저작자표시 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. l 이차적 저작물을 작성할 수 있습니다. l 이 저작물을 영리 목적으로 이용할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다.
Modulation of Alzheimer’s Disease
Pathology by Collagen-Induced
Rheumatoid Arthritis in APP/PS1 Mice
by
Sun Mi Park
Major in Neuroscience
Department of Medical Sciences
The Graduate School, Ajou University
Modulation of Alzheimer’s Disease
Pathology by Collagen-Induced
Rheumatoid Arthritis in APP/PS1 Mice
by
Sun Mi Park
A Dissertation Submitted to The Graduate School of Ajou
university in Partial Fulfillment of the Requirements for
the Degree of Ph.D.in Neuroscience
Supervised by
Byoung Joo Gwag, Ph.D
Eun-Hye Joe, Ph.D
Major in Neuroscience
Department of Medical Sciences
The Graduate School, Ajou University
This certifies that the dissertation
of Sun Mi Park is approved.
SUPERVISORY COMMITTEE
In Soo Joo
Byoung Joo Gwag
Eun-Hye Joe
Eun Young Kim
Yong Beom Lee
The Graduate School, Ajou University
December, 20th, 2011
i
-ABSTRACT-
Modulation of Alzheimer’s Disease Pathology by
Collagen-Induced Rheumatoid Arthritis in APP/PS1 Mice
Several evidences suggest that rheumatoid arthritis (RA) may enhance or reduce the progression of Alzheimer’s disease (AD). The present study was performed to directly explore the effects of collagen-induced rheumatoid arthritis (CIA) on cognitive function, amyloid plaque formation, microglial activation, and cerebrovascular pathology in the cortex and hippocampus of the double transgenic APP/PS1 mouse model for AD. Wild-type or APP/PS1 mice that received Wild-type II collagen (CII) in complete Freund’s adjuvant (CFA) at presymptomatic stage (2 months) or postsymptomatic stage (8.5 months) of age revealed characteristics of RA, such as joint swelling, synovitis, and cartilage and bone degradation 4 months later. Joint pathology was accompanied by sustained induction of IL-1β and TNF-α in plasma over 4 weeks after administration of CII in CFA.
In the presymptomatic stage, prior to the onset of plaque formation, CIA reduced levels of soluble and insoluble amyloid beta (Aβ peptides) and amyloid plaque formation in the cortex and hippocampus of APP/PS1 mice, which correlated with increased blood brain barrier disruption, Iba-1 or Mac-1 positive microglia, and CD45-positive microglia/macrophages. In contrast, CIA reduced survival and vessel density and length with features of cerebrovascular pathology including vascular segments, thinner vessels, and atrophic string vessels.
ii
In the postsymptomatic stage, after the onset of amyloid plaque formation, CIA mice improved cognitive function behavior based on the performance in the MWM and the Y-Maze and reduced amyloid plaque pathology. Additionally CIA mice increased cerebrovascular pathology and mortality.
The present findings suggest that RA may exert beneficial effects against Aβ burden and harmful effects on cerebrovascular pathology and mortality in AD. However, the chronic systemic inflammation causes cerebrovascular pathology, which likely aggravates pathology and neurological deficit in APP/PS1 and possibly AD patients.
Key words: Alzheimer’s disease (AD), Rheumatoid arthritis (RA), Inflammation, Blood-brain barrier (BBB), Amyloid beta (Aβ)
iii
TABLE OF CONTENTS
ABSTRACT ---i
TABLE OF CONTENTS ---iii
LIST OF FIGURES ---vi
LIST OF TABLES ---viii
LIST OF ABBREVATION ---ix
I.INTRODUCTION ---1
A. Overview ---1
B. Alzheimer’s disease (AD) and inflammation ---3
C. Neuroinflammation ---4
D. Systemic inflammation and AD ---5
E. Vascular dysfunction ---7
F. Aβ degradation enzyme ---8
G. Rheumatoid arthritis (RA) ---9
H. Specific aims of the study ---11
II.MATERIALS AND METHODS ---12
A.MATERIALS ---12
1. Animals ---12
2. Reagents ---12
B. METHODS ---13
1. Induction of CIA ---13
2. Analysis of plasma IL-1β and TNF-α ---13
iv
4. Aβ ELISA ---14
5. Immunohistochemistry ---14
6. Image analysis of amyloid plaque burden, activated microglia/macrophages, and vascular pathology ---15
7. Extravasation of Evans blue (EB) and IgG Western blotting---16
8. Aβ degradation enzyme ELISA ---16
9. Quantitative real-time PCR ---17
10. Morris water maze (MWM) ---17
11. Y-Maze ---18
12. Statistical analysis ---19
III. RESULTS ---20
A. Effects of CIA on presymptomatic stage of APP/PS1 mice ---20
1. Induction of collagen-induced arthritis (CIA) in APP/PS1 mice ---20
2. Effects of CIA on amyloid plaque pathology in APP/PS1 mice ---25
3. Enhanced activation of microglia/macrophages in APP/PS1 mice ---28
4. Effects of CIA on permeability of the blood-brain barrier (BBB) in APP/PS1 mice ---34
5. Effects of CIA on cerebrovascular pathology in APP/PS1 mice ---37
6. Effects of CIA on mortality in APP/PS1 ---40
B. Effects of CIA on postsymptomatic stage of APP/PS1 mice ---41
1. CIA improves spatial learning and memory in APP/PS1 mice ---41
(1) Morris water maze (MWM) ---41
(2) Y-Maze ---41
v
3. CIA reduces Aβ peptide levels ---47
4. CIA reduces microglial activation ---48
5. CIA increases tPA activity and decreases PAI-1 (tPA inhibitor) ---51
6. CIA reduces oxidative stress in APP/PS1 mice ---54
7. CIA enhances the permeability of the BBB and the pathology of the cerebrovessel---56
8. CIA enhances mortality in APP/PS1 ---59
IV. DISCUSSSION ---64
V. CONCLUSION ---72
REFERENCES ---73
vi
LIST OF FIGURES
Fig. 1. Experimental design of collagen-induced arthritis (CIA) in APP/PS1 mice ---21
Fig. 2. Characterization of collagen-induced arthritis (CIA) in wild-type and APP/PS1 mice ---22
Fig. 3. CIA reduces amyloid plaque burden in APP/PS1 mice ---26
Fig. 4. CIA enhances activation of microglia/macrophages in APP/PS1 mice ---30
Fig. 5. Transient increase of microglial cell proliferation in wild-type mice treated with CIA ---33
Fig. 6. CIA enhances BBB permeability and macrophages at the leptomeningeal and perivascular regions in APP/PS1 mice ---35
Fig. 7. CIA enhances cerebrovascular pathology in APP/PS1 mice ---38
Fig. 8. CIA increases mortality in APP/PS1 mice ---40
Fig. 9. CIA improves spatial learning and memory in postsymptomatic stage of APP/PS1 mice ---43
Fig. 10. CIA reduces amyloid plaque burden in postsymptomatic stage of APP/PS1 mice ---46
Fig. 11. CIA reduces Aβ peptide levels in postsymptomatic stage of APP/PS1 mice ---47
vii
Fig. 12. CIA reduces microglial activation in postsymptomatic stage of APP/PS1 mice ---49
Fig. 13. CIA increases tPA activity and decreases PAI-1 (tPA inhibitor) ---52
Fig. 14. CIA reduces oxidative stress in postsymptomatic stage of APP/PS1 mice ---55
Fig. 15. CIA enhances BBB permeability and cerebral vascular pathology in post
symptomatic stage of APP/PS1 mice ---57
viii
LIST OF TABLES
Table 1. Main transgenic mouse models of Alzheimer’s disease ---60
Table 2. Animal list ---61
Table 3. Primary antibodies used for immunohistochemistry ---62
Table 4. Primer sequence used for RT-PCR ---63
ix
LIST OF ABBREVIATION
AD: Alzheimer’s disease RA: Rheumatoid arthritis CIA: Collagen-induced arthritis CII: Bovine type II collagen CFA: Complete Freund’s adjuvant CNS: Central nervous system BBB: Blood brain barrier MWM: Morris water maze EB: Evans blue
TL: Tomato lectin IL-1β: Interleukin 1-beta
TNF-α: Tumor necrosis factor-alpha tPA: tissue plasminogen activator NEP: Neprilysin
1
I. INTRODUCTION
A. Overview
Systemic inflammation may be associated with increased risk for Alzheimer’s disease (AD) and extensive evidence supports a dynamic role for inflammation in the AD pathogenesis.
