차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 1 / 16 BRIC View 2016-T12
차세대 염기서열 데이터의 생물정보학적 분석
이 창 용
공주대학교 E-mail: [email protected] 요약문 유전체 연구에 획기적인 변화를 가져온 차세대 염기서열 분석 기술(NGS)은 다양한 전산학 및 통계학적 방법을 필요로 하며 생물정보학과 계산생물학의 새로운 연구 주제로 부각되고 있다. NGS로 생성된 원시 데이터에서 유전변이와 같은 유용한 정보를 추출하기 까지 여러 분석 단계를 거치고, 각 단계의 분석 결과들은 서로 연관이 있어 어느 단계의 결과는 다음 단계의 결과에 영향을 미친다. NGS 데이터를 재분석에 이용하는 경우, 분석 단계들은 원시 데이터인 리드의 생성 및 염기 해독, 염기 정확도 관리, 참조 염기서열에 리드 정렬, 정렬 후속 과정과 염기 정확도 재보정, 그리고 정렬된 결과를 사용한 유전변이 추출 및 필터링 등으로 대략 구분될 수 있다. 본 보고서에서는 재분석의 모든 단계에 사용되는 통계학 및 전산학 방법과 개념 그리고 이에 따른 컴퓨팅 자원에 대하여 논하고, 향후 NGS 데이터 분석에 따른 연구 전망을 언급하였다.Key Words: next generation sequencing, resequencing, base quality score, alignment/mapping,
variant calling, pipeline
목 차
1. 서론 2. 본론
2.1 염기 해독과 정확도 관리(base calling and initial quality control) 2.2 정렬(alignment/mapping)
2.3 정렬 후속 과정과 염기 정확도 재보정(alignment post processing and quality score recalibration)
2.4 변이 추출과 SNP 후보 필터링(variation calling and filtering SNP candidates) 2.5 NGS 데이터 분석과 컴퓨팅 환경(NGS data analysis and computing enviroment) 3. 결론 및 전망
4. 참고문헌
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 2 / 16
1. 서론
1970년대 생어(F. Sanger)에 의해 개발된 DNA 시퀀싱(sequencing) 기술은 인간 유전체 프로젝트(human genome project)가 완성된 2003년경까지 독보적으로 사용되었다. 이 후 2007년경 Sanger 시퀀싱 방법을 개선한 차세대 염기서열 분석 기술(next generation sequencing, NGS) [1, 2]의 등장으로 염기서열 해독(sequencing)에 걸리는 시간과 비용을 획기적으로 줄일 수 있게 되었다. HTS(high-throughput sequencing) 혹은 SGS(second-generation sequencing)라고도 불리는 NGS의 가장 두드러진 특징은 초 병렬(massively parallel)적으로 백만에서 십억 단위의 리드(reads, 염기서열 단편)를 단 몇 시간 혹은 몇 일 내로 해독할 수 있다는 것이다. NGS는 대량의 염기서열 데이터를 빠른 시간 내에 해독할 수 있을 뿐만 아니라 시퀀싱 비용도 급속히 감소하는 추세이기 때문에, 유전체 연구에 매우 효율적인 방법으로 자리 잡고 있다. 예를 들어, Sanger 방법은 한 개인(Craig Venter)의 유전체를 규명하는데 약 8,000만 달러가 소요된 반면, NGS는 같은 크기의 유전체 해독에 현재 약 2,000달러면 가능하고, 더구나 이 가격은 점차 낮아질 것으로 예상된다.
NGS 플랫폼(platform)을 생산하는 대표적인 회사로 Roche/454 [3], Illumina/Solexa [4], 그리고 Life/APG [5] 등을 들 수 있는데, 플랫폼은 PCR 증폭(amplification) 방식에 따라 크게 두 종류 (solid-phase amplification, emulsion PCR 등)로 나눌 수 있다. 또한 플랫폼은 다르더라도 염기서열을 해독하는 중요한 단계인 템플릿 DNA의 준비와 증폭, 이미지 작업 및 염기서열 해독, 염기서열 정확도 관리(quality control) 등은 공통적으로 거치는 과정이다. NGS는 비단 DNA뿐만 아니라 RNA, 특히 RNA 전사 수준(transcription level)을 측정하는데 적용할 수도 있다.
NGS로 생산된 데이터는 과거에는 생각할 수 없었던 크기로 테라 바이트(terabyte, 1012 bytes) 수준인데, 컴퓨터 기술의 발전으로 인해 저비용 고효율로 데이터 저장이 가능하게 되었다. 또한 대용량 염기서열 데이터는 전통적인 데이터 분석법에 새로운 도전과 변화를 야기하게 되어 NGS 데이터 분석은 생물정보학의 새로운 연구 분야로 자리매김하였다. 따라서 NGS는 단순히 시퀀싱 방법만을 바꿔놓은 것이 아니라, 염기서열 데이터 분석 방법의 발전과 유전체 연구의 새로운 토대를 마련하는 계기가 되었다. 특히 NGS를 사용한 대규모 유전체 연구(예를 들어, 1,000 인간 유전체 [6]와 3,000 벼 유전체 연구 [7] 등)의 결과는 다양한 후속 연구를 파생시키고 있고, NGS를 통해 규명한 유전변이를 유전체 수준에서 형질과 연관분석(association analysis)에 적용 가능하게 되어 전장유전체연관분석(genome-wide association analysis, GWAS)을 포함한 비교유전체(comparative genomics) 연구가 현재 활발하게 진행되고 있다. 이에 따라 차세대 생물정보 분석 방법 개발, 전장유전체 연관분석을 위한 기반 구축, 그리고 이러한 기술 및 개념을 이해하고 응용할 수 있는 인력 양성 또한 중요한 과제로 부상하고 있다.
NGS 데이터는 드 노브 어셈블리(de novo assembly) [8]와 참조 어셈블리(reference assembly) 혹은 재분석(resequencing) 등 크게 두 가지 분야에 적용될 수 있다. 두 적용 분야의 가장 큰 차이점은 참조 염기서열(reference sequence)의 유무이다. 드 노브 어셈블리는 해당 종의 유전체 염기서열을 처음으로 해독하는 것을 말하며, 참조 어셈블리란 이미 규명된 염기서열(참조 염기서열)에 샘플의 리드를 정렬(alignment/mapping)하는 것으로, 샘플 간의 유전변이(genetic variation) 연구에 주로 사용된다.
