZH2: enhancer of zeste homolog 2 HDAC1: histone deacetylase 1 DDR: DNA damage response (DDR) MRN: MRE11-RAD50-NBS1 ATM: ataxia telangiectasia mutated ATR: ATM and Rad3-related
DNAPK: DNA-dependent protein kinase DSB: double-strand break
MDC1: mediator of DNA damage checkpoint protein 1 BRCA1: breast cancer susceptibility 1
53BP1: p53-binding protein 1 CHK2: cyclin-dependent kinase 2 CHK1: cyclin-dependent kinase 1 NHEJ: non-homologous end-joining HR: homologous recombination
DNA-PKcs: catalytic subunit of DNA-PK PARP: Poly (ADP-ribose) polymerase
xi CtIP: CTBP interacting protein
MMS: methyl methanesufonate RPA: replication protein A
SWI/SNF: switch/sucrose non-fermentable (SWI/SNF) CHD: chromodomain-helicase DNA binding protein (CHD) WSTF: Williams syndrome transcription factor (WSTF) ACF1: ATP-dependent chromatin assembly factor 1 (ACF1) TNM: tumor size and tumor, node, metastasis (TNM) CBP: CREB-binding protein
KAP1: KRAB-Interacting Protein 1
NuRD: nucleosome remodeling and deacetylase CSB: Cockayne syndrome protein B
RNAPII: RNA polymerase II BrdU: 5-bromo-2-deoxyuridine NCS: Neocarzionstain
ChIP: chromatin immunoprecipitation MNase: Micrococcal Nuclease
NaB: Sodium butyrate
FRAP: fluorescence recovery after photobleaching 5EU: 5-Ethynyl Uridine
RMFI: relative mean fluorescence intensity
SMARCC1: SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin Subfamily C Member 1
ActD: Actinomycin D
xii IPA: Ingenuity Pathway Analysis
qPCR: quantitative PCR BAX: BCL2 Associated X
FACS: Fluorescence-activated cell sorting PI: Propidium iodide
1
I. INTRODUCTION
A. DNA damage response
Upon DNA damage, cells strive to sustain genomic integrity by coordinating with activation of DNA damage checkpoint signals and the proper repair machineries. The DNA damage response (DDR) is a signal transduction pathway that activates DDR factors in the right place at the right time. DNA lesion is initially recognized by sensor proteins, which are Poly (ADP-ribose) polymerase (PARP), KU70/80 and MRE11-RAD50-NBS1 (MRN), followed by transducing DNA damage signaling. There are three PI3-kinases that are known as the major transducers; ataxia-telangiectasia mutated (ATM) kinase, ATM and Rad3-related (ATR) kinase, and DNA-dependent protein kinase (DNAPK). These kinases phosphorylate their substrates and recruit repair proteins at the sites of DNA damage. DNA double-strand breaks (DSBs) repair is initiated by ATM, which phosphorylates H2AX (γH2AX), as known for histone marker in DDR. In response to DNA damage, γH2AX spreads immediately for 1-2 mega-bases away from DNA lesions (Bonner et al., 2008; Rogakou et al., 1999). In turn, γH2AX offers a platform to propagate and sustain DNA damage signaling cascades, such as mediator of DNA damage checkpoint protein 1 (MDC1), breast cancer susceptibility 1 (BRCA1), or p53-binding protein 1 (53BP1) and damage repair machineries (Lukas et al., 2004; Panier and Durocher, 2009; Stucki et al., 2005). Next, these signals serve to recruit DNA damage repair factors. Eventually, DDR signaling by major kinases spreads away from the site of DNA damage and activates downstream kinases, cyclin-dependent kinase 2 (CHK2) and cyclin-dependent kinase 1 (CHK1), to phosphorylate the effectors such as p53 and CDC25. These effectors determine the outcomes of DNA damage, either apoptosis or cell cycle arrest. Transient cell cycle arrest allows the damaged cells to repair the breaks by repair factors (Sulli et al., 2012).
2
B. DSB repair pathways
There are two different major DSB repair pathways: non-homologous end-joining (NHEJ), which is error-prone and homologous recombination (HR), which is error-free (Huertas, 2010; Jackson and Bartek, 2009). NHEJ occurs in all phases of the cell cycle, but mainly presents in G1 phase, because this repair pathway does not need sister chromatids or homologous templates. During NHEJ, KU heterodimer (Ku70/Ku80) binds to DSB ends, followed by activating the catalytic subunit of DNA-PK (DNA-PKcs). DNA-PK plays a critical role in preventing excessive DNA end resection through its serial autophosphorylation and stabilizing DSB ends. On the other hand, HR is dependent on the presence of homologous DNA templates or sister chromatids, which is the reason why HR is limitedly involved from S to G2 phase (Ciccia and Elledge, 2010). DSB is initially recognized by MRN complex, recruited by PARP. This initial recruitment is independent to γH2AX and MDC1. RAD50 in MRN complex binds to DNA end of the DSB and interacts with MRE11. MRE11 is another subunit of MRN complex and has endonuclease and exonuclease activities critical for the initial step of HR pathway. NBS1 associates with MRE11 and interacts with ATM to promote the recruitment of ATM at DSB sites. MRN complex and ATM kinase activity promote CTBP interacting protein (CtIP) recruitment at DSB, which is essential for DNA end resection. Since HR-deficient cells show hypersensitivity to DNA-damaging agents such as methyl methanesufonate (MMS) and radio-mimetic drug, phleomycin, defects in HR lead to accumulation of unrepaired chromatin breaks (Ciccia and Elledge, 2010). Furthermore, replication protein A (RPA) and Rad51 are important to intermediate the DNA template switch, and their reduced accumulation to DSBs and inactivation significantly reduce HR efficiency.
3
C. Remodeling and Spacing Factor 1 in ISWI family
There are four conserved families of chromatin remodeling factors in humans; the switch/sucrose non-fermentable (SWI/SNF), the Imitation SWItch (ISWI) ISWI, the chromodomain-helicase DNA binding protein (CHD), and the INO80 families (Clapier and Cairns, 2009). Among these families, the ISWI family includes the common ATPase SNF2h, which has different types of binding partners such as RSF1, Williams syndrome transcription factor (WSTF), and ATP-dependent chromatin assembly factor 1 (ACF1). The RSF complex is consisted of RSF1 and SNF2h (Aydin et al., 2014; Nair and Kumar, 2012). Previously, published clinical research has reported that RSF1 is overexpressed in various cancers, and that its overexpression is correlated with poor prognosis for overall survival (Maeda et al., 2011; Shih Ie et al., 2005; Tai et al., 2012; Zhang et al., 2017a). Furthermore, overexpression of RSF1 activates the DNA damage signaling pathway by activating the ATM-Chk2 kinases (Sheu et al., 2010). Recently, overexpression of RSF1 showed a positive correlation with p53 levels, and this combined expression of RSF1 and p53 was correlated to tumor size and tumor, node, metastasis (TNM) staging in breast cancer patients (Ren et al., 2014).