Several epidemiological studies have demonstrated that the relative risk of AD is significantly reduced in rheumatoid arthritis (RA) patients treated with nonsteroidal anti-inflammatory drugs (NSAIDs) for longer than 2 yrs (McGeer et al., 1996; Etminan et al., 2003). In meta-analysis of 17 epidemiological studies, protective effects of arthritis and anti-inflammatory drugs have been observed against AD (McGeer et al., 1996). These studies have raised the hypothesis that anti-inflammatory drug treatment protects against AD, but arthritis or RA itself could modulate AD risk.
In a recent study, amyloid plaque formation and cognitive impairment were reduced in AD mice that were subjected to subcutaneous administration of granulocyte-macrophage colony-stimulating factor (GM-CSF), an inflammatory cytokine shown to increase in RA, for 20 days (Boyd et al., 2010).
Despite numerous studies, there is no direct experimental evidence on the risk factors for arthritis and Alzheimer’s disease (AD). This experiment was conducted to verify whether RA is a risk factor for AD and whether it can directly modulate the pathology of AD divided into presymptomatic stage (prior to the onset of plaque formation) and postsymptomatic stage (after the onset of plaque formation).
2
collagen (CII) in complete Freund’s adjuvant (CFA), which has been widely used to induce RA in rodents (Brand et al., 2007). To determine whether RA directly modulates amyloidosis and microvascular pathology in AD, Aβ plaque formation, activation of microglia/macrophages, cerebral vascular pathology, cognitive impairment, and mortality were investigated in APP/PS1 mice 4 months after the injections of CII in CFA.
3
B. Alzheimer’s disease (AD) and inflammation
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder that is the most common form of dementia affecting people 65 years and older. It is characterized by extracellular Aβ deposition, intracellular neurofibrillary tangles, neuronal loss, and cerebral microvascular pathology, resulting in progressive learning and memory deficits (Masters et al., 1985; Selkoe, 1991; Farkas and Luiten, 2001).
Microglia, the macrophages that reside in the central nervous system, are activated around Aβ deposits in the brain of AD patients (McGeer et al., 1987). Levels of inflammatory mediators including TNF-α and C-reactive protein (CRP) are also increased in the peripheral blood of AD patients (Zaciragic et al., 2007; Holmes et al., 2009) and associated with increased risk of AD (Tan et al., 2007; Mancinella et al., 2009). Activated microglia/macrophages derived from the brain and blood produce chemokines, cytokines, and free radicals, which participate in Aβ plaque formation and the neurodegenerative process in AD (Dickson et al., 1993; Griffin et al., 1995; Meda et al., 1995; Pachter, 1997; Meda et al., 2001) .
Cyclooxygenase-2 (COX-2), an inducible cyclooxygenase known to mediated inflammation in RA and AD, has been proposed as a positive regulator for amyloid plaque formation and cognitive deficit in AD. In support of this hypothesis, administration of ibuprofen, a mixed COX-1/COX-2 inhibitor, reduces microglial activation, Aβ production, plaque burden, and cognitive impairment in Tg 2576 mice (Lim et al., 2000; Lim et al., 2001). Amyloid plaque formation is enhanced in APP/PS1 mice overexpressing human COX-2 (Kotilinek et al., 2008). However, the beneficial effects of NSAIDs have not been verified in randomized, double-blind,
placebo-4
controlled clinical trials for AD patient (Szekely and Zandi, 2010). In such clinical trials, NSAIDs were administrated for less than 1 yr due to increased risk of acute myocardial infarction in patients receiving NSAIDs for longer than 18 months (Bresalier et al., 2005).
In a current report from Alzheimer’s disease anti-inflammatory prevention trial (ADAPT) with NSAIDs, treatment of asymptomatic individuals with naproxen for 2yrs was shown to reduce incidence of AD after 2 to 3 yrs. (Breitner et al., 2011). Therefore, the preventive and disease-modifying potential of NSAIDs remains to be determined in AD patients treated with NSAIDs for a long period of time (>2yrs).
To date, several transgenic mouse models have been generated that over express APP and/or presenilins harboring one or more mutations associated with AD (Table 1). Although none of the AD transgenic mice models reproduces the human condition exactly, the AD transgenic mouse model may be useful for evaluating potential drug targets and developing therapeutic approaches.
C. Neuroinflammation
Traditionally, the CNS has been considered an immunologically privileged organ. However, the CNS is now known to have a field of neuroimmunology. The CNS differs from other organs in that it contains a blood−brain barrier, which is a system of tight junctions at the capillaries within the CNS that obstructs the entry of inflammatory cells, pathogens, and some macromolecules into the subarachnoid space. Classic signs of inflammation such as rubor (redness), tumor (swelling), calor (heat), and dolor (pain) are typically not seen in the CNS (Tuppo and Arias, 2005).
5
characterized by a complex cellular responses such as activation of microglia and astrocytes, cytokines ( IL-1, IL-6, TNF-α, TGF-β ) and chemokines release (IL-8, CXCR2, CCR3, CCR5, MIP-1β, MCP-1), complement proteins (C1q, C4-9, MAC), prostaglandins, free radicals and related molecular processes.
D. Systemic inflammation and AD
Inflammatory responses are regulated by bidirectional communication between the brain and the immune system. Many lines of research have established numerous routes by which the immune system and the CNS communicate. Systemic cytokines might influence the brain via several pathways, and lead to change in metabolism and behavioral alternations; these include active transport across the BBB, free diffusion through the leaky region of the BBB in the circumventricular organs, and activation of the vagus nerve (Goehler et al., 1999) and the humoral pathway (Laflamme and Rivest, 2001). The CNS can be influenced not only by inflammatory mediators produced within the brain, but also by the actions of mediators originating from the periphery.
The role of innate immunity in the brain is a critical issue and has both beneficial and deleterious impacts on peripheral immune cell traffic into the CNS depending on the stage of the disease. Blood-derived macrophages are activated by damage to or infection of the CNS and are recruited rapidly to the site of insult. Infiltration into the brain parenchyma is tightly regulated at the level of the blood−brain barrier. Peripheral immune cell migration into the CNS resembles typical leukocyte extravasations into other organs, involving chemoattraction, cellular rolling, adhesion, and diapedisis across the vascular wall (Engelhardt, 2008).
6
The peripheral immune system could play a role in the pathology of AD. Several studies support the idea that infiltrating peripheral monocytes/macrophages can act as an anti-amyloid force in AD. Town et al. found that genetic blockage of transforming factor beta (TGF-β)-Smad 2/3 signaling in peripheral macrophages promotes brain infiltration of these cells with subsequent phagocytosis and attenuation of cerebral amyloidosis (Town et al., 2008). In a mouse model of AD, Hawkers and McLaurin reported that the administration of liposome-encapsulated clodronated to TgCRND8 mice to deplete peripheral macrophages, significantly increased (approximately 5-fold) cerebral amyloid angiopathy (CAA) pathology (Hawkes and McLaurin, 2009). ElKhoury et al. reported that the recruitment of both peripheral monocyte/macrophages and resident microglia in AD mice deficient in the CC chemokine receptor 2 was significantly reduced and that they showed accelerated amyloid deposition (El Khoury et al., 2007). The peripheral innate immune cells, CD11c+ cells, served as an important anti-amyloid force. In addition, selective ablation of bone marrow−derived dendritic cells by systemic injection of diphtheria toxin resulted in significantly increased amyloid plaque in AD mice (Butovsky et al., 2006; Butovsky et al., 2007).
Several clinical and animal studies have suggested that systemic inflammation, which contributes to the exacerbation of acute symptoms of chronic neurodegenerative disease, is a risk factor for AD. Cunningham et al. showed that trainsient systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease following a challenge with LPS in a prion disease model (Cunningham et al., 2009). Systemic infection in patients with AD may lead to delirium. In addition, exposure to various infectious pathogens such as the herpes virus and Chlamydia pneumonia may be a possible risk factor for AD. Amber et al. reported that
7
bacterial and viral infections commonly associated with periodontal disease (PDD) may affect the brain and contribute to the development of AD either directly by pathogenic products or by impacting on vascular integrity, or indirectly by systemic signaling (Watts et al., 2008).