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 3 / 16 재분석을 통해 추출할 수 있는 유전변이 중 가장 대표적인 것이 단일염기다형성(single nucleotide polymorphism, SNP)인데, 이를 이용하여 특정 질병과 관련된 새로운 돌연변이, 인간 및 작물의 집단 역사의 구성, 돌연변이 과정의 이해 등이 가능할 뿐 아니라 형질과 SNP/Indel 사이의 연관분석을 통하여 질병 치료 및 작물 육종(breeding) 등에 응용할 수 있다 [9-11]. SNP/Indel 규명의 정확도는 추 후 생물학적 분석 및 해석에 막대한 영향을 미치기 때문에, NGS 데이터에서 출발하여 SNP/Indel 규명에 이르는 전 과정인 SNP/Indel 추출 파이프라인(SNP/Indel calling pipeline)에 대한 기본적인 이해는 매우 중요하다. 본 보고서에서는 NGS 데이터를 재분석에 활용하기 위하여 원시 데이터(raw data)에서 시작하여 유전변이 추출에 이르는 단계들에서 사용되는 전산학 및 통계학적 방법과 개념, 그리고 이에 따른 컴퓨팅 자원 등을 살펴보고자 한다. 단, 외부 데이터(예를 들어, 가계도 정보, 집단 구조, 이미 알려진 변이와 유전자형 등)를 사용하여 확인된 유전변이와 유전자형의 정확도를 높이거나 확인된 유전변이에 대한 평가를 수행하는 부분은 본 보고서에 포함되지 않았다.
2. 본론
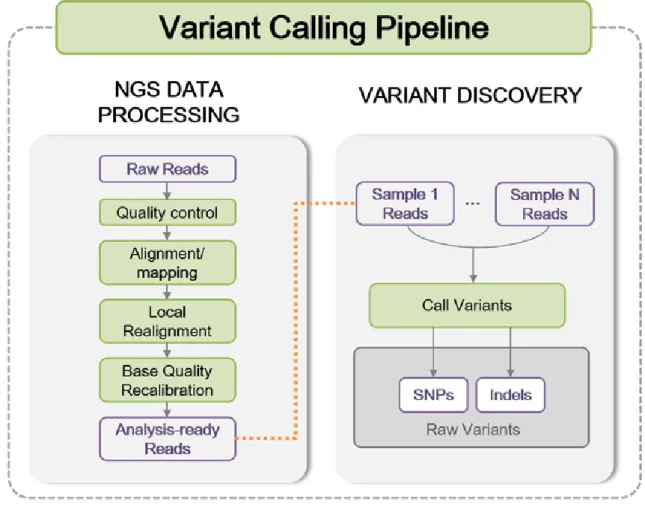
NGS는 보통 100개 정도의 염기로 구성된 짧은 서열 조각인 리드를 생성하여 염기서열을 해독한다. NGS는 해독한 염기서열을 FASTQ 형식 [12]의 파일로 저장하는데, 이를 보통 원시 데이터라 부른다. FASTQ 파일에서 SNP/Indel calling에 이르는 과정은 다양한 통계학 및 전산학적 방법을 사용하는 여러 분석 단계를 거치는데, 이를 프로그램화한 상업용 혹은 공개 분석 파이프라인들이 제공되고 있다 [13]. 원시 데이터에서 SNP/Indel calling에 이르는 과정은 크게 3단계, 즉 원시 데이터 생성(raw data generation), 참조 염기서열에 리드의 정렬(alignment/mapping), 그리고 유전변이 추출(variant calling) 등으로 구분할 수 있고 [14, 15], 각 단계의 결과물로 세가지 형식(FASTQ, SAM/BAM, VCF)의 파일이 있다. 또한 원시 데이터 생성과 리드의 정렬은 각 리드의 염기서열 정보를 처리하는 단계 [그림 1의 왼쪽]에 해당하며, 유전변이 추출은 모든 리드에 대한 정보 처리 결과를 통합하는 단계 [그림 1의 오른쪽]에 해당한다.대부분의 파이프라인은 위의 3단계를 좀 더 세밀하게 구분(base calling and initial QC, alignment to reference, post-processing of alignment, quality score recalibration, SNP calling, filtering of SNP candidate 등)하여 데이터 분석을 진행한다 [13, 16, 17]. 특히 모든 단계는 순차적으로 진행 [그림 1]됨으로 이전 단계 결과물은 다음 단계 실행을 위한 데이터가 되고, 따라서 초기 단계의 분석 결과가 추후 모든 단계에 지속적으로 영향을 미치는 특징이 있다. Broad Institute에서는 GATK(Genome Analysis Toolkit) [18]라 불리는 소위 ‘모범실무’(best practice) SNP/Indel calling 파이프라인 환경을 공개하고 있는데, GATK는 현재 NGS 데이터 분석에 가장 많이 사용되고 있는 파이프라인이다.
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 4 / 16
그림 1: SNP/Indel calling 파이프라인 예
2.1 염기 해독과 정확도 관리(base calling and initial quality control)
NGS 리드의 길이는 약 100 bp 정도로 기존 Sanger 타입의 500-1,000 bp에 비하여 길이가 짧고, 시퀀싱 오류가 상대적으로 크며, 플랫폼에 의존하는 오류도 포함될 수 있다. NGS 플랫폼들이 생성하는 FASTQ 파일은 기존의 DNA 염기서열을 나타내는 텍스트 기반의 표준 염기 데이터 형식인 FASTA 형식 [19]에 해독한 염기의 정확도(quality score 혹은 error rate)를 포함시킨 것이다. 각 리드 당 생성되는 FASTQ 파일은 4 줄로 구성되는데, 첫째 줄은 @으로 시작하며 사용한 플랫폼과 염기서열 길이 등에 대한 정보를 포함하고 있고, 둘째 줄은 해독한 염기서열, 셋째 줄은 + 기호로 시작하며 기타 설명, 그리고 마지막 줄은 둘째 줄의 염기서열에 대한 정확도(quality score)를 표시한다 [그림 2]. 따라서 둘째 줄과 넷째 줄은 같은 개수 정보로 구성된다. 또한 Illumina 플랫폼은 리드의 양쪽 끝에서 각각 염기서열을 해독하여 리드 당 같은 크기의 FASTQ 파일 한 쌍(forward and reverse)을 생성한다. 이것을 paired-end reads라 부른다 [그림 2].