D. Chromatin Remodeling Factor in DNA damage response
Recently, several studies show that chromatin remodeling factors are important for recruiting DNA repair proteins to access into DNA in DDR (Lans et al., 2012; Osley et al., 2007; Price and D'Andrea, 2013). Most of them have been reported to be recruited at DNA lesions to mobilize the nucleosome and remodel the chromatin structure. INO80 was initially found to be required for DNA repair in yeast (Morrison et al., 2004; van Attikum et al., 2007; van Attikum et al., 2004). Later, it was reported that mammalian INO80 is also involved in DSB repair through its role
4
in DNA end resection (Gospodinov et al., 2011). The mammalian SWR1-related TRRAP-Tip60 complex in INO80 is also recruited at DSB sites and contributes to decrease nucleosome stability and to activate ATM for efficient repair. SWI/SNF complexes are shown to promote the DSB repairs by facilitating γH2AX induction (Lee et al., 2010; Park et al., 2006). BRG1 and BRM in mammals SWI/SNF are recruited at the sites of DSB and interact with acetylated H3 and H4 by the activities of acetyltransferases GCN5, CREB-binding protein (CBP), and p300 upon DNA damage. In addition, BRM is required to recruit KU70 at DSB sites and promote the efficient NHEJ. CHD family also regulates both DNA damage signaling and repair pathways (Larsen et al., 2010; Polo et al., 2010; Smeenk et al., 2010). CHD3 in CHD family is important for chromatin relaxation in heterochromatic DSB repair by dispersing itself from DSB via KRAB-Interacting Protein 1 (KAP1) phosphorylation by ATM (Goodarzi et al., 2011). CHD4, another member of mammalian CHD family is also recruited to DSB sites in PARP dependent manner and facilitates DSB checkpoint activation. In the ISWI family, multiple members form in a complex with SNF2h ATPase. SNF2h ATPase, the catalytic subunit in ISWI family, is also known to be recruited at DSBs in a PARP-dependent manner and involved in chromatin relaxation. It was also reported that ACF1 is required for DNA repair by interacting with KU70/80 complex (Lan et al., 2010). In addition, WSTF, another binding partner of SNF2h, also interacts with H2AX and phosphorylates H2AX on tyrosine 142, which is important for the maintenance of γH2AX and DSB checkpoint signaling pathway (Xiao et al., 2009).
E. DNA damage response in the context of chromatin
In eukaryotes, DNA is wrapped around histone octamer, called nucleosome, and organized in chromatin structure. This structure comes into the barrier for DNA repair proteins to get access to
5
DNA to repair the breaks. In mammalian cells, chromatin is composed of active or inactive genes, related to transcription status, and this complexity is a challenge to DNA repair proteins. These challenges upon DNA damage are overcome by the most representative model, “Access-repair-restore” model, to explain how chromatin is dealt with DNA repair proteins (Price and D'Andrea, 2013; Soria et al., 2012).
“Access-Repair-Restore” model proposes the minimal steps to repair DNA in chromatin organization, focusing on how chromatin remodeling factors and epigenetic modulators regulate DDR. Briefly, DSB repair machinery must recognize DSB sites in chromatin structure and remodel the structure to get access to the site. After the break is repaired, chromatin structure is re-organized to restore the chromatin organization. Previously, access step in this model was proposed that all proteins were displaced from the chromatin to access DNA for repair proteins (Price and D'Andrea, 2013). However, recent publications uncovered that chromatin proteins such as chromatin remodelers and histone modifiers are recruited at the sites of DNA damage to create open chromatin environment for efficient repair. Thus, Soria et al suggested “Prime-Repair-Restore” model rather than “Access-Repair-Restore” model established in 1991. In priming step, chromatin remodelers and histone modifiers are recruited and create the platform to assemble the signaling and repair machineries simultaneously. The best characterized proteins in chromatin remodelers contributing to chromatin relaxation is Tip60 complex. Chromatin remodeling factors are required to decondense and slide the nucleosome nearby the breaks and epigenetic modulators are required to modify the chromatin structure. Thus, the access step in the early response is critical to detect the damage in the context of chromatin and repair the breaks (Price and D'Andrea, 2013).
6
F. DSB-induced transcriptional silencing
Recently, it had been reported that pre-existed chromatin structure makes a decision for DNA repair proteins to repair DNA by HR or NHEJ. Transcriptionally active chromatin activates HR repair pathway, while transcriptionally inactive chromatin activates NHEJ repair pathway (Aymard et al., 2014). It was also reported that transcription is silenced in cis to the break sites, and ATM is the major player to determine the transcription silencing at the DSB site and recruit polycomb repressive complex. Shanbhag et al. demonstrated that ATM inhibition causes chromatin decondensation, leading to failure of transcription silencing and finally leads to the defects in DNA repair (Shanbhag et al., 2010). However, ATM kinase is also reported to relax chromatin structure globally (Ziv et al., 2006). According to this opposite mechanism of ATM kinase upon DNA damage, recent review explains that ATM may work differently in transcriptionally active and inactive regions. In other words, in heterochromatin, ATM-dependent chromatin relaxation is critical step for repair proteins to get access to DNA, while in euchromatin, a large scale of chromatin condensation by ATM kinase promotes the efficient DNA repair by preventing DNA broken ends from falling apart (Shanbhag and Greenberg, 2010). The following result from another group supporting DSB-induced transcriptional silencing suggests that ATM-dependent phosphorylation of ENL, the transcriptional elongation factor, enhances its interaction with polycomb repressive complex 1 (PRC1) in order to rapidly switch from transcriptional elongation to transcriptional pausing at DSB sites (Ui et al., 2015). In addition, PBAF, the chromatin remodeling factor, was also targeted by ATM kinase to promote DSB-induced transcriptional silencing to recruit PRC1 at DSB sites (Kakarougkas et al., 2014). Recent work also screened bromodomain proteins and found ZMYND8 promoting DSB-induced transcriptional silencing by recruiting nucleosome remodeling and deacetylase (NuRD) complex to damaged chromatin (Gong et al., 2015).
7
silencing at DSB sites, another report suggested that ongoing transcription producing non-coding RNA is also important for DSB repair such as 53BP1 foci formation (Francia et al., 2012). Recently, it had been also reported that HR factors are preferentially recruited at transcriptionally active sites and RNA-templated recombination mechanism of DSB repair at transcriptionally active sites is mediated by cockayne syndrome protein B (CSB) ATPase, associated with RNA polymerase II (RNAPII) (Wei et al., 2015). Given the importance of transcription status at DSB sites, additional evidences may exist to explain these two different transcriptional activities at DSBs.
G. Aim of this study
Chromatin remodeling factors are a key determinant of transcriptional regulation. To gain the access to DNA, chromatin remodeling factors are required to translocate or remove the nucleosomes. In addition to transcriptional regulation, it is suggested that chromatin remodeling factors are also required for remodeling the chromatin structure upon DNA damage for efficient repair. Cellular DNA is exposed to endogenous and exogenous source of DNA breaks per a day. Thus, the maintenance of genomic integrity from endogenous and exogenous source of DNA break is critical for cells’ surveillance.
“Access-Repair-Restore” model implicates the significance of chromatin remodeling factors upon DNA damage. Thus, understating the role of chromatin remodeling factors at DSB sites is crucial for understanding the model. However, in mammalian cells, it is still unclear which chromatin remodeling factors are involved in each stage in the model. In addition, most of chromatin remodeler is reported to be involved as the downstream of ATM kinase.