E. Vascular dysfunction
Numerous studies have linked vascular risk factors with cognitive decline and dementia in the elderly. In AD, the brain endothelial wall often degenerates, and Aβ and amyloid deposits accumulate on the outer side of the basement membrane. These deposits promote local neuroinflammatory vascular responses, including the activation of the brain endothelium, perivascular microglia, pericytes, and astrocytes. Abnormalities in the vascular system of the brain could contribute to the onset or progression of neurodegenerative events in AD. Elevated levels of markers of endothelial dysfunction have been determined in the plasma of older subjects with late onset AD and vascular dementia (Zuliani et al., 2008). Recently, data from clinical imaging and epidemiological studies have indicated that vascular changes play an important role early in the pathogenesis of AD. Magnetic resonance imaging (MRI), transcranial doppler measurements, and single photon excitation computed tomography (SPECT) in humans have established that the resting CBF is significantly reduced in AD patients and that this may be an early event in the pathogenesis of AD (Grammas, 2011).
Alternations of the cerebral microvasculature have also been reported to be associated with aging and AD. The microvascular abnormalities observed include irregularities of arterioles and capillaries, a decrease in microvascular density, increased
8
numbers of atrophic string vessels, vascular tortuosity, twisted vessels, disruption of the basement membrane, and glomerular loops (Bell and Zlokovic, 2009) (Buee et al., 1994).
F. Aβ degradation enzyme
The accumulation of the Aβ is thought to play a central role in the pathogenesis of AD. The level of Aβ within the brain depends not only on the rate of Aβ production, but also on the rate of its removal through various clearance pathways and by enzyme-mediated degradation. A number of proteases have been implicated in the proteolytic clearance of Aβ from the CNS, including neprilysin, insulin-degrading enzyme, endothelin converting enzyme, and tPA/plasminogen/plasmin.
Neprilysin (NEP) is a 90−110 kDa plasma membrane glycoprotein of the neutral zinc metalloendopeptidase family (Turner et al., 2001). The level of NEP in post-mortem brains has been shown to decline with age, and NEP enzyme activity in the CSF has been shown to be significantly reduced in AD patients and in patients with mild cognitive impairment compared with controls (Maruyama et al., 2005).
The insulin-degradation enzyme (IDE), a zinc metalloednopeptidase, has been shown to play a key role in Aβ peptide degradation both in vitro and in vivo. IDE has also been shown to be selective for the Aβ monomer (Vekrellis et al., 2000).
The plasmin cascade initiate with the tissue plasminogen activator (tPA) cleaving plasminogen to the active serine plasmin, which, cleaves the Aβ at multiple sites and prevents the aggregation of Aβ 1-42 into β-pleated sheets (Van Nostrand and Porter, 1999) (Tucker et al., 2000; Exley and Korchazhkina, 2001). In particular, tPA is widely expressed in the mouse hippocampus, amygdala, hypothalamus, and cerebellum and
9
participates in synaptic plasticity, memory and learning.
G. Rheumatoid arthritis (RA)
Rheumatoid arthritis (RA) is an autoimmune disease with a worldwide incidence of approximately 1%. The disease is characterized by chronic systemic inflammation and destruction of the cartilage and bone in the joints (Harris, 1990). Additional features include an erosive inflammatory attack on cartilaginous joints, resulting in synovitis, pannus formation and progression, cartilage and bone destruction, and eventually joint deformity (Feldmann et al., 1996), in addition to synovial lining hyperplasia and chronic infiltration of inflammatory cells including T lymphocytes, B lymphocytes, monocytes/macrophages and granulocytes. Both humoral and cellular immune mechanisms are considered to be responsible for the induction of RA (Holmdahl et al., 2002). In RA, the primary site of inflammation is the synovial tissue, from which cytokines are released into the systemic circulation where they exert many effects on distant organs including the liver, adipose tissue, skeletal muscle, immune system, and vascular endothelium (Sattar et al., 2003).
Collagen-induced arthritis (CIA) is a widely used murine model of RA and has helped delineating the role of cellular and molecular mediators in the pathogenesis of inflammatory joint disease. The disease is genetically controlled by the class II major histocompatibility complex (MHC) (Nepom et al., 1989). CIA is induced in susceptible mouse strains following intradermal immunization with collagen type II (CII) emulsified in an adjuvant. CII is the major protein constituent of joint cartilage, and immunization provokes an autoimmune response that attacks the joint (Trentham et al., 1977; Myers et
10
11
H. Specific aims of the study
The purpose of the present study is to delineate the effects of RA on presymptomatic and postsymptomatic stages of AD pathology in APP/PS1 mice. The present study was performed to address following specific aims:
1) To verify the induction of effect of RA in APP/PS1 mice by the administration of collagen type II with CFA
2) To investigate whether the induction of CIA in the presymptomatic stage of APP/PS1 mice might influence the pathology of AD in terms of Aβ levels, amyloid plaque, microglial activation, BBB dysfunction, vascular pathology, and mortality.
3) To investigate whether the induction of CIA in the postsymptomatic stage of APP/PS1 mice might influence the pathology of AD in terms of Aβ levels, amyloid plaque, microglial activation, BBB dysfunction, vascular pathology, mortality, Aβ degradation enzyme, and cognitive impairment.
12
ll. MATERIALS AND METHODS
A. MATERIALS
1. Animals
All experiments were performed in accordance with the Guideline for Animal Experiments of Ajou University and GNT Pharma. APP/PS1 mice (APP/PS1dE9) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). These mice were generated by co-injection of chimeric APPswe (K595N, M596L) and an exon-9-deleted PS1 variant (dE9) vector controlled by a mouse prion promoter (Jankowsky et al., 2001). Male APP/PS1 transgenic mice were cross-bred with B6C3F1/J background females. APP/PS1 mice (Tg+) and wild-type littermates (Tg-) were determined by PCR analysis of tail DNA.
2. Reagents
Bovine type II collagen (CII) and complete Freund’s adjuvant (CFA) were purchased from Chondrex (Redmond, WA, USA). IL-1β, TNF-α, and Aβ ELISA kits were purchased from Biosource (Camarillo, CA, USA). tPA ELISA kit was purchased from Molecular Innovations (Novi, MI, USA), Neprilysin ELISA kit was purchased from R&D systems (Minneapolis, MN), and insulysin/insulin degadation enzyme (IDE) ELISA kit was purchased from Calbiochem (San Diego, CA).
13
B. METHODS
1. Induction of CIA
Bovine CII (Chondrex) was dissolved in 0.1 M acetic acid at a 4 mg/ml by stirring overnight at 4°C, added to an equal volume of CFA (Chondrex), and homogenized as described previously (Brand et al., 2007). To induce CIA, female Tg+ and Tg- mice were injected intradermally at the tail base with 0.1 ml CII in CFA or 0.1 ml mineral oil as a vehicle. The animals received another injection of CII in CFA or mineral oil at the right hind paw 14 d after the primary immunization. Paw edema was measured using a plethysmometer (Ugo Basile, Comerio, Italy) monthly after the second immunization. After 4 months, CIA mice were sacrificed and examined for AD pathogenesis on the basis of amyloid plaques and cerebrovascular pathology. Experimental design is shown in Fig. 1.
2. Analysis of plasma IL-1β and TNF-α
Plasma cytokine levels were analyzed at various time points after the secondary injections of CII in CFA. Blood was collected via cardiac puncture and then transferred into a heparin-coated tube (Becton, Dickinson, USA). The tube was placed on ice and centrifuged at 5000 rpm (1500 g) for 3 min. The supernatant was collected and stored at −80°C. Plasma levels of IL-1β and TNF-α were measured with a commercial ELISA kit (Biosource, Camarillo, CA, USA) according to the manufacturer’s instructions.
14
Animals were anesthetized with an intraperitoneal injection of chloral hydrate (400 mg/kg) and transcardially perfused with 0.9% NaCl. The right hind paw injected with CII in CFA was removed, fixed in 10% formalin solution at 4°C for 24 h, and decalcified in 10% EDTA solution at 4°C for 3 weeks. The tissues were then embedded in paraffin and cut into 8−μm−thick sections. The right brain hemisphere was immersion-fixed with 4% paraformaldehyde for 24 h, cryoprotected in 30% sucrose for 48 h, and cut into 18−μm−thick coronal cryosections. The hippocampal formation and cortex were dissected out from the left hemisphere and frozen at −80°C for protein assay.