NGS로 해독한 각 염기의 정확도는 프레드 수치(Phred score) [20]로 나타내는데, quality score라고도 불리는 프레드 수치 𝑄𝑃ℎ𝑟𝑟𝑟는
𝑄𝑃ℎ𝑟𝑟𝑟 = −10 log10𝑝𝜀 (1)
로 정의되며, 여기서 𝑝𝜀는 염기를 잘못 해독할 확률로 오류율(error rate)라 부른다. FASTQ 파일에서
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 5 / 16 Sanger 형식과 동일하며 0-93까지의 프레드 수치를 ASCII 코드 33에서 126까지 문자로 표현하며, 리드의 𝑄𝑃ℎ𝑟𝑟𝑟는 0에서 41 사이의 값을 가진다. 식(1)에서 알 수 있듯이 프레드 수치가 클수록 염기의 정확도는 높으며, 오류율은 그 반대이다. 예를 들어, 𝑄𝑃ℎ𝑟𝑟𝑟= 20 은 오류율 𝑝𝜀 = 1% 에 해당하며, 이것은 염기를 잘못 해독할 확률이 1%임을 의미한다. 그림 2: Paired-end FASTQ 파일 예
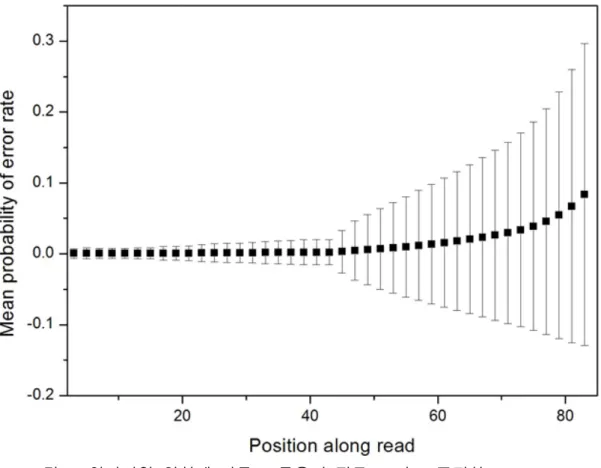
FASTQ 파일이 포함하고 있는 염기서열은 SNP/Indel calling을 위한 후속 분석 과정들에 지속적으로 영향을 미치기 때문에 염기서열의 정확도는 매우 중요하다. 또한 SNP는 인간의 경우 전체 지놈(genome)의 약 0.1% (약 1,000bp 중 1개) 정도 밖에 나타나지 않으므로 이를 확인하는 기술은 대단히 정확해야 하며, 시퀀싱 오류는 부정확한 SNP/Indel calling으로 이어질 수 있다. 일반적으로 리드 염기서열의 길이가 커짐에 따라 해독한 염기의 오류율이 증가함이 알려져 있다 [9]. 그림 3에서 불 수 있듯이 리드의 염기 위치(혹은 sequence cycle)가 커짐에 따라 평균 오류율은 증가하며, 특히 표준편차는 평균 오류율 보다 더 가파르게 증가한다. 이것은 sequence cycle이 증가함에 따라 오류율의 변동이 심하고, 후속 분석을 위한 데이터로 사용하기 어려운 매우 큰 오류의 염기도 포함됨을 의미한다.
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 6 / 16
그림 3: 염기서열 위치에 따른 오류율의 평균(■)과 표준편차(error bars).
표 1: 염기 정확도 관리 소프트웨어
소프트웨어 특징 URL
FastQC 염기정확도 관리, JAVA, GUI http://www.bioinformatics. babraham.ac.uk/projects/fastqc/ PRINSEQ 리드 가지치기 http://prinseq.sourceforge.net/ qrqc 염기정확도 관리, 가지치기, R언어 http://bit.ly/quick-qc
SolexaQA 주로 Illumina 데이터의 염기정확도 관리 http://solexaqa.sourceforge.net
대부분의 분석 파이프라인은 후속 분석의 정확도를 높이기 위해 NGS 염기서열의 정확도를 관리한다. 염기서열의 정확도 관리는 어댑터(adaptor) 서열을 제거하거나 정확도가 현저히 낮은 염기 (예를 들어, 𝑄𝑃ℎ𝑟𝑟𝑟< 10 인 염기)를 제거하는 것을 포함한다. Illumina 플랫폼의 경우에는 플랫폼 자체적으로 부분적이나마 정확도 관리를 하고, SOLiD 플랫폼의 경우에는 추후 정렬 과정에서 정확도가 낮은 염기는 걸러질 수 있기 때문에 특별한 정확도 관리를 하지 않는 것으로 알려져 있다 [16]. 현재 염기의 정확도 관리를 위한 다양한 소프트웨어가 개발되어 사용되고 있는데 [표 1], 자바(JAVA) 언어로 구현된 FastQC는 현재 가장 널리 사용되는 소프트웨어이며, qrqc은 R 언어로 구현한 패키지이다. 특히 정확도가 낮은 염기를 FASTQ 파일에서 제거하는 작업을 가지치기(trimming)라 하는데, 가지치기는 기준 염기서열에 정렬하기 전에 실시할 수도 있으며, BWA 알고리즘과 같이 정렬 과정에서 가지치기를 할 수도 있다.