Even though SNF2h ATPase has been studied in DNA damage response as chromatin remodeling factor, the function of RSF1 in ISWI complex is still uncovered. Based on RSF1 mass
8
spectrometry in the previous study in our group, it was found that RSF1 interacts with chromatin-bound proteins involved in DDR. In addution, RSF1 is reported as one of the substrates of ATM/ATR kinases, the major kinases in DDR. Furthermore, during the course of this study, it was also found that ATM directly interacts with RSF1 via its C-terminal FATC domain.
Thus, in this study, I hypothesized that RSF1 functions in DDR and DSB repair as chromatin remodeling factor and transcriptional regulator. To prove the function of RSF1 in DDR and DSB repair as chromatin remodeling factor, I depleted RSF1 in U2OS cell line and examined DDR signaling pathway and DSB repair efficiency. In addition, chromatin modification was monitored by MNase, ChIP, Nucleosome stability assay, and FRAP in RSF1-depleted cells. In PART I, I propose the role of RSF1 as chromatin remodeling factor. RSF1 is required for γH2AX propagation to activate ATM-dependent DDR checkpoint signaling pathway and also for the efficient repair by remodeling the compact structure of chromatin to recruit DSB repair proteins in the context of chromatin as the upstream of ATM kinase.
In addition to DDR signaling, I hypothesized that RSF1 functions with ATM kinase in DSB-induced transcriptional silencing as platform of chromatin modifying enzymes. DSB-induced transcriptional silencing is also prerequisite for the efficient repair upon DNA damage. To prove the function of RSF1 in transcriptional regulation, I used inducible transcription system at DSB sites. In order to investigate the mechanism of DSB-induced transcriptional regulation by RSF1, I screened RSF1-binding partners from mass spectrometry and found the interacting proteins that are recruited at DSB sites by RSF1. Based on the screening results, it was found that histone modifiers, EZH2 and HDAC1, are recruited in RSF1 dependent manner and promotes histone modification for DSB-induced transcriptional silencing. Thus, in PART II, I suggest another role of RSF1 as the scaffold protein that RSF1 promotes DSB-induced transcriptional silencing by recruiting histone modifiers, such as EZH2 and HDAC1. Eventually, RSF1 regulates the global transcription that promotes the
9
10
II. MATERIALS AND METHOD
1. Cell culture, reagents, and treatment
Human U2OS osteosarcoma and 293T embryonic kidney cells were grown in DMEM containing 10% fetal bovine serum (Gibco). RPE1 retinal pigment epithelial cells were cultured in F12 (Gibco) containing 10% FBS and 10 µg/ml hygromycin. yz5 (human fibroblast) and S7 (human A-T fibroblast) cells were grown in DMEM with 15% FBS and 100 µg/ml hygromycin. To make the stable cell lines, U2OS cell lines and HeLa cells were infected with plasmid RSF1-shRNA, using lentiviral system and selected under puromycin (2 µg/ml) for 4 weeks. Hydroxyurea, methyl methanesufonate and phleomycin were purchased from Sigma-Aldrich. ATM inhibitor (KU55933) was from Selleckchem.
2. Plasmids and RNA interference
pcDNA6-RSF1-V5, kindly gifted from Dr. Ie-Ming Shin (Johns Hopkins Medical Institutions), was subcloned into pEGFP-N1 (GenBank Accession) to generate RSF1-GFP. The siRNA for RSF1 purchased from Ambion was transfected with Neon electroporation (Invitrogen) or Lipofectamine 2000 (Invitrogen) and incubated for 72 hours. H2AX-GFP, SNF2h-GFP, FokI wild type and mutant D450A, Mre11-YFP, shATM/shATR, or K230-ZFN was provided by Dr. Jongbum Kwon (Ewha Womans Univeristy), Akira Yasui (Tohoku University), Brendan D. Price (Dana-Farber Cancer Institute), Ho Chul Kang (Ajou University School of Medicine), Young-Joo Jang (Dankook University), or Jin-Soo Kim (Seoul National University), respectively. Inducible transcription system at DSB sites (IFII cell line) and inducible DSB system (ASiSI cell line) were supported from Roger Greenberg (University of Pennsylvania) and Gaëlle Legube (Université de Toulouse), respectively. EZH2 construct was purchased from Harvard.
11
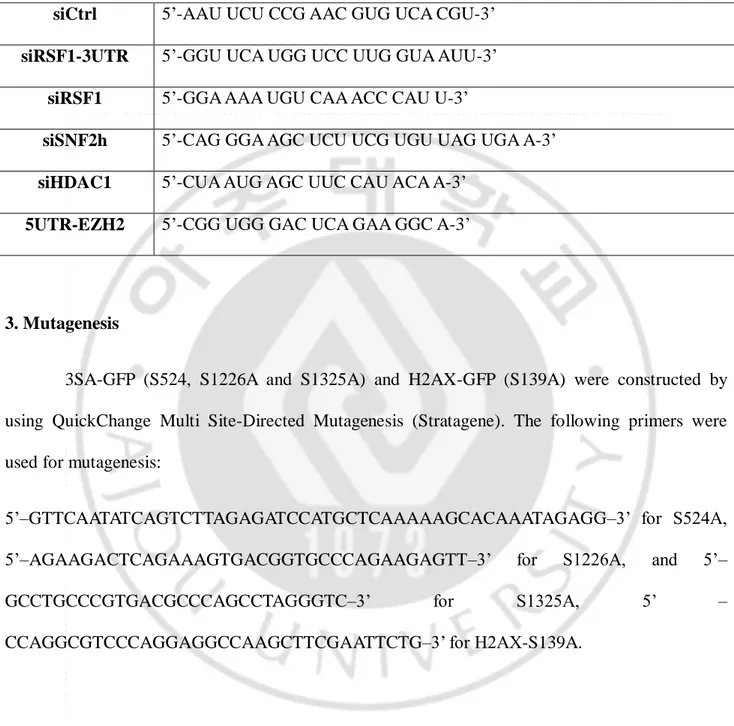
Table 1. siRNA sequences
siCtrl 5’-AAU UCU CCG AAC GUG UCA CGU-3’
siRSF1-3UTR 5’-GGU UCA UGG UCC UUG GUA AUU-3’
siRSF1 5’-GGA AAA UGU CAA ACC CAU U-3’
siSNF2h 5’-CAG GGA AGC UCU UCG UGU UAG UGA A-3’
siHDAC1 5’-CUA AUG AGC UUC CAU ACA A-3’
5UTR-EZH2 5’-CGG UGG GAC UCA GAA GGC A-3’
3. Mutagenesis
3SA-GFP (S524, S1226A and S1325A) and H2AX-GFP (S139A) were constructed by using QuickChange Multi Site-Directed Mutagenesis (Stratagene). The following primers were used for mutagenesis:
5’–GTTCAATATCAGTCTTAGAGATCCATGCTCAAAAAGCACAAATAGAGG–3’ for S524A, 5’–AGAAGACTCAGAAAGTGACGGTGCCCAGAAGAGTT–3’ for S1226A, and 5’– GCCTGCCCGTGACGCCCAGCCTAGGGTC–3’ for S1325A, 5’ – CCAGGCGTCCCAGGAGGCCAAGCTTCGAATTCTG–3’ for H2AX-S139A.