4. Aβ ELISA
Combined cortical and hippocampal tissues were homogenized in 0.5 ml Tris-buffered saline (TBS) containing protease inhibitor cocktail (Calbiochem, San Diego, CA, USA) and centrifuged at 14,000 rpm for 30 min at 4°C. The supernatant was collected and stored at −80°C for Western blot analysis of IgG. The pellet was sonicated in 0.5 ml of 2% SDS in TBS and centrifuged at 14,000 rpm for 40 min at 4°C. The supernatant was used for analysis of soluble Aβ. For analysis of insoluble Aβ, the pellet was sonicated with 0.5 ml 70% formic acid in water and then neutralized by 1:40 dilution into 1 M Tris-phosphate buffer (pH 11.0). Levels of Aβ40 and Aβ42 were analyzed using an ELISA kit (Biosource, Camarillo, CA, USA) according to the manufacturer’s instructions.
5. Immunohistochemistry
Brain sections were washed in phosphate-buffered saline, incubated in 0.3% H2O2 and 0.25% Triton X-100 for 10 min at room temperature, and reacted with 10% horse
15
serum for 1 h. Sections were then reacted overnight at 4°C with one of the following primary antibodies. Primer antibodies for the following murine genes can be found in Tabel 2. For 4G8 immunostaining, sections were pretreated with 70% formic acid for 10 min to expose the Aβ epitope. The sections were reacted with anti-mouse, anti-rat, or anti-rabbit IgG fluorescent or biotin-conjugated (Vector, Burlingame, CA, USA) secondary antibody for 2 h. The biotin-labeled sections were incubated with avidin-biotin-peroxidase complex (ABC Elite kit, Vector) for 1h and then visualized using 3,3′-diaminobenzidine tetrahydrochloride dehydrate (DAB,Vector). The primary antibodies used for immunohistochemistry are summarized in Table 2.
6. Image analysis of amyloid plaque burden, activated microglia/macrophages, and vascular pathology
Brain sections containing the hippocampal formation and cortex were immunolabeled with an antibody to 4G8 (amyloid plaque burden), Mac-1, CD45 (activated microglia/macrophage), or collagen type IV (vascular pathology). To identify amyloid plaques using thioflavine-S staining, the brain sections were incubated in 1% aqueous thioflavine-S (Sigma) for 10min, followed by dehydration with 70% ethyl alcohol, and cover slipped with mounting medium. Three sections on the anatomically comparable plane for each animal were analyzed using MetaMorph 6.1 software (Universal Imaging Corp, Downingtown, PA, USA). The immunostained sections were captured using an Olympus BX 51 microscope (Olympus, Japan) at a magnification of 200 ×. The areas positive for 4G8, thioflavine-S, or CD45 in the hippocampal formation and cortex were subtracted from nonspecific signal from the same section and scaled to the vehicle group (=100). The vascular pathology was analyzed in the area including the
16
CA1 pyramidal layer and cortex by measuring vessel density and length immunolabeled with collagen type IV.
7. Extravasation of Evans blue (EB) and IgG Western blotting
BBB permeability was investigated by monitoring the extravasation of EB
and IgG western blotting. Animals were injected with 0.1 ml of 2 % EB (Sigma)
in 0.9% NaCl through a tail vein. After 24 h, animals were killed by transcardial
perfusion using 0.9 % NaCl. The extravasation of Evans blue was observed under
a fluorescent microscope (excitation 557 nm, emission 576 nm).
For IgG western blotting, brain TBS-supernatant samples were electrophoresed on 8~10 % SDS polyacrylamide gel and transferred to a nitrocellulose membrane. The membrane was preincubated with 5% nonfat dry milk, reacted biotinylated anti-mouse lgG (1:500, Vector) for overnight at 4 °C. Next the membrane was incubated with avidin-biotin-peroxidase complex (ABC Elite kit, Vector) for 2 h and detected with enhanced chemiluminescence reagents (Amersham, Buckinghamshire, UK) on X-ray film. The intensity of the bands was quantified using Image Gauge 3.12 (Fuji PhotoFilm Co.). Membranes were stripped, and re-probed with anti β-actin (1:1000, Sigma) to ensure equal protein loading.
8. Aβ degradation enzyme ELISA
The TBS supernatant was collected and total protein was determined by the BCA method (Pierce Biotechnology, Rockford IL). ELISA for free catalytic site tPA (Molecular Innovations, USA) was performed on 100 μg of protein extract. Briefly,
17
biotinylated human PAI-1 was adsorbed on a streptavidin coated plate and used to capture “free catalytic site tPA”, corresponding to active tPA. A polyclonal antibody for mouse tPA was used to detect the captured tPA (Molecular Innovations, USA). Neprilysin activity was measured by mouse nerpilysin ELISA kit (R&D Systems, USA) and insulysin/insulin degradation enzyme (IDE) was measured by using a commercial ELISA kit (Calbiochem, USA) according to the manufacturer’s instruction.
9. Quantitative real-time PCR (qPCR)
RNA fraction was performed by using Easy-Blue (Intron, Korea), total RNAs from each sample were reverse-transcribed using the bioneer RT-Kit (Bioneer, Korea) according to the manufacturer’s instruction. The qPCR was performed with the Rotor– Gene 6000 unit (Corbett Life Science, USA) using SYBR Green (QIAGEN, USA) to detect the amplification products. The following cycles were performed: initial denaturation cycle of 95 ℃ for 10min, followed by 40 amplification cycle of 95 ℃ for
15s and 60 ℃ for 1 min and ending with one cycle at 25 ℃ for 15s. Analysis was
performed on the data output from the Rotor-Gene 6000 unit software (Corbett, USA) using relative quantification for mRNA expression was calculated by the comparative cycle method described by the manufacturer. Primer sequences for the following murine genes can be found in Table 3.
10. Morris water maze (MWM)
The Morris water maze was used to measure spatial learning and memory. The water maze was a circular 100 cm pool filled with water at 25–27 °C and made opaque
18
by the addition of nontoxic white paint. An escape platform (14 X 14 cm) was located 1.0 cm below (hidden) the water surface. The temperature of the water was kept constant throughout the experiment and a 10-min recovery period was allowed between the training trials. The hidden platform test consisted of 5 consecutive days of testing, with three trials per day. If the mouse failed to find the escape platform within the maximum time (60 sec), the animal was placed on the platform for 10 sec by the experimenter. During the first 5 days of testing, the mice were trained with a hidden platform. The platform location was kept constant, and the starting position fixed opposite arm. After hidden platform test, a probe trial was conducted in which the platform was removed from the pool and mice were allowed to search for the platform for 60 sec. A computer connected to an EthoVision 3.0 video-tracking system (Noldus, USA) and monitored the swim pattern. During training trials, the escape latency to the platform and path length was measured. During the probe trial, we measured the percentage of time spent in the located platform quadrants.
11. Y -Maze
To measure spontaneous alternation behavior and exploratory activity, a white Y-Maze, with 21 cm (long) by 4cm (wide) with 40-cm walls, was used. Each animal was tested in a single-5 min session, in the course of which the animal was placed in the central platform and allowed free exploration of the maze. Alternations and total number of arm choices were recorded. The percentage of spontaneous alternation was calculated as the ratio of the actual-to-possible alternations (defined as the total number of arm entries minus 2), multiplied by 100: as shown in the following equation:
19
Thus, if an animal made the following sequence of arm choices( 3,2,1,2,3,2,1,3) , the total number of alternation opportunities would be six (total entries minus two) and the percentage alternation would be 67 % (four of six).
12. Statistical analysis
All values are expressed as mean ± S.E.M. An independent-samples t-test was used to compare two samples. Analysis of variance (ANOVA) and the Student-Newman-Keuls test were used for multiple comparisons. All analyses were calculated using SPSS version 12.0 for Windows. Statistical significance was assumed at P < 0.05.
20
II. RESULTS
A. Effects of CIA on presymptomatic stage of APP/PS1 mice
1. Induction of collagen-induced arthritis (CIA) in APP/PS1 mice
APP/PS1 mice and wild-type littermates injected with CII in CFA revealed pathologic features of RA. Paw swelling and redness of the joint were manifest immediately after the second immunization, reached a peak 1 month later, and significantly persisted over the next 4 months when animals were sacrificed (Fig. 2A, P < 0.05). In contrast to CII in CFA induced paw swelling in all four paws in DBA/1 mice (Brand et al., 2007), paw swelling was confined to the injection site in wild-type and APP/PS1 mice. Plasma levels of the pro-inflammatory cytokines, IL-1β and TNF-α were increased within 1 d after the second immunization and remained elevated over the next 4 weeks in wild-type and APP/PS1 mice (Fig. 2B, P < 0.05). Inflammatory organ, splenic weight remarkably increased 2~3 folds in CIA mice compared with the vehicles (Fig. 2C). Paraffin-embedded joint sections stained with hematoxylin and eosin revealed severe bone erosion, cartilage/chondrocyte degradation, and proliferative synovitis in wild-type (Fig. 2D) and APP/PS1 (data not shown) at 4 months after the second immunization. The induction of RA has a few limitations by the difference in the APP/PS1 strains compared with the gold standard model. However, following the induction of RA, the pathology were similarly observed. The APP/PS1 mice showed various charaterisitcs associated with the collagen-induced arthritis model, including an increase in edema, spleen weight, and plasma cytokines as well as a change in a joint histology. This result means that the induction of CIA is succesfully established in APP/PS1 mice.