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 7 / 16 2.2 정렬(alignment/mapping) 정렬(alignment/mapping)은 유전변이와 시퀀싱 오류를 포함하고 있는 106~109 개의 리드들을 참조 염기서열과 비교하여 리드의 염기서열과 일치하는 위치를 참조 염기서열에서 찾는 과정이다. 정렬에서 가장 큰 문제는 리드의 길이가 짧고 참조 염기서열 자체에 반복 영역(repetitive regions)이 있음으로 참조 염기서열의 여러 곳에 동시에 정렬될 수 있는 가능성이다. 두 염기서열을 비교하는데 일반적으로 동적 프로그래밍(dynamic programming) 알고리즘에 기초한 BLAST(basic local alignment search tool)와 같은 소프트웨어를 사용할 수 있으나, BLAST는 계산량이 너무 많아서 NGS 데이터의 정렬에는 사용하지 않는다. NGS 데이터의 정렬을 위한 알고리즘은 크게 BWT(Burrows-Wheeler transformation)와 해싱(hashing)에 기반한 알고리즘 등 두 가지 유형으로 구분할 수 있는데, 두 알고리즘 모두 BLAST에 비해 계산 속도가 1,000-10,000배 정도로 빠른 알고리즘이다. BWT는 원래 데이터 압축에 사용되는 변환으로 참조 염기서열(전체 문자열에 해당함)을 접미사(suffix)의 특별한 순열(permutation) 형태로 변환하고, 변환된 형태에는 동일한 염기가 연속적으로 나타나는 특징을 이용하여 리드 염기서열(부분 문자열에 해당)이 포함된 구간을 검색한다. 이에 반하여 해싱 방법은 참조 염기서열과 리드 염기서열을 더 짧은 길이로 자른 후 해시 함수(hash function)를 이용하여 해시 테이블의 값으로 표현하는 것으로, 두 염기서열의 유사성이 해시 테이블의 같은 값으로 표현됨을 이용한 것이다. BWT를 사용하는 소프트웨어로 Bowtie, Bowtie2 [21], BWA [22] 등이 있으며, 해싱 방법을 사용하는 소프트웨어로 MAQ [23], Stampy [24], SOAP [25] 등이 있다 [표 2].
표 2: 리드 정렬을 위한 소프트웨어
소프트웨어 특징 URL
Bowtie BWT 사용, 가장 빠름 http://bowtie-bio.sourceforge.net BWA BWT 사용, 모든 플랫폼 지원 http://bio-bwa.sourceforge.net MAQ Hashing 사용, SNP calling에도 사용 http://maq.sourceforge.net SOAP Hashing 사용, quality recalibration에도 사용 http://soap.genomics.org.cn
Stampy BWT와 Hashing 결합 http://www.well.ox.ac.uk/project-stampy
정렬 알고리즘의 성능은 처리량(throughput)과 정확도(accuracy)로 나타낼 수 있다. 처리량은 정렬 속도로 같은 시간 내에 보다 많은 리드를 정렬하는 능력을 말하며, 정확도는 시퀀싱 과정에서의 오류와 실제 염기서열의 변이에 적절히 대응하면서 리드의 정확한 정렬 위치를 찾아내는 능력을 가리킨다. 처리량 측면에서 보면 BWT에 기반한 정렬 알고리즘이 해쉬 테이블을 사용한 알고리즘 보다 약 10배 정도 빠르나, 정확도 측면에서는 해싱 알고리즘이 우수하다. 즉, BWT 기반 알고리즘은 변이가 적은 데이터에서는 동등한 수준의 정확도를 가지면서도 더 빠른 처리
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 8 / 16
속도를 보이는 반면, 해싱 기반 알고리즘은 염기 변이가 많은 데이터에서 더 많은 리드를 성공적으로 정렬한다 [24]. 따라서 정렬 소프트웨어는 처리량과 정확도 사이의 균형(tradeoff)을 고려해서 선택한다. 참고로 GATK는 BWA를 정렬 알고리즘으로 채택하고 있다. 특히 paired-end read의 경우에는 forward 리드와 reverse 리드 각각을 독립적으로 정렬하고 각 리드가 정렬된 위치에 대한 정보(.sai 파일)를 구한 후, 이를 이용하여 두 리드의 정렬 결과를 합친다. 정렬 알고리즘 선택보다 더 중요한 것은 매개변수(parameter) 값의 결정이다. 중요한 매개변수로 참조 염기서열과 리드 사이의 불일치(mismatch) 염기 개수를 들 수 있다. 불일치를 허용하지 않고 완벽한 일치만 허용한다면 후속 분석에서는 SNP/Indel은 검출할 수 없게 된다. 이와 반대로 너무 큰 불일치를 허용하면 잘못된 정렬 결과를 나을 수 있고, 그 결과로 많은 거짓양성(false-positive) SNP/Indel을 얻게 된다. 따라서 적절한 불일치 염기 개수를 결정하는 것이 중요한 문제로 부각된다. 유전체는 리드의 길이와 유사하거나 더 긴 길이의 반복 염기서열(repetitive sequence)이 존재하기 때문에, 하나의 리드가 참조 염기서열의 여러 위치에 동일한 정확도로 정렬될 수 있다. 또한 몇 개의 돌연변이(mutation) 혹은 오류로 인하여 다른 위치에 정렬될 수 있다. 따라서 정렬 과정이 끝난 다음 정렬 결과를 다양한 통계치를 사용하여 검토하는 것이 필요하다. 예를 들면 기준 염기서열에 정렬된 리드의 비율, paired-end 리드의 경우에는 성공적으로 쌍(pair)을 이룬 비율, 그리고 paired-end의 삽입 크기(insert size)에 대한 빈도 등을 들 수 있다. 이러한 통계치는 정렬을 위한 매개변수 결정에 도움을 줄 수 있으며, 정확도를 높이는데 기여할 수 있다.