4. Laser micro-irradiation
For laser micro-irradiation, cells were seeded onto 35-mm round glass bottom dishes (SPL, Korea) and 5-bromo-2-deoxyuridine (BrdU, final 10 μM) was added to the medium for 30 h prior to
12
micro-irradiation. For confocal imaging, an A1 confocal microscope (Nikon) with a 60X oil objective was used in a temperature-controlled chamber (37°C, 5% CO2). Damage sites were induced by micro-irradiation with a 405 nm laser and 1.2 sec irradiation time (32 lines/sec). Images were acquired every 1 second for 10 min, 30 min, or 2 h. At least ten cells were irradiated in every experiment, and representative data are shown.
5. Immunofluorescence microscopy
Cells were grown on cover-slips or 35-mm round glass bottom dishes, and then treated with phleomycin or carried out micro-irradiation experiments as indicated, respectively. Coverslips or glass bottom dishes were washed once with PBS, fixed with 4% paraformaldehyde and 0.1% TritonX-100 in PBS, and blocked for 30 min at room temperature in blocking buffer (3% BSA in PBS). Primary antibodies were incubated for 2 h at room temperature or overnight at 4°C in the same buffer. Cells were then washed off three times in PBS before incubation with Alexa 488, 594 (Molecular Probes) or cy3 (Jackson) in blocking buffer for 40 min at room temperature. Cells were again washed three times in PBS and mounted onto glass slides with DAPI-containing mounting solution. Images were analyzed using an Eclipse Ti (Nikon) or an A1 confocal microscope (Nikon) with a Nikon PlanApo 60x oil objective lens.
6. Antibodies
Antibodies used in this study are as follow: RSF1 (Abnova or abcam), pATM (pS1981; abcam), MDC1 (abcam), RPA32 (abcam), γH2AX (Millipore), SNF2h (Millipore), pATR (pS428; Cell signaling), pSQ/pTQ (Cell signaling), p53BP1 (Cell signaling), 53BP1 (Cell signaling), pChk1
13
(pS296; Cell signaling), pChk2 (pT68; Cell signaling), pNBS1(pS343; Cell signaling), H2AX (Cell signaling), Rad51 (Santa cruz), GAPDH (Santa cruz), GFP (Santa cruz), V5 (Invitrogen), Flag (Santa cruz), p53 (Santa cruz), p21 (Santa cruz), p53 acetyl-K382 (Cell signaling), H2Aub (Millipore and Cell signaling), RNA Polymerase II (abcam) RNA Polymerase II phosphor-S2 (abcam), H3 (abcam), Rad50 (Cell signaling), ATM (Calbiochem), BrdU (Calbiochem), SPT16H (Santa cruz), H2AX (Cell signaling). The HRP-conjugated anti-mouse IgG and anti-rabbit IgG secondary antibodies were purchased from Invitrogen. The following antibodies were used as secondary antibodies in immunofluorescence microscopy: Alexa Fluor 488, Alexa Fluor 594, Alexa Fluor 647 (Invitrogen), Cy3 (Jackson).
7. Purification of recombinant protein in vitro
SF9 cells were transfected with bacmid DNA using Cellfectin II Reagent (Invitrogen) and incubated at 27°C incubator for 72 h. Cells were lysed with NETN buffer and incubated with glutathione-Sepharose 4B resin (GE Healthcare Life Science) at 4°C for 1 h. Resin was washed with NETN buffer four times, and protein expression was tested by Coomassie Brilliant Blue (Sigma) staining and immunoblot assay with anti-GST antibody.
8. Immunoblotting and protein membrane overlay assay
Cells were washed with PBS three times and directly lysed in sample buffer (0.3M Tris, pH 6.8, 12% SDS, 30% β-mercaptoethanol, 0.6% Bromophenol blue, 40% glycerol) with protease inhibitor cocktail (Roche). Proteins were separated by the gradient gel (4%-20% acrylamide gel) and transferred onto nitrocellulose membrane (Whatman) or PVDF (Millipore). The membranes
14
were blocked for 1hr with 3% BSA in TBST (Tris-buffered saline with 0.1% triton X-100, pH 7.6) and blotted with the appropriate primary antibodies at 4°C for overnight. After washing the membranes with TBST for 1hr, the membranes were incubated with the appropriated secondary antibodies (Invitrogen). Proteins were detected with ECL reagents (GE Healthcare). For protein membrane overlay assay, EZH2 deletion mutants purified in vitro from insect cells were blotted in NC membrane and incubated with RSF1 purified in vitro from insect cells 4°C for overnight.
9. Immunoprecipitation and chromatin fractionation
Cells were harvested and lysed in NETN buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% NP-40, and 5 mM EDTA) with protease and phosphatase inhibitors. The lysate was sonicated and centrifuged at 13,000rpm for 15 min at 4°C. The supernatant was measured by Bradford assay and the equal amount of protein lysate was incubated with primary antibody for overnight at 4°C. The immunoprecipitates were captured by Protein A sepharose Fast-Flow (GE Healthcare). The beads were washed four times with NETN buffer and boiled in 2X sample buffer.
For chromatin fractionation, cells were harvested and lysed in NETN buffer excluding 5mM EDTA with protease and phosphatase inhibitors on ice for 20min at 4℃. The lysate was centrifuged at 13,000 rpm for 15 min 4℃. The pellet was incubated with MNase (25U) and Benzonase (25U) for 20 min at 37℃ shaking incubator. The supernatant was collected by centrifugation at 4℃ and processed to western blotting.
10. Micrococcal Nuclease (MNase) assay
15
day, cells were harvested after treatment with NCS for 30 min and lysed in lysis buffer A (10mM HEPES (pH 7.4), 10mM KCl, 1.5mM MgCl2, 0.34M sucrose, 10% glycerol, 0.1% Triton X-100,
1mM DTT) for 5 min on ice. Pellet was collected after centrifugation at 1,300g at 4℃ and suspended with micrococcal nuclease digestion buffer (15mM Tris-HCl (pH 7.4), 60mM KCl, 15mM NaCl, 0.25M sucrose, 1mM CaCl2, and 0.5mM DTT). The appropriate concentration of MNase (Thermo) was added in digestion buffer. The reaction was stopped by adding MNase stop buffer (20mM EDTA). Protease K and SDS were added and incubated in the samples overnight at 37℃. Genomic DNA was purified by using Nucleospin PCR clean up kit (Macherey-Nagel) and processed to running on agarose gel.
11. Homologous recombination (HR) and Non-homologous end joining (NHEJ) repair assay
U2OS-DR-GFP (HR) and U2OS-EJ5-GFP(NHEJ) reporter cells were seeded in 12 well plate on the day before transfection. Cells were transfected with siRNA by Lipofectamine RNAiMax (Invitrogen). Next day, cells were transfected with I-sceI and incubated for 48h. After incubation, cells were trypsinized and washed with PBS, and GFP-positive cells were counted by FACS.