21
Fig. 1. Experimental design of collagen-induced arthritis (CIA) in APP/PS1 mice. (A) Pre sypmptomatic stage and (B) post symptomatic stage of CIA in wild-type and APP/PS1 mice. Animals were sacrificed 4 months after CIA, and analyzed AD pathology.
24
Fig. 2. Characterization of collagen-induced arthritis (CIA) in wild-type and APP/PS1 mice. (A) Wild-type (Tg-) and APP/PS1 (Tg+) mice were subjected to
injection with vehicle, or with CII in CFA to induce CIA. Edema was observed at the right hind paw, which received the second injection of CII in CFA and was measured at indicated time points after injection. (B) ELISA of plasma IL-1β and TNF-α over 4 weeks after injection with vehicle or CII in CFA in wild-type mice (n = 6 per group). * p<0.05, t-test, significant difference from relevant vehicle. (C) Weight of spleen after injection of CII in CFA at indicated time points in wild-type mice (n = 6 per group). (D) Bright field photomicrographs showing ankle joints stained with hematoxylin and eosin in 6-month-old wild-type mice treated with vehicle (top row) or CIA (bottom row): left, low power of ankle joint (Arrows, bone; Scale bar = 2 mm); middle, high power of cartilage (Arrow heads, chondrocyte; Scale bar = 80 μm); right, high power of synovium (Scale bar = 200 μm). Note severe bone erosion, degradation of cartilage and chondrocytes, and intensive proliferation of synovial tissue in ankle joint 4 months after injection of CIA.
25
2. Effects of CIA on amyloid plaque pathology in APP/PS1 mice
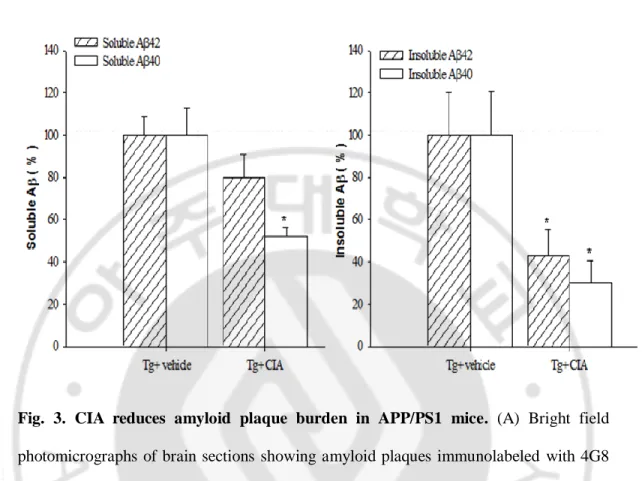
We examined the effect of CIA on Aβ deposition in APP/PS1 mice. APP/PS1 mice received CII in CFA at 2 months of age before Aβ deposition was evolved. When Aβ deposition was analyzed 4 months after CIA, levels of Aβ deposition were moderate in APP/PS1 mice (Jankowsky et al., 2004). Aβ plaques were widespread in the cerebral cortex and hippocampus of 6-month-old APP/PS1 mice (Fig. 3A). Interestingly, CIA substantially reduced Aβ plaque accumulation in APP/PS1 mice. Image analysis of Aβ deposition showed that Aβ plaques that were positive for 4G8 or thioflavine-S were significantly reduced in the cerebral cortex and hippocampus of 6-month-old APP/PS1 mice treated with CII in CFA, compared with vehicle-treated 2APP/PS1 mice (Fig. 3B). The effect of CIA on Aβ levels was analyzed in the opposite hemisphere using an ELISA that detects SDS-soluble and SDS-insoluble Aβ 40 and Aβ 42. Levels of soluble Aβ 40 and Aβ 42 appeared reduced by 48 % (P = 0.056) and 20 % (P = 0.026) in APP/PS1 mice 4 months after CIA, respectively, compared with vehicle-treated APP/PS1 mice. APP/PS1 mice treated with CIA revealed substantially significant reduction in levels of insoluble Aβ 40 and Aβ 42 (Fig. 3C, P < 0.05). This result also suggests that the reduction of amyloid plaque is caused by Aβ clearance.
27
Fig. 3. CIA reduces amyloid plaque burden in APP/PS1 mice. (A) Bright field
photomicrographs of brain sections showing amyloid plaques immunolabeled with 4G8 in 6-month-old wild-type mice (Tg- vehicle) and APP/PS1 mice treated with vehicle (Tg+ vehicle) or CIA (Tg+ CIA). Scale bar = 2 mm. (B) Plaque burden was analyzed by measuring areas positive for 4G8 (n = 3 for Tg+ vehicle and 4 for Tg+ CIA) or thioflavine-S (n = 7 for Tg+ vehicle and 8 for Tg+ CIA). (C) ELISA of soluble and insoluble forms of Aβ 42 and Aβ 40 in combined cortical and hippocampal tissues (n = 7 for Tg+ vehicle; n = 8 for Tg+ CIA).
28
3. Enhanced activation of microglia/macrophage in APP/PS1 mice
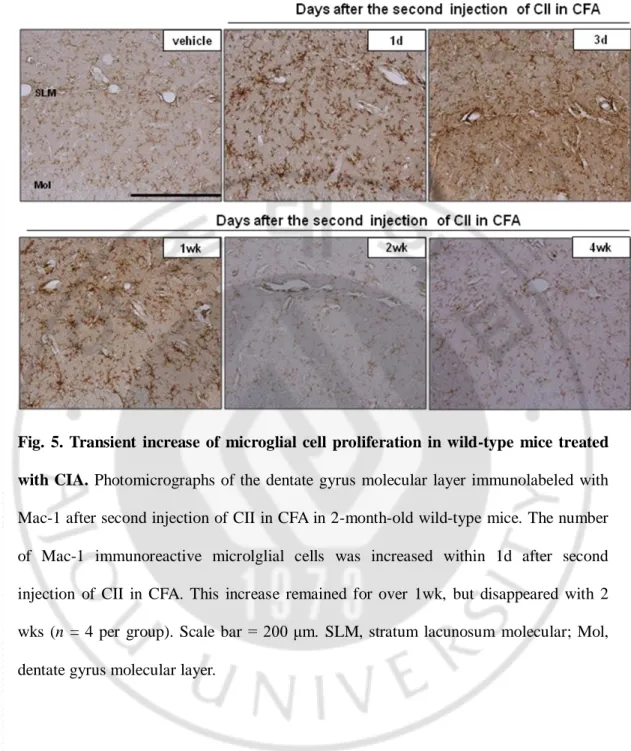
We analyzed microglia activation to determine whether these cells clear Aβ via phagocytosis. CIA-induced systemic inflammation likely triggers brain inflammation that contributes to internalization and degradation of Aβ through activated microglia. Iba-1 or Mac-1-immunoreactive microglial cells were observed in the brains of wild-type mice and increased in the brains of APP/PS1 mice at 6 months of age (Fig. 4A). Immunoreactive Mac-1 positive cells were transiently increased in the cortex and hippocampus of wild-type APP/PS1 mice over the course of 1 d after the second immunization injection. However, by 2 weeks after CIA Mac-1 immunoreactivity had returned to levels that were comparable to the levels in vehicle-injected mice (Fig. 5). In APP/PS1 mice treated with CIA, the number of Iba-1 or Mac-1-immunoreactive cells in the cerebral cortex and hippocampus increased 4 months later. CD45-positive activated microglia or macrophages were not observed in wild-type mice at 4 months after CIA (Fig. 4A). Activated microglia or macrophages were observed in the cortex of 6-month-old APP/PS1 mice and increased in the cerebral cortex and hippocampus of APP/PS1 mice treated with CIA up to approximately 210 % (the number of CD-45 immunoreactivity) and 230 % (the area of CD-45 immunoreactivity) (Fig. 4C, P < 0.05). Immunoreactivity to IL-1β and TNF-α was increased in microglia and astrocytes in the
molecular layer of the dentate gyrus of APP/PS1 mice compared to wild-type as previously reported (Benzing et al., 1999; Mehlhorn et al., 2000; Jimenez et al., 2008). CIA further increased expression of IL-1β and TNF-α in APP/PS1 mice (Fig. B). In
CIA-treated APP/PS1 mice, Iba-1-positive or CD45-positive cells were observed adjacent to Aβ plaques (Fig. 4D).