그림 4: SAM/BAM 파일 예
리드들의 정렬 결과는 SAM(sequence alignment map) 형식 [26]으로 저장된다. SAM 파일은 최소 11개의 필드는 반드시 가지고 있는데, 리드이름, 리드가 참조 염기서열에 정렬된 결과를 나타내는 bitwise flags, 참조 염기서열 이름, 정렬된 위치, 정렬 정확도(mapping quality), CIGAR 등의 정보를 포함한다 [그림 4]. 정렬 여부에 대한 중요한 정보는 bitwise flags에 포함되어 있는데, 모든 리드가 기준 염기서열에 유일하고 정확하게 정렬되는 것은 아님으로 다양한 유형의 정렬 결과가 가능하고 중요한 유형을 표 3에 요약하였다. CIGAR는 정렬된 서열에 대한 정보를 부호화한 것으로, 정렬된 염기의 개수, insertion, deletion, skipped region에 관한 정보 등을 표현한다. 또한 SAM 파일을 이진(binary) 형식으로 압축한 BAM 파일은 SAM 파일과 더불어 현재 정렬 결과를 저장하는
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 9 / 16
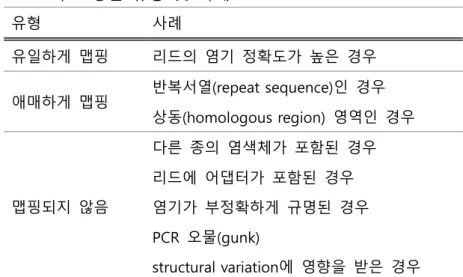
표준형식으로 자리잡고 있다. SAM/BAM 파일을 효율적으로 처리할 수 있는 소프트웨어로 SAMtools [26], GATK [27], Picard [28] 등이 현재 많이 사용되고 있다. BAM 파일은 IGV(Integrative Genome Viewer) 소프트웨어 [29]를 통해 시각화할 수 있으며, CRAMTools 패키지 [30]을 사용하여 CRAM 형식으로 압축할 수도 있다. 표 3: 리드 정렬 유형 및 사례 유형 사례 유일하게 맵핑 리드의 염기 정확도가 높은 경우 애매하게 맵핑 반복서열(repeat sequence)인 경우 상동(homologous region) 영역인 경우 맵핑되지 않음 다른 종의 염색체가 포함된 경우 리드에 어댑터가 포함된 경우 염기가 부정확하게 규명된 경우 PCR 오물(gunk) structural variation에 영향을 받은 경우
2.3 정렬 후속 과정과 염기 정확도 재보정(alignment post processing and quality score recalibration)
BAM/SAM 파일을 유전변이 추출(variant calling) 단계에 적용 하기 전에 일반적으로 두 가지 선행 처리, 즉 정렬 후속 과정(alignment post processing)과 염기 정확도 재보정(quality score recalibration)을 수행한다. 정렬 후속 과정은 정렬된 결과를 염색체 별로 분류하고, PCR 과정에서 발생하는 인공적 결함(artifact)를 제거하며, 참조 염기서열의 여러 위치에 동시에 정렬된 리드들을 제거하는 것을 포함하고, Indel을 중심으로 지역적 재정렬(local realignment)을 실행하기도 한다. 정렬 후속 과정은 소프트웨어 SAMtools 혹은 Picard [표 4]등으로 처리할 수 있다.
표 4: 정렬 후속 과정을 위한 소프트웨어
소프트웨어 특징 URL
Picard SAM 파일 처리, 다양한 통계치 계산 http://picard.sourceforge.net SAMtools SAM 파일 처리 http://samtools.sourceforge.net
SOAPsnp 정확도 재보정, SOAP의 패키지 http://soap.genomics.org.cn/soapsnp.html GATK 유전변이 추출의 거의 모든 과정을 포함 http://www.broadinstitute.org/gatk/
염기 정확도 재보정은 시퀸싱 플랫폼에서 해독한 염기의 오류율이 실제 오류율과 상이할 수 있음 [25]에 기초하여 염기의 오류율로 보정하는 과정이다. 실제 오류율을 추정하는 이유는 이후 분석에서 유전자형을 최종 결정할 때 리드의 각 위치 별 염기의 오류율이 사용되기 때문이다. 이를
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 10 / 16
위하여 가장 널리 사용되는 소프트웨어로 SOAPsnp [25]와 GATK를 들 수 있다 [표 4]. SOAPsnp는 시퀸싱 플랫폼이 제공한 염기 정확도, 리드에서 염기의 위치(sequencing cycle), 대체 유형(substitution type, 예: 참조 염기서열에서는 A 인데 리드에서는 G인 경우) 등을 사용하여 참조 염기서열에 대하여 평균 불일치 비율(mismatch rate)를 계산하고, 그 결과를 보정된 염기 정확도(recalibrated quality score)를 결정하는데 사용한다. GATK도 유사한 방법을 사용하는데, 염기를 정확도와 dinucleotide content 등으로 실험적인 오류 모델을 사용하여 분류한 후, 각 분류 군에 대하여 실험적인 불일치 비율을 계산하여 보정된 염기 정확도를 결정하는 자료로 사용한다. 정렬과 후속 과정을 마친 염기서열 데이터의 특성은 너비(breath)와 깊이(depth 혹은 coverage) 등 크게 두 가지 척도로 표현할 수 있다. 너비란 규명한 유전체 정도를 나타내며, 깊이란 유전체에서 각 염기가 리드에 의해 평균적으로 규명된 정도를 나타낸다. 깊이는 염기에 대하여 대략적으로 정규분포를 따른다.
2.4 변이 추출과 SNP 후보 필터링(variation calling and filtering SNP candidates)
변이 추출(variant calling)이란 정렬, 정렬 후속 과정, 그리고 염기 정확도 재보정 등을 거친 리드들의 BAM/SAM 파일들을 통합하여 SNP/Indel 영역을 찾는 과정이다. 변이 추출은 염기서열의 위치를 SNP/Indel로 확인될 확률로 표현하며, 주로 베이지안 방법(Bayesian method) [25]으로 계산한다. 베이지안 방법이란 사전확률(prior probability)과 우도(likelihood)를 사용하여 사후확률(posterior probability)을 계산하는 방법으로, 단순히 우도만 사용하여 추정하는 것보다 더 많은 정보를 얻을 수 있다. 이때 사전확률은 선험적으로 각 염기 위치(base position)가 유전변이가 될 확률이며, 우도는 그 위치의 염기가 서로 다른 유전자형이 될 빈도(frequency)이다. 즉, 어느 locus에서 샘플 𝑖의 유전자형이 𝐺𝑖가 될 확률은 NGS 데이터 집합 𝐷와 베이즈 규칙(Bayes’ rule)에
의하여 𝑝(𝐺𝑖|𝐷) =∑𝑆𝑝(𝐺𝑝(𝐺𝑖) 𝑝(𝐷|𝐺𝑘) 𝑝(𝐷|𝐺𝑖) 𝑘) 𝑘=1 (2) 가 된다. 여기서 𝑆 는 유전자형 개수, 𝑝(𝐺𝑖) 는 사전확률, 그리고 𝑝(𝐷|𝐺𝑖) 는 우도를 나타낸다. 사전확률은 주로 dbSNP [31]와 같이 이미 알려진 SNP 데이터베이스를 사용하여 추정한다. 변이 추출을 위해 가장 널리 사용되는 소프트웨어로 SAMtools와 GATK를 들 수 있다 [표 4].