12. Cell cycle analysis
HeLa RSF1 stably knocked down cells were harvested after treatment phleomycin at the indicated time points and fixed with 80% ethanol overnight at 20℃. The fixed samples were washed with PBS three times and stained with propidium iodide (PI) and analyzed by Fluorescence-activated cell sorting (FACS).
16
13. Chromatin Immunoprecipitation (ChIP)
Cells were harvested after the indicated treatment (4-OHT or Shield1) and cross-linked with 1% formaldehyde for 20 min. Cells were lysed with SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.1) supplemented with protease and phosphatase inhibitor cocktail (Thermo) for 10 min on ice. Cell lysates were sonicated by Bioruptor (Diagenode) and centrifuged at 13,000 rpm for 15 min. The supernatants were collected and diluted for overnight incubation with primary antibody. After overnight incubation, 20μl of Protein A agarose/salmon sperm DNA (Millipore) was added to each sample and incubated with rotation at 4℃. After 1h incubation, beads were washed with the following washing buffer: low salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl), high salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl), LiCl immune complex wash buffer (0.25 M LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid (sodium salt), 1 mM EDTA, 10 mM Tris, pH 8.1), and TE buffer (10 Mm Tris-HCl, 1mM EDTA, pH 8.0). After washing the beads, the immune complex was eluted in elution buffer (1% SDS, 0.1 M NaHCO3) for 30 min at RT. The eluates were incubated with 10μl of 5M NaCl to reverse
histone-DNA crosslinks by heating at 65℃ for 4 h, followed by incubation with proteinase K at 45℃ for 1 h. DNA was purified with Nucleospin PCR clean up kit (Macherey-Nagel) and processed for quantitative PCR.
Table 2. ChIP-qPCR primers
Chr22_prox
FW : CCTTCTTTCCCAGTGGTTCA RV: GTGGTCTGACCCAGAGTGGT
17 Chr22_distal FW: CCCATCTCAACCTCCACACT RV: CTTGTCCAGATTCGCTGTGA DSB_I_800bp FW: TATGGGACCAAGCGAGTAGG RV: GCCTCACACACACACCCATA DSB_I_80bp FW: GTCCCTCGAAGGGAGCAC RV: CCGACTTTGCTGTGTGACC DSB_V_800bp FW: GTCAGTATGGCCCCAGAGTC RV: ACGGCTGATGGACTTAGACG DSB_V_80bp FW: CCTAGCTGAGGTCGGTGCTA RV: GAAGAGTGAGGAGGGGGAGT DSB_1_800bp FW: GGAGAAGTGGCAGGACAATG RV: CAAGGCAAATTTGGGGACTA DSB_1_80bp FW: TCCCCTGTTTCTCAGCACTT RV: CTTCTGCTGTTCTGCGTCCT DSB_2_800bp FW: TTTTTGGGGGAAAGAGGTG RV: AGTGGGTGAGCCATTCAAAG DSB_2_80bp FW: ATCGGGCCAATCTCAGAGG RV: GCGACGCTAACGTTAAAGCA PUMA_+1313 FW: TCA GTG TGT GTG TCC GAC TGT C RV: GGC AGG GCC TAG CCC A PUMA_+746
FW: GTC GGG CGA ATG TCA CTT TC RV: CCT GGA TAC ACG GCC AAA TC
BAX_p53RE
FW: AGA TCA TGA AGA CAG GGG CCC TTT RV: TGG AGT GAG GGT GCA GAA TCA GAA
18
14. FokI assays
For the recruitment of RSF1-GFP, HDAC1-EGFP, H2AX-GFP, or Mre11-YFP, 2-6-5 cells were seeded on coverglass bottom dish (SPL) on the day before transfection and transfected with siRSF1 using Lipofectamine RNAiMax (Invitrogen). Next day, the indicated GFP-tagged construct was transfected with Lipofectamine 2000 (Invitrogen) and incubated for 48 h. Cells were harvested after 48 h transfection and fixed with 4% paraformaldehyde (Sigma), followed by imaging analysis.
15. RNA isolation and RNA sequencing
Total RNA was isolated from U2OS cells, treated with control siRNA and RSF1 siRNA, using RNeasy Plus Mini kit (Qiagen). The quality of RNA was checked using Bioanalyzer RNA ChIP (Agilent Technologies). mRNA library was prepared using Truseq stranded mRNA kit (Illumina), and RNA sequencing running was performed using Nextseq 500 (Illumina).
16. Reverse transcription and quantitative RT-PCR
For quantitative RT-PCR, total RNA was isolated using RNeasy Plus Mini kit (Qiagen). 2μg of total RNA was used for cDNA synthesis by amfiRivert cDNA Synthesis Platinum Master Mix (GenDEPOT). Quantitative PCR was performed on Roter-GeneQ (Qiagen) using Maxima SYBR Green qPCR master mix (Thermo) with the following primers.
19
p21
FW: TTG TAC CCT TGT GCC TCG CT RV: TTG GAG AAG ATC AGC CGG C
MDM2
FW: AAG AGA CCC TGG TTA GAC CAA AGC RV: TTT CTT CTG TCT CAC TAA TTG CTC T
Gadd45α
FW: ATG ACT TTG GAG GAA TTC TCG RV: CAT TGA TCC ATG TAG CGA CTT
BTG2
FW: GAA AAG CCG TCC AAG GGC RV: CTT GTG GTT GAT GCG AAT GC
BAX
FW: AAG AAG CTG AGC GAG TGT RV: GGC GGC AAT CAT CCT CTG
PUMA
FW: GCT GCT GTA GAT ACC GGA ATG AA
RV: AAA AAA ATT AAC CAA ACA TGT ACA GAA AAT
GAPDH
FW: CTC TGC TCC TCC TGT TCG AC RV: ACG ACC AAA TCC GTT GAC TC
β-ACTIN
FW: AGC CGG GCT CTT GCC AAT RV: AGT TAG CGC CCA AAG GAC CA
17. ATM retention assay
Cells were resuspended in fractionation buffer (50mM Hepes pH 7.5, 150mM NaCl, 1mM EDTA, 0.2% NP-40) and incubated on ice for 5 min. The lysate was centrifuged at 1,000 g for 5 min. Supernatant (Fraction I) was isolated, and pellet was further extracted in fractionation buffer containing 0.5% NP-40 and incubated on ice for 40 min. The extract was centrifuged at 16,000 g for
20
15 min. Supernatant (Fraction III) was separated, and pellet was resuspended in 1X sample buffer and boiled for 10 min. Equal aliquots of each fraction were separated on 4-12% SDS-PAGE.
18. Nucleosome stability assay
Cells were suspended in lysis buffer A and incubated for 5 min on ice. The lysate was centrifuged at 1,300 g for 4 min. Pellet was resuspended in lysis buffer B (20mM Tris pH 8.0, 15% glycerol, 1.5% Triton X-100, 0.8M NaCl) and incubated for 30 min on ice. The extract was centrifuged at 13,000 rpm for 10 min. Insoluble and soluble fractions were boiled in sample buffer.
19. Statistical analysis
Statistics and graphs were performed using GraphPad Prism (version 5.0). Unpaired student’s t test was applied to compare two individual groups, while one-way ANOVA was applied to compare multiple groups. Asterisks indicates each p-values (* P < 0.05; ** P < 0.01, *** P < 0.005).