29
The increase in microglia activation in the CIA-treated mice was associated with a decrease in amyloid plaques. In particular, an increase in the leukocyte antigen marker, CD45, led to the recruitment and infiltration of immune cells through the BBB from the periphery.
32
Fig. 4. CIA enhances activation of microglia/macrophages in APP/PS1 mice. (A)
Photomicrographs of the dentate gyrus molecular layer immunolabeled with Iba-1 (top row) or CD45 (bottom row) 4 months after injection of 2-month-old wild-type (Tg-) and APP/PS1 mice (Tg+) with vehicle or CIA. Scale bar = 200 μm. SLM, stratum lacunosum molecular; Mol, dentate gyrus molecular layer. (B) Photomicrographs of the dentate gyrus molecular layer immunolabeled with IL-1β (top row) or TNF-α (bottom row) 4 months after injection of 2-month-old wild-type (Tg-) and APP/PS1 mice (Tg+) with vehicle or CIA. Scale bar = 80 μm. SLM, stratum lacunosum molecular; Mol, dentate gyrus molecular layer. (C) Activation of microglia/macrophages was analyzed by measuring CD45-immunoreactivity (CD45 burden) in the hippocampal and cortical regions (Tg+ vehicle, n = 3, Tg+ CIA, n = 4). (D) Fluorescence photomicrographs showing Iba-1 and CD45 signal around 4G8-positive amyloid plaques in the cortex of CIA-treated APP/PS1 mice. Top row, triple staining with Iba-1, 4G8, and DAPI (a nuclear marker); bottom row, triple staining with CD45, 4G8, and DAPI. Scale bar = 20
33
Fig. 5. Transient increase of microglial cell proliferation in wild-type mice treated with CIA. Photomicrographs of the dentate gyrus molecular layer immunolabeled with
Mac-1 after second injection of CII in CFA in 2-month-old wild-type mice. The number of Mac-1 immunoreactive microlglial cells was increased within 1d after second injection of CII in CFA. This increase remained for over 1wk, but disappeared with 2 wks (n = 4 per group). Scale bar = 200 μm. SLM, stratum lacunosum molecular; Mol, dentate gyrus molecular layer.
34
4. Effects of CIA on permeability of the blood-brain barrier (BBB) in APP/PS1 mice
We examined the possibility that CIA would increase the permeability of the BBB by monitoring the extravasation of Evans blue and IgG. Levels of both Evans blue (Fig 6A) and IgG (Fig 6B) remarkably increased in the CIA-treated APP/PS1 mice compared with the vehicle mice. Next, to verify macrophage infiltration through the BBB in the CIA-treated mice, immunohistochemistry using antibodies against CD206 (mannose receptor, a marker of M2 macrophage) was carried out.
Immunoreactivity to CD206 antibodies was observed sparsely in the perivascular and leptomeningeal regions of wild-type and APP/PS1 mice. CD206-positive macrophages were increased in the vicinity of vessels in wild-type and APP/PS1 mice subjected to CIA (Fig. 6D), suggesting the possibility that CIA induces migration of macrophages into the brain parenchyma through the disrupted BBB. In support of this hypothesis, levels of IgG were significantly increased in the brains of wild-type and APP/PS1 mice 1.5 and 4 months after CIA compared with relevant vehicle (Fig. 6, B and C). The results indicate that the CIA treatment enhanced BBB permeability and that increase in CD206-immunopositive macrophages could contribute to Aβ clearance.
36
Fig. 6. CIA enhances BBB permeability and macrophages at the leptomeningeal and perivascular regions in APP/PS1 mice. (A) Photomicrographs of Evans blue
extravasation of cortical area 4 months after vehicle or CIA injection into wild-type mice. Evans blue (red), tomato lectin (green), DAPI (blue) and merged images. (B) Western blot of IgG in combined cortical and hippocampal samples 4 months after CIA injection into and Tg+ mice. (C) IgG levels were analyzed 1.5 months ( vehicle, n = 3; Tg-CIA, n = 3; Tg+ vehicle, n = 3; and Tg+ Tg-CIA, n = 4) and 4 months after vehicle or CIA injection into Tg- and Tg+ mice (Tg- vehicle, n = 3; Tg-CIA, n = 4; Tg+ vehicle, n = 4; and Tg+ CIA, n = 4). (D) Fluorescence photomicrographs of cortical areas immunolabeled with rat monoclonal anti-CD206 4 months after injection of 2-month-old wild-type (Tg-) and APP/PS1 (Tg+) mice with vehicle or CIA. Note increased leptomeningeal (top row) and perivascular (bottom row) macrophages in Tg- and Tg+ mice treated with CIA. Scale bar = 200 μm.
37
5. Effects of CIA on cerebrovascular pathology in APP/PS1 mice
Next, we examined whether CIA would cause cerebrovascular pathology. Cerebral microvessels were immunolabeled with an anti-collagen type IV antibody, a marker of the endothelial basement membrane. CIA significantly reduced vascular density and length in the cerebral cortex and hippocampus of APP/PS1 mice 4 months later (Fig. 7, A and B). CIA also induced cerebrovascular pathology, such as vascular segments and atrophic string vessels (Fig. 7C).
39
Fig. 7. CIA enhances cerebrovascular pathology in APP/PS1 mice. (A) Bright-field
photomicrographs showing microvascular pathology in the cerebral cortex (Cor, top row) and hippocampus (Hip, bottom row) immunolabeled with collagen-IV 4 months after treatment of 2-month-old wild-type (Tg-) and APP/PS1 (Tg+) mice with vehicle or CIA. Scale bar = 200 μm. (B) Analysis of vessel density and length in the cerebral cortex and hippocampus (Tg- vehicle, n = 3; Tg- CIA, n = 3; Tg+ vehicle, n = 3; and Tg+ CIA, n = 4). (C) High-power photomicrographs showing microvascular pathology in the cortex of CIA-treated Tg+ mice. Note thinner vessel (arrow), string vessel (arrowhead), and degenerating vascular segments (open arrow) in Tg+ CIA compared with vehicle. Scale bar = 80 μm.
40 6. Effects of CIA on mortality in APP/PS1
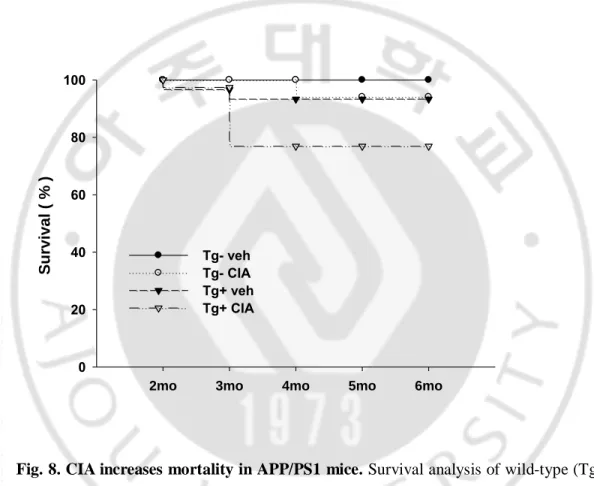
Finally, we exaimed the effect of CIA on mortality in APP/PS1 mice. At 4 months after the injection procedure, the collagen-induced arthritis in APP/PS1 transgenic mice had increased mortality compared with the vehicles (Fig.8, Survival, Tg-, Tg-CIA, Tg+, and Tg+CIA; 100 %, 94.4 %, 92.8 %, and 77.2 %, respectively, n = 30~39).