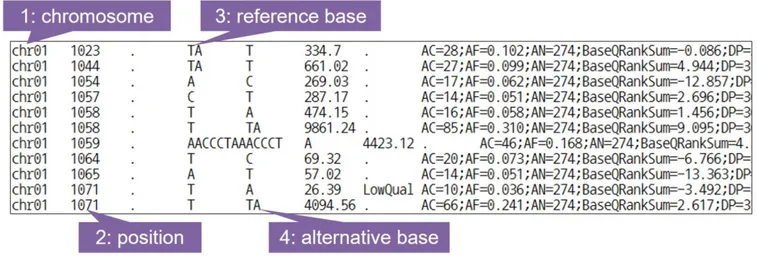
변이 추출 결과는 VCF(variant call format) [32] 형식의 파일에 저장된다. VCF 파일은 SNP/Indel로 확인된 유전변이 후보에 대한 정보, 즉 염색체 위치, 참조 염기, 치환/삽입/삭제 등이 일어난 표본 염기, 변이 정확도(variation quality), 변이로 판명된 샘플의 빈도 등을 포함하고 있다. 또한 변이 빈도(variation frequency)에 의해 SNP는 homozygous와 heterozygous 등 두 가지 유형으로 나뉜다. Homozygous는 해당 위치에 정렬된 거의 모든 샘플에서 변이가 일어난 경우이고, heterozygous는 일부 샘플에서만 변이가 일어난 경우이다. 그림 5는 SAMtools를 사용하여 구한 VCF 형식의 파일이다.
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 11 / 16 그림 5: VCF 파일 예 변이 추출을 통해 얻어진 SNP/Indel은 잠재적인 것으로 모두 유의한 유전변이라고 간주하기 어렵기 때문에 필터링을 통해 거짓양성(false-positive)에 해당하는 SNP/Indel을 제거하고 유의한 SNP/Indel만 골라내는 과정을 거친다. 보통 사용하는 방법은 Hardy-Weinberg 평형에서 벗어난 정도, 리드의 최대 및 최소 깊이, 인접한 Indels 등을 기준으로 조사한다. 특별히 정해져 있는 기준이 없기 때문에 정렬된 리드들의 깊이, 변이로 판명된 샘플의 빈도 등을 고려한 적정 기준을 세워 필터링을 진행한다. 필터링을 위한 소프트웨어로 GATK의 VariantFiltration, vcfutil.pl, SAMtools, 그리고 VCFtools [32] 등이 있다. 특히 VCFtools은 VCF 파일을 다양한 방면으로 다루는 역할을 할 수 있는데, 여러 파일을 합치거나 필요한 영역에 속하는 SNP만 추출할 수 있다.
2.5 NGS 데이터 분석과 컴퓨팅 환경(NGS data analysis and computing enviroment)
NGS로 생성한 FASTQ 파일의 크기는 압축을 해도 샘플 당 기가 바이트(gigabyte, 109 bytes)
수준에 이른다. BAM/SAM 파일과 VCF 파일의 용량을 고려하면 샘플 100개의 파일 용량은 테라 바이트(terabyte, 1012bytes) 수준이 된다. 이러한 빅 데이터(big data)를 저장하고 처리하기 위해서는
대용량 데이터 저장 장치와 고성능 서버 컴퓨터가 필요한데, NGS 데이터 분석을 위한 컴퓨팅 환경을 살펴보면 다음과 같다.
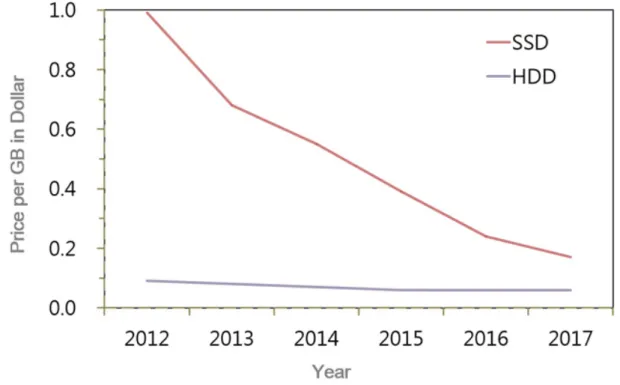
NGS 데이터를 저장하는 대표적인 수단으로 하드디스크 드라이버(hard-disk driver, HDD)를 들 수 있다. HDD는 여러 개의 디스크를 합친 디스크 배열(disk array)로 HDD의 용량은 최근 10년간 매년 50%-100%의 증가를 보이고 있으나, 데이터를 읽는 속도는 용량의 증가 추세를 따라가지 못하는 실정이다. 최근에는 HDD를 대체할 수 있는 SSD(Solid State Devices)가 등장하였는데, SSD는 일종의 비휘발성(non-volatile) 메모리로 HDD와 같은 저장 장치 역할을 하며 데이터를 읽는 속도는 HDD에 비하여 매우 빠른 장점이 있다 [표 5]. 이런 장점에 기초하여 기존의 하드디스크에 비하여 데이터 전송 속도가 매우 빠른 장점으로 여러 개의 SSD를 결합한 SSD 배열이 등장하였다. SSD의 가격은 현재는 HDD에 비하여 상대적으로 높은 편이나 곧 기존 HDD의 가격과 차이가 없을 것으로 예상됨 [그림 6]으로 조만간 HDD를 대체할 것으로 전망된다 [33].
