저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Bortezomib via Induction of
Paraptosis-associated Cell Death
by
Dong Min Lee
Major in Cancer Biology
Department of Biomedical Sciences
The Graduate School, Ajou University
Bortezomib via Induction of
Paraptosis-associated Cell Death
by
Dong Min Lee
A Dissertation Submitted to The Graduate School of Ajou University in Partial Fulfillment of the
Requirements for the Degree of
Master of Biomedical
Sciences
Supervised by
Kyeong Sook Choi, Ph.D.
Major in Cancer Biology
Department of Biomedical Sciences
The Graduate School, Ajou University
i
Nutlin-3 sensitizes cancer cells to bortezomib via induction of
paraptosis-associated cell death
The ubiquitin-proteasome system is a complex and tightly controlled system in charge of degrading 80-90% of proteins, and is central to regulating cellular function and keeping protein homeostasis. Since proteasome inhibition can lead to the accumulation of the misfolded proteins and subsequent proteotoxicity in tumor cells, the proteasome is considered a target for cancer therapies. Bortezomib, the first FAD approved proteasome inhibitor, is now in the clinic to effectively treat multiple myeloma and mantle cell lymphoma, but its efficacy in solid tumors is not satisfactory. In this study, we show that the nutlin-3, a small-molecule inhibitor of murine double minute 2 (MDM2), effectively overcomes the resistance of MDA-MB 435S breast cancer cells to bortezomib. While bortezomib treatment did not markedly alter cellular morphologies, nutlin-3 treatment induced mitochondrial dilation without induction of cell death. Alternatively, combined treatment with bortezomib and nutlin-3 effectively induced cell death, which was accompanied by the simultaneous dilation of mitochondria and the endoplasmic reticulum (ER). These results suggest that nutlin-3 may overcome the resistance of these breast cancer cells to bortezomib via induction of paraptosis-like cell death. We found that the combined treatment yielded in MCU channeled calcium increase. This is supported with inhibition of MCU by Ru360 or knockdown of MCU significantly attenuated the cell death by the combined treatment. We also found that nutlin-3 co-treatment remarkably potentiated the bortezomib-mediated upregulation of ER marker proteins, including ATF4 and CHOP. Knockdown of CHOP effectively inhibited the cellular vacuolation and subsequent cell death by the combined treatment, suggesting that upreugulation of CHOP plays an important role in this
ii
Ca²⁺ overload and CHOP-mediated ER dilation may critically contribute to paraptosis-like cell death induced by the combination of bortezomib and nutlin-3 in breast cancer cells.
iii
ABSTRACT --- i
TABLE OF CONTENTS --- iii
LIST OF FIGURES --- v
I. INTRODUCTION --- 1
II. MATERIALS AND METHODS --- 7
A. Chemicals and antibodies --- 7
B. Cell culture of various cancer cell lines --- 7