21
III. RESULTS
PART I. ATM-dependent chromatin remodeler RSF1 facilitates DNA damage checkpoints and homologous recombination repair
1. RSF1 is recruited at DNA double-strand break sites
To understand the role of RSF1 in DDR, I first examined whether RSF1 is recruited at DSBs upon DNA damage. RSF1 formed DNA damage foci with DNA damaging agent phleomycin (radio-mimetic drug making DSBs), which is co-localized with γH2AX and pATM, as the major hallmarks of DSBs (Fig. 1A and B). Furthermore, to demonstrate whether its localization is specific to DSBs, 2-dimentional (2D) and 3-dimentional (3D) images were obtained by workstation program (Nikon). As expected, RSF1 and γH2AX are co-locolizaed at DSBs (Fig. 1A and B). It has been shown that RSF1 is over-expressed in cancer cells rather than in normal cells.(Fang et al., 2011; Hu et al., 2012; Liu et al., 2012; Mao et al., 2006; Shih Ie et al., 2005) To confirm clearly what the recruitment of RSF1 at DSBs is not specific in cancer cells, the endogenous RSF1 was immunostained at 10 min after micro-irradiation and found that RSF1 was accumulated at the micro-irradiated sites in normal (human RPE1) and cancer cells (U2OS) (Fig. 1C). Again, in order to exclude the possibility that the recruitment of RSF1 at micro-irradiated sites is caused by non-specific damaging effects other than by DNA double-strand breaks, the recruitment of RSF1 was validated in stably integrated LacO-LacI nuclease system (Shanbhag et al., 2010). Briefly, the fused mCherry-LacI with FokI endonuclease (mCherry-LacI-FokI) generates a single double-strand break on chromosome 1 integrated LacO repeats. To detect a single focus at DSB in a single cell, RSF1-GFP was transfected with mCherry-LacI-FokI (WT or inactive nuclease (D450A)). As expected, RSF1-GFP was recruited at the site of FokI-induced single break, whereas the nuclease-deficient
22
mutant form of FokI (D450A) did not make a focus of RSF1-GFP at the site (Fig. 1D). The recruitment of RSF1 at DSBs was again confirmed using K230-ZFN, which is zinc finger nuclease generating unique DSB on chromosome 3 specifically (Xu et al., 2012). Accumulation of RSF1 at DSB together with γH2AX was validated again with Chromatin immunoprecipitation assay (Fig. 1E). Furthermore, SNF2h ATPase, given that SNF2h is recruited in PARP1 dependent manner (Smeenk et al., 2013), was also recruited at DSB on chromosome 3. Thus, these results strongly indicate that RSF1 is recruited at DSBs upon DNA damage.
24
Figure 1. RSF1 is recruited at DNA double-strand break site(s). (A and B) U2OS cells were
immunostained with anti-RSF1 and anti-γH2AX at 2 h after treatment of phleomycin (+Phleo) (50 μg/ml) or untreatment of phleomycin (-Phleo). Scale bar, 10 μm. The damage foci were analyzed in 2-dimentional (2D) and 3-dimentional (3D) image with Workstation (Nikon). (C) U2OS and RPE1 cells were pre-sensitized with BrdU (10 μM) for 30 h, followed by laser micro-irradiation. Both cell lines were immunostained with anti-RSF1 and anti-γH2AX at 10 min after micro-irradiation. (D) Diagram of the recruitment of GFP-fused proteins and mCherry-LacI-FokI (wild type and D450A mutant) on chromosome 1p3.6, stably integrated with LacO repeats, and co-localization of GFP-fused proteins and FokI wild type (WT) or FokI mutant (D450A) in live cells. H2AX-GFP and Mre11-YFP used as controls. (E) Enrichment of RSF1, SNF2h and γH2AX on site-specific DSB on chromosome 3. DSB was induced by transfection with K230-ZFN in U2OS cells, and fold enrichment by ChIP was divided by input.
25
2. ATM-dependent accumulation of RSF1 at DSBs
Based on the large scale proteomic analysis of ATM and ATR substrates and mass spectrometry-based quantitative proteomics, RSF1 is one of the potential substrates for ATM/ATR and its phosphorylation is upregulated upon DNA damage (Beli et al., 2012; Matsuoka et al., 2007). Since the recruitment of RSF1 is rapidly occurred at DSBs after micro-irradiation, the effect of ATM on the function of RSF1 upon DNA damage was investigated. The foci formation of RSF1 and pATM was examined after treatment with ATM inhibitor, KU55933, prior to adding to phleomycin. The average number of foci formation of RSF1 and pATM in pre-incubated cells with KU55933 was compared to the average number of foci in control cells without KU55933. Consistently, the number of RSF1 and pATM foci was increased in the treatment with phleomycin., while the foci formation of RSF1 and pATM upon DNA damage was impaired after treatment with KU55933 (Fig. 2A and B). These results suggest that ATM inhibition diminished the foci formation of RSF1 upon DNA damage.
In order to confirm again whether the foci formation of RSF1 depends on ATM upon DNA damage, human A-T cells (ATM knockout fibroblast) was immunostained with RSF1 and γH2AX after treatment with phleomycin. As expected, the percentage of foci formation of RSF1 (containing over 10 foci) was reduced in ATM-deficient cells after treatment with phleomycin rather than in ATM wild type cells (Fig. 2C and D). Since it had been reported that IR can induce the foci formation of γH2AX by all three PIKKs (ATM, ATR, and DNA-PK) (Wang et al., 2005), γH2AX positive cells were counted in ATM wild type and ATM deficient cells. As expected, γH2AX-positive cells were not changed dramatically in ATM wild type cells versus ATM deficient cells upon DNA damage, whereas the foci formation of RSF1 is significantly reduced in ATM knock-out cells (Fig. 2C and D). Next, the accumulation of RSF1 was examined by micro-irradiation in ATM- or ATR-depleted cells using short hairpin RNA (shRNA) transiently expressed. The accumulation of
26
RSF1 was impaired in ATM-depleted cells, while ATR depletion did not affect the accumulation of RSF1 (Fig. 2E and F). These data therefore prove that ATM, rather than ATR, is required for accumulation of RSF1 at DSB sites in DDR.
28
Figure 2. The accumulation of RSF1 at DSBs is dependent on ATM activity. (A) U2OS cells
were untreated (Ctrl) or treated with ATM inhibitor (KU55933) (20 μm) prior to phleomycin treatment (+Phleo) or untreatment (-Phleo) and immunostained with pATM (pS1981) and anti-RSF1. (B) Quantitative analysis of result in (A). ‘Before’ means untreatment of phleomycin; ‘After’ means treatment of phleomycin. The average number (Mean ± S.E.M.) of foci per cell was counted from over 100 cells. ***P < 0.005 by Student’s t-test. (C) Human fibroblast wild type (WT) or ATM knock-out (KO) cell was untreated (-Phleo) or treated with phleomycin (+Phleo) for 2 h and immunostained with anti-γH2AX and anti-RSF1. Scale bar, 10μm. (D) Quantitative analysis of result in (C). The percentage of foci positive cells (>10 foci per cell) was counted. **P < 0.01, ***P < 0.005 by Student’s t-test. (E) U2OS cells were transfected with EGFP, shATM, or shATR and pre-sensitized with BrdU for 30 h, followed by laser micro-irradiation. Cells were immunostained with anti-RSF1 and anti-pATM at 1 hour after micro-irradiation. Scale bar, 10 μm. (F) The level of pATM (pS1981) and pATR (pS428) was analyzed by western blot.