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 12 / 16
그림 6: SSD와 HDD의 가격 변동 추이
연산을 담당하는 CPU(central processing unit)의 연산 속도는 1975년경 이전까지는 매년 2배씩 증가하였고, 이 후 2012년경까지는 18개월에 2배씩 증가(Moore’s law)하였으나, 2013년 이후는 거의 정체되고 있다. CPU의 연산 속도가 한계점에 거의 도달함에 따라 2000년경부터 CPU 제조회사들은 멀티코어 칩(multi-core chip)을 생산하기 시작했다. 멀티코어란 하나의 칩에서 캐시 메모리 등을 공유하면서 여러 연산을 동시에 실행할 수 있도록 한 것으로 멀티 프로세서와 유사한 효과를 나타낼 수 있는 CPU를 말한다. 요즘 생산되는 대부분의 CPU는 멀티코어 칩이며, 칩에 들어가는 코어의 숫자는 지속적으로 증가하고 있다. 표 5: HDD와 SDD의 성능 비교 유형 bandwidth (MB/sec) I/O 성능 (operations/sec) 반응 시간 (millisecond) Laptop HDD 125 100 12 Desktop HDD 200 300 6 SSD 550 100,000 0.05
CPU와 유사한 기능을 할 수 있는 GPU(graphical processing unit)는 최근 들어 고속 연산과 연계되어 매우 관심을 끌고 있는데, 그 이유는 GPU가 그래픽 처리를 하지 않을 때 CPU처럼 사용할 수 있기 때문이다. GPU는 초기에는 일반 텍스트를 모니터에 나타내는 역할을 했는데, 이 후 병렬 처리 등의 기능이 포함되어 현재는 3차원 CAD(computer-aided design) 소프트웨어, 비디오 게임 등을 위해 사용되고 있다. CPU는 직렬 처리에 최적화된 몇 개의 코어로 구성된 반면, GPU는 병렬 처리용으로 설계된 수 천 개의 소형이고 효율적인 코어로 구성된다. 특히 GPU는 가속 컴퓨팅에
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 13 / 16
적용할 수 있는데, GPU 가속 컴퓨팅이란 GPU와 CPU를 함께 이용하여 데이터 및 연산 처리속도를 높이는 것으로, CPU의 멀티코어를 사용한 병렬 처리 능력을 이용하여 연산 집약적인 부분을 GPU로 넘기고 나머지만 CPU에서 처리하는 것을 말한다. GPU를 사용한 예로 최근 알파고(AlphaGo)가 사용한 알고리즘으로 유명해진 딥 러닝(deep learning)에 GPU를 사용하여 CPU보다 약 50배 정도의 계산 속도를 향상시킨 결과가 있다 [34]. 아직 생물정보학 분야에는 널리 적용되고 있지는 않지만 GPU를 사용하여 계산 시간을 단축하려는 노력도 진행되고 있는데, Nvidia CUDA를 사용하여 BWA 알고리즘을 적용한 정렬 프로그램인 Barracuda [35]를 예로 들 수 있다.
1990년대까지 개인용 컴퓨터의 메모리(random access memory, RAM)는 주로 32-bits 운영체제에서 사용되었기 때문에 용량은 최대 약 4GB까지 허용되었다. 따라서 운영체제 및 실행중인 기타 소프트웨어를 위한 메모리를 포함하여 4GB 이상의 메모리를 요구하는 유전체 데이터의 분석은 불가능하였다. 2004년경 64-bits 컴퓨팅을 지원하는 CPU가 등장함에 따라 운영체제 역시 최대 약 8TB의 메모리를 지원할 수 있기 되어, NGS 데이터 전체를 메모리에 탑재하여 분석할 수 있게 되었다. 메모리 가격 또한 급격히 하락하는 추세인데, 2000년에 1 MB RAM이 약 1.12달러였으나, 2005년에는 평균 0.185달러로 하락하였고, 2010년에는 0.0122달러로 급속하게 하락하였다. 이에 따라 요즘은 테라 바이트 급의 메모리를 가진 서버도 쉽게 발견할 수 있다. 또한 컴퓨팅 자원이 부족하거나 관리하기 힘든 경우에는 인터넷에 기반한 클라우드 컴퓨팅(cloud computing)을 통하여 사용한 시간만큼 비용을 지불하는 방법도 있다. 예를 들어 Amazon Web Services Cloud [36]의 경우를 보면, 1,000 Genome Project로부터 생성된 유전체 데이터와 같은 공개된 데이터를 보유하고 있을 뿐만 아니라 데이터 분석도 실행할 수 있도록 되어 있다.
3. 결론 및 전망
NGS로 대용량의 유전정보를 생산할 수 있음에 따라 인간뿐만 아니라 다양한 동식물의 유전체 해독이 보편화 되고 있으며, 질병 진단 및 예측과 유용 유전정보 발굴 및 육종에 응용 등이 가능하게 되었다. NGS 데이터의 재분석을 통한 생물학적 해석과 활용은 원시 데이터의 분석 결과인 SNP/Indel calling의 정확도가 전제되어야 하며, 정확도는 분석 방법론과 알고리즘에 영향을 받는다. 따라서 FASTQ 파일에서 SNP calling에 이르는 분석 파이프라인에 대한 지속적인 연구가 필요하며, 생물학적 연구 목적에 따라 기능 추가/삭제 등을 포함한 파이프라인의 구조를 변화시킨 맞춤형 파이프라인을 구축할 수 있는 데이터 분석 기술이 요구된다. 현재 SNP calling을 위한 파이프라인은 다양한데, GATK를 비롯하여 공개된 소프트웨어, 공개 소프트웨어를 수정한 파이프라인, NGS 서비스를 제공하는 기업체 혹은 소규모 실험실에서 자체적으로 개발한 파이프라인 등 여러 형태가 사용되고 있다. 중요한 점은 분석 파이프라인의 선택에 따라 SNP calling 결과가 예상보다 큰 정도의 차이가 있다는 사실이다 [16]. 이것은 여러 파이프라인을 사용한 SNP calling의 결과를 다양한 각도에서 분석하고 비교할 필요성이 있음을 나타내며, 파이프라인의 표준화 작업이 필요한 것으로 추론할 수 있다. 현재는 1,000 genome project와 같은 대형 유전체 프로젝트에서 사용하는 파이프라인을 표준으로 간주하고 있으나, 아직 의견 일치를 보이는 것은 아니며, 또한 파이프라인이 워낙 복잡하여 ‘표준 파이프라인’이 가능한지도차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 14 / 16
현재로는 의문이다. 한편 “The Genome in a Bottle Consortium” [37, 38]에서 DNA 시퀀싱의 표준 데이터를 제공하고 있음으로, 향 후 다양한 소프트웨어 및 파이프라인을 비교 검토하는데 사용할 수 있을 것으로 기대된다. 유전체 데이터가 증가함에 따라 대용량의 데이터를 처리 할 수 있는 컴퓨팅 기술의 중요성이 더욱 높아지고 있고, 특히 NGS 데이터의 효율적 활용을 위해서는 생물학, 통계학, 컴퓨터과학 등을 아우르는 융복합적 생명정보 분석 기술이 필요하다. NGS 데이터는 테라 바이트급 대용량 데이터로 개인용 컴퓨터는 대용량 데이터를 처리하는데 한계가 있을 것 임으로 중대형 컴퓨터와 다중 사용자 운영체제인 유닉스(UNIX) 혹은 리눅스(Linux)의 사용이 필수가 될 것으로 전망된다. 또한 대용량 데이터를 저장하기 위한 데이터 스토리지 서버(storage server)에 대한 발전도 이미 시작되었으며, 특히 SSD를 장착한 스토리지 서버는 조만간 대세를 이루고 활성화될 것으로 예상된다. 향후 NGS는 집단유전학을 포함한 생명과학뿐만 아니라 컴퓨터과학의 다양한 연구 분야를 활성화시킬 것으로 짐작되며, 시퀀싱 가격 역시 급속히 하락하는 추세임으로 머지않아 PCR 기술처럼 보편화된 방법으로 자리잡을 것으로 예상된다.