C. Examination of the stable cell lines expressing the fluorescence specifically in mitochondria or endoplasmic reticulum --- 8
D. Measurement of cellular viability --- 8
E. MTT assay --- 9
F. Western blotting --- 9
G. Immunocytochemistry --- 9
H. Measurement of cytosolic and mitochondrial Ca²⁺ levels --- 10
I. Small interfering RNAs --- 10
J. shRNA-mediated knockdown of proteins --- 10
K. Transmission electron microscopy --- 11
iv
III. RESULTS --- 13
1. Bortezomib and nutlin-3 synergistically induce cell death in various cancer cell lines with defective p53 --- 13 2. Vacuolation induced by bortezomib plus nutlin-3 is derived from the
dilation of both the ER and mitochondria --- 27 3. Protein synthesis is required for vacuolation and subsequent cell death by bortezomib plus nutlin-3 --- 33
4. CHOP induction critically contributes to the ER dilation and the cell death by bortezomib plus nultin-3 --- 36 5. Mitochondria Ca2+ uniporter-mediated mitochondrial Ca²⁺ overload is
important for the cell death induced by bortezomib plus nutlin-3 --- 41
IV. DISCUSSION --- 50 V. REFERENCES --- 68
v
Figure 1. Anti-cancer effect of bortezomib on various cancer cells --- 17
Figure 2. Bortezomib and nutlin-3 synergistically induce the cell death in diverse cell lines --- 18
Figure 3. Synergistic induction of cell death by bortezomib and nutlin-3 --- 19
Figure 4. Bortezomib and nutlin-3 synergistically decrease the number of viable cells in diverse cell lines --- 20
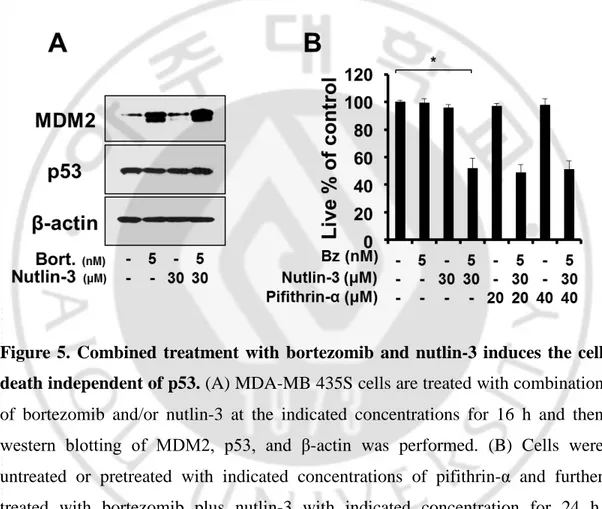
Figure 5. Combined treatment with bortezomib and nutlin-3 induces the cell death independent of p53 --- 21
Figure 6. High expression of p53 in the nuclei is not further accumulated by bortezomib and/or nultlin-3 --- 22
Figure 7. Cell death induced by bortezomib plus nutlin-3 is not associated with cell death activity of p53 --- 23
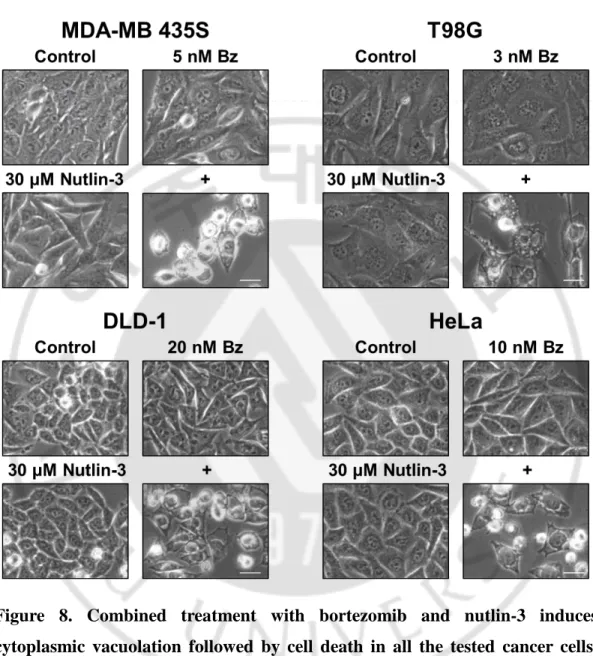
Figure 8. Combined treatment with bortezomib and nutlin-3 induces cytoplasmic vacuolation followed by cell death in all the tested cancer cells --- 24
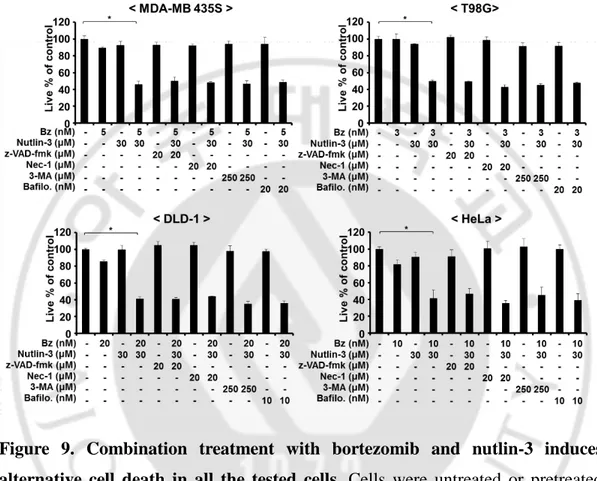
Figure 9. Combination treatment with bortezomib and nutlin-3 induces alternative cell death in all the tested cells --- 25
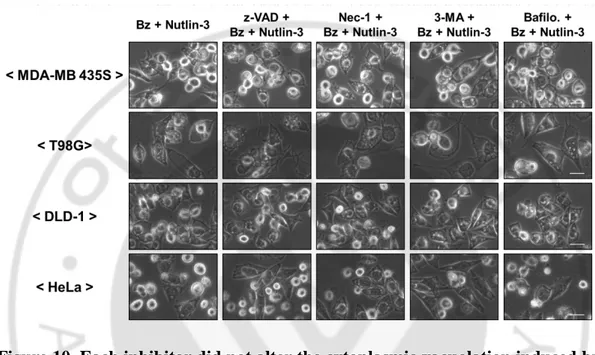
Figure 10. Each inhibitor did not alter the cytoplasmic vacuolation induced by bortezomib plus nutlin-3 in all the tested cells --- 26
vi
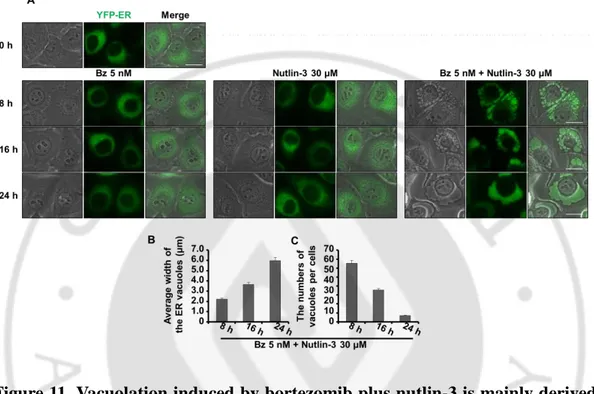
Figure 11. Vacuolation induced by bortezomib plus nutlin-3 is mainly derived from dilation of the ER --- 29
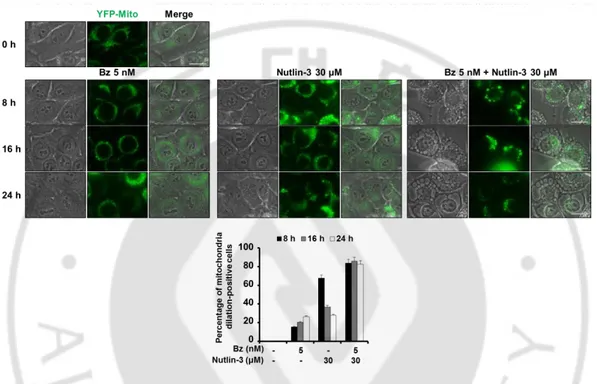
Figure 12. Combined treatment with bortezomib and nutlin-3 induces the formation of mitochondria-derived vacuoles --- 30
Figure 13. Cytoplasmic vacuolation is derived from dilation of both the ER and mitochondria --- 31
Figure 14. Dilation of the ER and mitochondria is induced by bortezomib plus nutlin-3 in carious cancer cells with defective p53 --- 32