Fig. 8. CIA increases mortality in APP/PS1 mice. Survival analysis of wild-type (Tg-)
and APP/PS1 (Tg+) mice treated with vehicle or CIA at 2 months of age
2mo 3mo 4mo 5mo 6mo
Surviva l ( % ) 0 20 40 60 80 100 Tg- veh Tg- CIA Tg+ veh Tg+ CIA
41
B.Effects of CIA on postsymptomatic stage of APP/PS1 mice
1. CIA improves spatial learning and memory in APP/PS1 mice (1) Morris water maze (MWM)
We examined the possibility that CIA could mediate cognitive function behavior in APP/PS1 mice. The Morris water maze (MWM) is a widely used to test of spatial learning and memory in rodents. For the training trial, latency to find the platform location is recorded. For the probe trials, the platform is removed, we measured the percentage of time spent in the located platform quadrants. In the MWM task, the mice were trained to escape on to a submerged platform for a maximum of 60 sec using a protocol of three trials per day for 5 consecutive days. As previously reported, there was a significant difference in the escape latencies between the 12.5 month-old APP/PS1 mice and their litter mate controls. Cognitive impairment in the APP/PS1 mice was improved following CIA treatment compared with the vehicles (Fig. 9). To further analyze memory retention, we measured probe test on the sixth day without the platform. In the probe test, CIA-treated APP/PS1 mice spent more time in the target quadrant compared with the vehicles. The difference was statistically significant (Fig. 9).
(2) Y -Maze
The Y-Maze is a behavior test used to assess memory function and the willingness of rodents to explore new environments. The Y-Maze is particularly useful in measuring cognitive deficits in transgenic mice, assessing hippocampal damage and evaluating the effects of drugs on cognition. The animal is placed in the centre of the Y Maze, spontaneous alternation is calculated by the total number of individual arm entries as
42
well as the sequence of entries are recorded ( Refer to method 11 ). Compared with wild-type mice, the 12.5-month-old APP/PS1 mice showed impaired spontaneous alternation behavior. The reduced alternation in the APP/PS1 mice was statistically significant. The CIA-treated APP/PS1 mice showed a significantly improved alternation rate compared with the vehicle mice. The percentage of spontaneous alternation was as follows: Tg-vehicle 65 %, Tg+Tg-vehicle 50 %, and Tg+CIA 70 %, respectively. As shown in Fig. 9, cognitive impairment was improved in the CIA-treated APP/PS1 mice compared with the vehicles based on their performance in the MWM and Y-Maze.
44
Fig. 9. CIA improves spatial learning and memory in postsymptomatic stage of APP/PS1 mice. (A) Escape latencies of APP/PS1 mice as assessed by MWM.
Acquisition of the water maze task measured 5 consecutive days of testing, with three trials per day. Group means ± SEM are shown for the escape latency to the hidden platform (top). After hidden platform test, a 60-sec probe trial was conducted for memory retention. During the probe trial, we measured the frequency of located platform quadrants (bottom). (B) Spontaneous alternation performance in Y-Maze, expressed as a percentage of possible alternations. Values are mean ± S.E.M. p <0.05, *,Tg- vehicle vs Tg+ vehicle; #, Tg+ vehicle vs Tg+CIA.
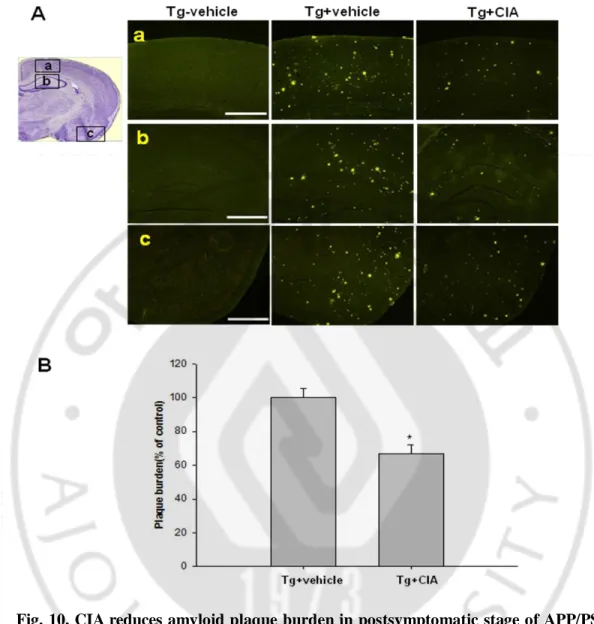
45 2. CIA reduces amyloid plaque burden
We examined the effect of CIA on Aβ deposition in postsymptomatic stage of APP/PS1 mice. Interestingly, CIA substantially reduced Aβ plaque accumulation in these APP/PS1 mice. Brain amyloid deposits were measured by thioflavine-S staining, and the plaque burden was quantified with the Metamorph program. Quantitative histochemical analysis revealed that CIA-treated APP/PS1 mice showed 40 % reduction in the plaque burden compared with the vehicle mice (Fig. 10B). The effect of CIA on the Aβ levels was analyzed in the opposite hemisphere using an ELISA that detects SDS-soluble and SDS-insoluble Aβ 40 and Aβ 42.
46
Fig. 10. CIA reduces amyloid plaque burden in postsymptomatic stage of APP/PS1 mice. (A) Fluorescence photomicrographs of brain sections showing amyloid plaques
immunstained with thioflavine-S in the 12.5-month-old wild-type mice (Tg- vehicle) and APP/PS1 mice with vehicle (Tg+ vehicle) or CIA (Tg+ CIA). (a) top row; association cortex, (b) middle row; hippocampus, (c) bottom row; pyriform cortex. Scale bar = 2 mm. (B) Plaque burden was analyzed by measuring positive areas for thioflavine-S staining (n = 8 per group).
47 3. CIA reducesAβ peptide levels
The amounts of Aβ 40 and Aβ 42, both soluble and insoluble, in the hippocampal samples were determined by a commercial ELISA kit after extraction with 2 % SDS (SDS-soluble) and 70 % formic acid (SDS-insoluble). Concentrations of the soluble Aβ 40 and Aβ 42 decreased by 23 % and 22 %, respectively, whereas levels of insoluble Aβ 40 and Aβ 42 decreased by 51 % and 41 %, respectively, in the brains of the CIA-treated APP/PS1 mice compared with the vehicle mice.
Fig. 11. CIA reduces Aβ peptide levels in postsymptomatic stage of APP/PS1 mice.
The amounts of Aβ 40 and Aβ 42, both soluble (left panel) and insoluble (right panel), in hippocampus was determined by ELISA and calculated based on the total protein.
48 4. CIA reduces microglial activation
To assess the potential role that microglial activation may play in the reduction of amyloid plaque pathology, we measured the expression of Mac-1 by quantitative RT-PCR and immunohistochemistry. In the presymptomatic AD stage, we found that microglial activation was markedly increased in the CIA-treated APP/PS1 mice as a result of Aβ clearance. In contrast, in the postsymptomatic stage, the expression of Mac-1 in the APP/PS1 mice following treatment with CIA was significantly decreased (Fig. 12). This result suggests that microglia activation induced by CIA-treated mice can act as different mechanism depending on presymptomatic stage and postsymptomatic stage.
50
Fig. 12. CIA reduces microglial activation in postsymptomatic stage of APP/PS1 mice. (A) Bright-field photomicrographs showing microglial activation in the cerebral
cortex (Cor, top panel) and hippocampus (Hip, bottom panel) immunolabeled with Mac-1 4 months after treatment of 8.5 month-old wild-type (Tg-) and APP/PSMac-1 (Tg+) mice with vehicle or CIA. Scale bar = 200 μm. SLM, stratum lacunosum molecular; Mol, dentate gyrus molecular layer. (B) Quantitative RT- PCR analysis of Mac-1 expression in the cerebral cortex and hippocampus (Tg- vehicle, n = 3; Tg+ vehicle, n = 3; and Tg+ CIA, n = 4). p <0.05, *, Tg- vs Tg+; #, Tg+ vs Tg+CIA.