29
3. The putative motifs pSQ of RSF1 by ATM is required for its accumulation at DSBs
As mentioned previously, RSF1 has the putative motifs (pS524, pS1226, pS1325) for ATM. I thus focused on whether these motifs are important for accumulation of RSF1 at DSBs. To evaluate whether the motifs of RSF1 by ATM is important in DDR, the level of RSF1 phosphorylation was examined by phspho-specific antibody (α-pSQ/TQ). The phosphorylation level of RSF1 is increased in DNA-damaging condition (Fig. 3A). Next, the recruitment of RSF1 at DSB sites was examined after treatment with ATM inhibitor KU55933 prior to micro-irradiation. Consistently, accumulation of RSF1 was reduced in ATM inhibition, compared to its recruitment in normal condition (Fig. 3B and C). Thus, these data suggested that the recruitment of RSF1 at micro-irradiated sites is dependent on ATM activity. Next, the effect of putative motifs of RSF1 on its recruitment at DSB sites was examined. After substituting three serine residues (S524, S1226, and S1325) to alanine, I tested the accumulation of RSF1 wild type and 3SA mutant after micro-irradiation. Interestingly, over-expression of the 3SA mutant showed the reduced accumulation at the DSBs, whereas RSF1 WT was accumulated gradually at DSBs. Again, after 3SA mutant was transfected into shRSF1 stable cell line, the accumulation of this mutant is declined at DNA lesions compared to wild type (Fig. 3B and C). These results suggest that RSF1 is phosphorylated by ATM upon DNA damage and these motifs by ATM are important for its accumulation at DSBs.
31
Figure 3. The putative pSQ motifs of RSF1 by ATM are required for its accumulation at DSBs.
(A) U2OS cells were irradiated at 10Gy and harvested at 1 hour after irradiation. Whole cell extracts were lysed and immunoprecipitated with RSF1 antibody. The immunoprecipitated was detected with pSQ/TQ antibody. γH2AX antibody used as a DNA damage control. (B) RSF1-GFP (WT) or RSF1 mutant (3SA) was transfected in U2OS cells and pre-sensitized with BrdU for 30 h, followed by laser micro-irradiation. ATM inhibitor KU55933 was pre-treated for 12 hours prior to micro-irradiation. Scale bar, 10 μm. (C) Quantitative analysis of fluorescence intensity at DSBs. The accumulated fluorescence intensity at micro-irradiated sites was normalized the background around the sites. NFU stands for normalized fluorescence unit. The values (Mean ± S.E.M.) were calculated and plotted against time from at least 10 individual cells.
32
4. Depletion of RSF1 attenuates DNA damage checkpoint signals
In general, DNA damage induces ATM/ATR activation, followed by the phosphorylation of γH2AX and recruitment of the mediators, including MDC1, BRCA1, 53BP1 and others (Ciccia and Elledge, 2010). Thus, the accumulation of RSF1 in ATM-dependent manner upon DNA damage led us to examine whether RSF1 affects in DNA damage checkpoint signaling. Stably RSF1-depleted HeLa cell line was used to check the DNA damage checkpoint signaling. Surprisingly, the depletion of RSF1 attenuated DNA damage checkpoints (pNBS1, pChk1/2), especially in the propagation of γH2AX (pS139) (Fig. 4A). This result was confirmed again with U2OS cell line that was transiently knock-downed RSF1 by siRNA (Fig. 4B). Next, the retention of γH2AX and pATM after micro-irradiation was monitored. The γH2AX retention was dramatically abrogated in RSF1-depleted cells compared to control cells, whereas signals of pATM seemed to be reduced at little in RSF1 depleted cells (Fig. 4C). Next, the cell cycle progression was analyzed to check whether the depletion of RSF1 affects the cell cycle progression upon DNA damage. FACS analysis demonstrates that cell cycle progression in RSF1 depleted cells with treatment of phleomycin for 12 hours showed the abolished DNA damage checkpoint at G2/M phase, whereas half of control cells were arrested at G2/M phase through DNA damage checkpoint. (Fig. 4D) I next checked the accumulation of the down-stream cascades of ATM after DNA damage. The foci formation of MDC1 and 53BP1, similar to those of γH2AX, was significantly reduced in RSF1 knocked-down cells (Fig. 4E). Furthermore, reintroduction of RSF1 WT restored the foci formation of γH2AX and 53BP1, whereas reintroduction of RSF1 3SA mutant was unable to restore the foci formation of γH2AX (Fig. 5). Unexpectedly, reintroduction of RSF1 3SA mutant also restored the foci formation of 53BP1 (Fig. 5C). Taken together, these results demonstrate that RSF1 is involved in ATM-dependent DNA damage checkpoint signaling.
34
Figure 4. Depletion of RSF1 attenuated DNA damage checkpoints. (A) Stably RSF1-depleted
(shRSF1) or control (shCtrl) cells (HeLa) were treated with phleomycin (50 μg/ml) and harvested at each indicated time points. Whole cell extracts were directly lyzed with sample buffer. The activation of DNA damage checkpoints was examined by western blot with the indicated antibodies. U, untreated. (B) U2OS cells were transfected with siRNA for Rsf-1 (siRsf-1) or for control (siCtrl) and harvested at the indicated time points after treatment with phleomycin (Phleo) (50 μg/ml). Activation of DNA damage checkpoints were estimated by western blot with the indicated antibodies. (C) Stably RSF1-knock-downed or control U2OS cells were immunostained with anti-RSF1 and anti-γH2AX, or anti-anti-RSF1 and anti-pATM at 1 hour after laser micro-irradiation. Scale bar, 10 μm. (D) FACS profiles of stably RSF1-depleted (shRSF1) or control (shCtrl) cells during continuous DNA damage with phleomycin. (E) Stably RSF1-depleted cells (U2OS) were treated with phleomycin for 2 h and immunostained with anti-γH2AX, anti-MDC1 and anti-53BP1. The average number (Mean ± S.E.M.) of foci per cell was counted from over 100 cells. **P < 0.01, ***P < 0.005 by Student’s t-test.
36
Figure 5. Depletion of RSF1 attenuated DNA damage checkpoints. Stably RSF1-knock-downed
U2OS cells were transfected with EGFP, RSF1 WT and RSF1 3SA mutant and immunostained with anti-γH2AX (A), anti-MDC1 (B), and anti-53BP1 (C) after treatment with phleomycin (50 μg/ml). The number of foci formation per cell was calculated and graphed (D). ***P < 0.005 by Student’s t-test. N.S., not significant.