4. 참고문헌
[1] M. Metzker, “Sequencing technologies-the next generation," Nature Genetics Reviews, 2010, Vol. 11, pp. 31-46.
[2] E. Berglund, et al. “Next-generation sequencing technologies and applications for human genetic history and Forensics,” Investigative Genetics, 2011, Vol. 2, pp. 23/1-23/15.
[3] https://lifescience.roche.com 04, 2016 [4] http://www.illumina.com 04, 2016 [5] http://www.454.com 04, 2016
[6] http://www.1000genomes.org/ 04, 2016
[7] http://iric.irri.org/resources/3000-genomes-project/ 04, 2016
[8] M. Chaisson, et al, “Genetic variation and the de novo assembly of human genomes,” Nature Reviews Genetics, 2015, Vol. 16, pp. 627-640.
[9] R. Khaja, et al., "Genome assembly comparison identifies structural variants in the human genome," Nature Genetics, 2006, Vol. 38, pp. 1413-1418.
[10] L. Barreiro, et al, "Natural selection has driven population differentiation in modern humans," Nature Genetics, 2008, Vol. 40, pp. 340–345.
[11] M. Ganal, T. Altmann, M. Röder, “SNP identification in crop plants,” Current Opinion in Plant Biology, 2009, Vol. 2, pp. 211-217.
[12] P. Cock, et al, "The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants," Nucleic Acids Research, 2009, Vol. 38 pp.1767–1771.
[13] R. Nelson, et al, “Genotype and SNP calling from next-generation sequencing data,” Nature Reviews Genetics, 2011, Vol. 12, pp. 443-451.
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 15 / 16 Bioinformatics,” 2011, UNIT 11.1
[15] P. Flieck and E. Birney, “Sense from sequence reads: methods for alignment and assembly,” Nature Methods, 2009, Vol. 6, pp. S6-S12.
[16] A. Altmann, et al, “A beginners guide to SNP calling from high-throughput DNA-sequencing data,” Human Genetics, 2012, Vol. 131, pp. 1541–1554.
[17] M. DePristo, et al, “A framework for variation discovery and genotyping using next-generation DNA sequencing data,” Nature Genetics, 2011, Vol. 43, pp. 491–498.
[18] Van der Auwera, et al, “From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline,” 2013, Current Protocol in Bioinformatics, Vol. 11, pp. 10.1–10.33.
[19] https://en.wikipedia.org/wiki/FASTA_format 04, 2016
[20] E. Hillier, M. Wendl, P. Green, “Base-calling of automated sequencer traces using phred. I. Accuracy assessment”, Genome Research, 1998, Vol. 8, pp. 175–185.
[21] B. Langmead, et al, “Ultrafast and memory-efficient alignment of short DNA sequences to the human genome,” Genome Biology, 2009, Vol. 10, pp. R25.
[22] H. Li and R. Durbin, “Fast and accurate short read alignment with Burrows-Wheeler transform,” Bioinformatics, 2009, Vol. 25, pp. 1754–1760.
[23] H. Li, et al, “Mapping short DNA sequencing reads and calling variants using mapping quality scores,” Genome Research, 2008, Vol. 18, pp. 1851–1858.
[24] G. Lunter and M. Goodson, “Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads,” Genome Research, 2011, Vol. 21, pp. 936–939.
[25] R. Li, et al, “SNP detection for massively parallel whole-genome resequencing,” Genome Research, 2009, Vol. 19, pp. 1124–1132.
[26] H. Li, et al, “The sequence alignment/map format and SAMtools,” Bioinformatics, 2009, Vol. 25, pp. 2078– 2079.
[27] A. McKenna, et al, ”The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data,” Genome Research, 2010, Vol. 20, pp. 1297–1303.
[28] http://broadinstitute.github.io/picard/ 04, 2016 [29] https://www.broadinstitute.org/igv/ 04, 2016
[30] http://www.ebi.ac.uk/ena/software/cram-toolkit 04 04, 2016
[31] S. Sherry, et al, “dbSNP: the NCBI database of genetic variation,” Nucleic Acids Research, 2001, Vol.29, pp. 308–311.
[32] P. Danecek, et al, “The variant call format and VCFtools,”, 2011, Bioinformatics, Vol. 27, pp. 2156–2158. [33] http://www.pcworld.com/ 04, 2016
[34] A. Krizhevsky, et al, “ImageNet Classification with Deep Convolutional Neural Networks, 2012, in NIPS. [35] http://sourceforge.net/projects/seqbarracuda 04, 2016
[36] http://aws.amazon.com/ec2 04, 2016 [37] https://sites.stanford.edu/abms/giab 04, 2016
[38] J. Zook, et al, “Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls,” 2014, Nat. Biotechnol., vol. 32, pp. 246–251
차세대 염기서열 데이터의 생물정보학적 분석 이창용 Page 16 / 16 The views and opinions expressed by its writers do not necessarily reflect those of the Biological Research Information Center.
이창용(2016). 차세대 염기서열 데이터의 생물정보학적 분석. BRIC View 2016-T12. Available from http://www.ibric.org/myboard/read.php?Board=report&id=2527 (Jun 21, 2016) Email: [email protected]