51
5. CIA increases tPA activity and decreases PAI-1 (tPA inhibitor)
To investigate whether the Aβ degradation enzyme was involved in amyloid plaque pathology, we measured the activity of this enzyme by ELISA, as well as tissue plasminogen activator (tPA), neprilysin (Nep), and the insulin degradation enzyme (IDE) in brain homogenates. As previously reported in AD transgenic mice, the activity of the Aβ degradation enzyme was significantly decreased compared with the wild-type mice (Melchor et al., 2003; Madani et al., 2006; Poirier et al., 2006). The CIA-treated mice showed increasing NEP and IDE levels compared with the vehicle mice, but the difference was not significant. In particular, the CIA-treated mice showed significantly increased tPA activity compared with the vehicle mice (Fig. 13A). By increasing the activity of tPA, which is a major type of plasminogen activator expressed in the brain and plays an important role in hippocampal long-term potentiation (Mizutani et al., 1996; Nakagami et al., 2000), CIA may enhance memory function and reduce amyloid plaque pathology. We then measured the levels of the plasminogen activator inhibitor 1 (PAI-1) (tPA inhibitor) using semi-quantitative real-time PCR and immunohistochemistry. In agreement with the finding for tPA, we observed a significant decrease in PAI-1 mRNA levels in the CIA-treated APP/PS1 mice compared with the vehicles. This result indicates that increased Aβ degradation enzyme activity (especially tPA) in the brain of the APP/PS1 mice following treatment with CIA, is associated with a reduction in amyloid plaque pathology and an improvement in memory function. However, in the presymptomatic stage, the early stage of plaque deposition, there was no difference in tPA expression (data not shown) .
52 C Tg- Tg+ Tg+LJI P A I-1 m R N A / ac tin (% ) 0 20 40 60 80 100 120 140 160 180 *
PAI-1 mRNA levels
Tg-vehicle Tg+vehicle Tg+CIA
# B Tg-vehicle Tg+vehicle Tg+CIA A Cor Hip Cor
53
Fig. 13. CIA increases tPA activity and decreases PAI-1 (tPA inhibitor). (A)
Measurement ELISA of tissue plasminogen activator (tPA) or neprilysin (NEP) and insulin degradation enzyme (IDE) was performed on protein extracted from cerebral cortex and hippocampus of 12.5-month-old APP/PS1 mice. Data are normalized vehicle mice (=100%) and represented as mean ± SEM. (B) Bright-field photomicrographs showing tPA inhibitor, PAI-1, in the cerebral cortex (Cor, top panel) and hippocampus (Hip, bottom panel) immunolabeled with PAI-1 4 months after treatment of 8.5-month-old wild-type (Tg-) and APP/PS1 (Tg+) mice with vehicle or CIA. Scale bar = 2.0 mm. (C) Quantitative RT- PCR analysis of PAI-1 expression in the cerebral cortex and hippocampus (Tg- vehicle, n = 3; Tg+ vehicle, n = 3; and Tg+ CIA, n = 4). p <0.05, *, Tg- vehicle vs Tg+ vehicle ; #, Tg+ vehicle vs Tg+CIA.
54 6. CIA reduces oxidative stress in APP/PS1 mice
To explore whether CIA influenced oxidative stress in the APP/PS1 mice, we analyzed the immunohistochemistry of 8-hydroxydeoxyguanosine (8-OHdG) and 4-hydroxy-2-nonenal (HNE) in brain tissues. 8-OHdG is an oxidative DNA marker, and HNE is the major product of lipid peroxidation. The 12.5-month-old APPP/PS1 mice showed an increase in 8-OHdG and HNE immunoreactivity compared with the wild-type mice, and these signals were mainly expressed in the neurons (Fig. 14). However, the staining intensities of 8-OHdG and HNE were decreased in the CIA-treated APP/PS1 mice compared with the vehicle mice. Decreased oxidative stress in the CIA-treated mice can be interpreted as a secondary effect of the reduction in the amyloid plaque burden.
55
Fig. 14. CIA reduces oxidative stress in postsymptomatic stage of APP/PS1 mice.
Photomicrographs of cortical areas immunolabeled with anti-8-OHdG antibody (Left panel) and anti-HNE antibody (right panel) 4 months after injection of 8.5-month-old wild-type (Tg- vehicle) and APP/PS1 (Tg+ vehicle) mice with vehicle or CIA. The staining intensities of 8-OHdG and HNE in the CIA treated APP/PS1 mice were decreased compared with the vehicle mice (Top panel: Tg-vehicle, Middle panel: Tg+vehicle, Bottom panel: Tg+CIA). Scale bar = 200 μm.
56
7. CIA enhances the permeability of the BBB and the pathology of the cerebro vessel
We then examined the effect of CIA on the permeability of the BBB and on the pathology of the cerebrovessels. The permeability of the BBB was measured by IgG Western blotting, and the vascular pathology was immunostained with an anti-collagen type IV antibody, a marker of the endothelial basement membrane. CIA significantly increased IgG expression (Fig. 15A) and reduced the vascular length and the density in the cerebral cortex and the hippocampus 4 months later in both the wild-type and the APP/PS1 mice (Fig. 15B).
58
Fig. 15. CIA enhances BBB permeability and cerebrovascular pathology in postsymptomatic stage of APP/PS1 mice. (A) Western blot of IgG in combined cortical
and hippocampal samples 4 months after CIA injection into 8.5-month-old wild-type (Tg- vehicle) and APP/PS1 (Tg+ vehicle) mice. CIA enhances BBB permeability in wild-type and APP/PS1 mice. (B) Bright-field photomicrographs showing cerebral vascular pathology in the cerebral cortex (Cor, top row) and hippocampus (Hip, bottom row) immunolabeled with collagen-IV 4 months after treatment of 8.5-month-old wild-type (Tg- vehicle) and APP/PS1 (Tg+ vehicle) mice with vehicle or CIA. Scale bar = 200 μm.
59 8. CIA enhances mortality in APP/PS1 mice
Finally, we exaimed the effect of CIA on mortality in postsymptomatic stage of APP/PS1 mice. At 4 months after the injection procedure, the collagen-induced arthritis in APP/PS1 transgenic mice had increased mortality compared with the vehicles (Fig. 16, Survival, Tg- vehicle, Tg-CIA, Tg+ vehicle, and Tg+CIA; 95.8 %, 95.2 %, 96.6 %, and 85.7%, respectively).
Fig. 16. CIA increases mortality in postsymptomatic stage of APP/PS1 mice.
Survival analysis of 4 months after treatment of 8.5-month-old wild-type (Tg-) and APP/PS1 (Tg+) mice with vehicle or CIA (Tg- vehicle, n = 29; Tg- CIA, n = 21; Tg+ vehicle, n = 30; and Tg+ CIA, n = 35).
8.5 mo 9.5 mo 10.5 mo 11.5 mo 12.5 mo S ur vival ( % ) 0 20 40 60 80 100 Tg - vehicle Tg - CIA Tg + vehicle Tg + CIA
60
Table 1. Main transgenic mouse models of Alzheimer’s disease
Mouse model Gene (mutation) Promoter Reference
Tg 2576 APP 695 (K670N/M671L) Hamster prion Hsiao et al. 1996
PDAPP APP 770 (V717F) PDGF-β Games et al. 1995
TgCRND8 APP(K670N/M671L,V717F) Prion Chishti et al.2001 APP23 APP 695 (K670N/M671L) Murine Thy 1 Sturchler-Pierrat
et al. 1997 APP/PS1 A246E Human/mouse APPP swe (K595N,M596L)
+PS1 mutation A246E
Not reported Borchelt et al. 1997
APP/PS1 M146L APP 695 (K670N/M671L ) +PS1 mutation M146L
Hamster prion Holcomb et al. 2002
*APP/PS1 dE9 APP 695 (K670N/M671L ) +PS1 exon 9 deletion
Mouse prion Jankowsky et al. 2001
3x Tg APP(K670N/M671L) + Tau (P301L) + PS1 (M146V)
Thy 1 Oddo et al.2003
*, The asterisk is a mouse model used in this experiment. APP, amyloid precursor protein, PS, presenilin, PDGF, platelet-derived growth factor, PDAPP, amyloid precursor protein controlled by PDGF promoter,
61 Table2. Animal list
Presymptomatic stage
Postsymptomatic stage
Start N (Female) Start N (Female)
Tg- (2M) 30 Tg- (8.5M) 29
Tg-CIA (2M) 34 Tg-CIA (8.5M) 21
Tg+ (2M) 30 Tg+ (8.5M) 30
62
Table 3. Primary antibodies used for immunohistochemistry
Antigen Antibody type Source Titer
4G8 Mouse monoclonal Serotec 1:1000
CD45 Rat monoclonal Serotec 1:500
Iba-1 Rabbit polyclonal Wako 1:1000
Mac-1 Rat monoclonal Serotec 1:500
CD206 Rat monoclonal Serotec 1:500
IL-1β Goat polyclonal R&D 1:200
TNF-α Goat polyclonal R&D 1:200
Collagen-typeIV Rabbit polyclonal Millipore 1:500
8-OHDG Goat polyclonal Alexis 1:500
HNE Goat polyclonal Alexis 1:500