37
5. Depletion of RSF1 caused the failure in chromatin relaxation upon DNA damage
To understand the mechanism of how RSF1 regulates ATM-dependent checkpoint signaling pathway, I examined the function of RSF1 as chromatin remodeling factor in response to DNA damage. In the context of chromatin, remodeling chromatin is the critical step to propagate DDR signaling pathway and recruit repair factors (Price and D'Andrea, 2013; Soria et al., 2012). Since γH2AX is not propagated in RSF1-depleted cells, I hypothesized that the reduction of γH2AX propagation was caused by chromatin condensation in RSF1 depleted cells. TSA treatment is well known drug that induces global chromatin relaxation by inhibiting class I and II HDACs and inducing hyper-acetylation at chromatin. Thus, I treated TSA in RSF1 depleted cells and examined whether the treatment with TSA rescued the failure of chromatin decompaction caused by RSF1 depletion. Interestingly, TSA treatment significantly rescued the defect in propagation of γH2AX upon DNA damage in RSF1 depleted cells, which indicates that the reduction of γH2AX propagation in RSF1-depleted cells was caused by defect in chromatin relaxation (Figure 6A). In order to visualize the chromatin status upon DNA damage, I performed Micrococcal Nuclease (MNase) assay. MNase assay is the conventional assay to measure the accessibility of chromatin. Chromatin relaxation was usually measured by MNase assay and the relaxation was induced by neocarzinostatin (NCS) treatment, which mimics ionizing radiation. This assay showed the level of di-nucleosome was increased in RSF1-depleted cells, and the level of mono-nucleosome was reduced in RSF1-depleted cells under DNA damage (Figure 6B). In addition to MNase assay, H3 eviction at the site of DSB is another method to examine the chromatin status under DNA damage. Histone eviction is commonly found at the sites of DSB in order to open and relax the chromatin structure (Goldstein et al., 2013). To examine the H3 eviction under DNA damage AsiSI cell line, which has the defined regions that are transcriptionally active and inactive, was used (Aymard et al., 2014). H3 eviction was analyzed by ChIP assay at DSB sites in RSF1 depleted cells in
38
transcriptionally active and inactive sites. In general, transcription regulation is determined by chromatin status. Thus, I assumed that H3 eviction will be mild in transcriptionally inactive site, compared to H3 eviction in transcriptionally active site. As I expected, the broad range of H3 in transcriptionally active sites was evicted in control cells upon DNA damage, while the narrow range of H3 in transcriptionally inactive sites was evicted upon DNA damage. Interestingly, RSF1 depletion prevented H3 eviction at DSB sites in both transcriptionally active and inactive sites (Figure 6C, D, and E). This result suggests that RSF1 is required to promote chromatin decondensation upon DNA damage.
Previous reports also showed that the loss of nucleosome stability at DSB sites is mediated by chromatin remodeling factor, p400 (Xu et al., 2010). Thus, I also tested if RSF1 depletion regulates nucleosome stability in response to DNA damage. In stably RSF1 knocked down cells, nucleosome was resistant to evict histones upon DNA damage (Figure 6F). In addition this resistance was rescued by the treatment of sodium butyrate (NaB), which induces chromatin relaxation (Figure 6G). Thus, these results indicate that the nucleosome is more stable and compacted in response to DNA damage in RSF1 depleted cells, compared to control cells.
In addition to chromatin remodeling factors, ATM kinase is also known as the major determinant of chromatin status in response to DNA damage. Previous studies showed that ATM kinase induces global chromatin relaxation upon DNA damage (Ziv et al., 2006). Since the above results showed that chromatin relaxation is mediated by RSF1, I tested if ATM is regulated by RSF1. Because the results in figure 4 showed the mild reduction of pATM in RSF1-depleted cells, the retention of ATM on chromatin in RSF1-depleted cells was examined. ATM retention on chromatin was significantly unchanged under DNA damage in RSF1-depleted cells, compared to its retention in control cells. As the downstream of ATM, Rad50 in MRN complex also showed the same pattern as ATM in RSF1-depleted cells (Figure 6H). Therefore, these data indicate that RSF1 propagates
39
41
Figure 6. RSF1 relaxes the chromatin structure at DSB sites, and the forced chromatin relaxation rescued the delayed propagation of γH2AX in response to DNA damage in RSF1 depleted cells. (A) U2OS cell line was transfected with siRSF1 and treated with TSA and
phleomycin. Chromatin was isolated and immunoblotted with the following antibodies. (B) MNase assay was performed in siCtrl and siRSF1 cells treated with NCS for 30 min. (C, D, and E) AsisI cells were transfected with siRSF1 and treated with 4-OHT for 4 h to induce DNA damage. Chromatin was isolated and immunoprecipitated with H3. H3 eviction was examined at transcriptionally active (C) and inactive (D) sites, including undamaged site (E). (F) HeLa shRSF1 stable cell line was lysed with 0.8M NaCl, and the supernatant released by high salt solution was immunoblotted with histones. (G) HeLa shRSF1 stable cell line was treated with NaB and phleomycin and lysed with 0.8M NaCl. (H) Chromatin-bound proteins were examined in HeLa shRSF1 stable cell line, compared to HeLa shCtrl stable cell line, by ATM retention assay.
42
6. Depletion of RSF1 decreased histone H2A and H2B exchange at the sites of DSB
Given that RSF1 is required for chromatin relaxation and nucleosome stability, I examined histone mobility upon DNA damage in RSF1-depleted cells. Previously, Dinant et al. found that SPT16 subunit of FACT is involved in H2A histone exchange at UV-induced DNA damage (Dinant et al., 2013). Interestingly, RSF complex is initially identified as the binding protein of FACT complex (LeRoy et al., 1998). Since chromatin is condensed in RSF1 depleted cells under DNA damage, the histone mobility upon DNA damage as a result of condensed chromatin in RSF1-depleted cells was examined. In order to visualize histone mobility at DSB site, I applied a fluorescence recovery after photobleaching (FRAP) to microirradiation in order to compare histone mobility at double strand breaks in living cells. First, the half of GFP signal in nucleus was photobleached by 488nm laser and local double strand break was introduced by 405nm laser. Interestingly, GFP-H2A and GFP-H2B showed the enhanced fluorescence recovery at DSB sites, compared to fluorescence intensity in undamaged sites, which indicates that H2A and H2B dimer exchange is enhanced at the local DSB sites (Figure 7A). Next, the fluorescence intensity in RSF1-depleted cells after FRAP was measured. γH2AX was used the indicator of double strand break. In control cells, H2A and H2B dimer was rapidly exchanged at DSB sites, while H3 was unchanged at DSB sites. As expected, histone exchange of H2A and H2B dimer, but not H3 was significantly decreased in RSF1 depleted cells (Figure 7B, C, and D). Thus, these results showed that RSF1 as chromatin remodeler is required for chromatin dynamics for efficient repair upon DNA damage.
44
Figure 7. histone H2A and H2B exchange was decreased in RSF1 depleted cells at the site of DSB. (A) Recovery of fluorescence of GFP-H2A and GFP-H2B after photobleaching with 488nm
laser was increased at local DSB sites. (B) Relative fluorescence intensity of H2A and GFP-H2B was normalized by the fluorescence intensity before local DSB with 405nm laser and quantified. (C and D) The recovery of fluorescence of GFP-H2A and GFP-H2B was decreased at the sites of DSB in RSF1-depleted cells. (E) The exchange of H3 was not changed in control and RSF1-depleted cells. ***P < 0.005 by Student’s t-test. N.S., not significant.