Figure 15. CHX pretreatment effectively blocks the cell death by bortezomib plus nutlin-3 --- 34
Figure 16. CHX pretreatment effectively blocks the dilation of both the ER and mitochondria induced by bortezomib plus nutlin-3 --- 35
Figure 17. Nutlin-3 enhances the accumulation of ubiquitin conjugated proteins and ER stress --- 38
Figure 18. Knockdown of CHOP attenuates cytoplasmic vacuolation and subsequent cell death induced by bortezomib plus nutlin-3 --- 39
Figure 19. Knockdown of CHOP inhibits ER-derived vacuolation, but not mitochondria-derived vacuolation --- 40
vii
mitochondrial calcium uptake --- 43
Figure 21. Ru360 pre-treatment effectively attenuates the mitochondrial calcium uptake induced by bortezomib plus nutlin-3 --- 44
Figure 22. Ru360 pre-treatment effectively blocks the cell death by bortezomib plus nutlin-3 --- 45
Figure 23. Mitochondria calcium uptake dose not contribute to the dilation of mitochondria and the ER --- 46
Figure 24. MCU-mediated mitochondrial Ca²⁺ overload is critical for the cell death induced by bortezomib plus nutlin-3 --- 47
Figure 25. MCU-mediated mitochondrial Ca²⁺ overload is critical for the cell death by bortezomib plus nutlin-3, but not for cytoplasmic vacuolation --- 48
Figure 26. Knockdown of MCU inhibits the cell death induced by bortezomib plus nutlin-3, but not dilation of the ER and mitochondria --- 49
Figure 27. MAPKs are not involved in the cytotoxicity induced by bortezomib plus nutlin-3 --- 57
Figure 28. Only Ru360, a mitochondrial calcium uptake inhibitor, attenuates the cytotoxicity induced by bortezomib plus nutlin-3 --- 58
viii
nutlin-3 --- 59
Figure 30. ROS are not involved in the formation of megamitochondria and the ER dilation --- 60
Figure 31. CHX completely blocks mitochondrial calcium uptake --- 61
Figure 32. Various proteins associated with mitochondrial dynamics are not involved in the cytotoxicity induced by bortezomib plus nutlin-3 --- 62
Figure 33. EtBr treatment induces the dilation of mitochondria --- 63
Figure 34. EtBr treatment sensitizes MDA-MB 435S cells to bortezomib-mediated vacuolation and cell death --- 64
Figure 35. Combined treatment with EtBr and bortezomib induces cytoplasmic vacuolation derived by both the ER and mitochondria. --- 65
Figure 36. EtBr treatment induces the accumulation of mtHsp70 within the mitochondria in MDA-MB 435S cells --- 66
Figure 37. Nutlin-3 treatment induces the accumulation of mtHsp70 within the mitochondria in MDA-MB 435S cells, similar to EtBr treatment ---- 67
- 1 -
I.
INTRODUCTION
[Paraptosis]
Cancer, a large mass of tissue called tumor, is the uncontrolled growth of abnormal cells anywhere in a body. In many cancer patients, chemotherapy is one of the reasonable choices for survival, but various cancer cells are resistant to chemotherapeutic-drug-induced apoptosis. Numerous data indicate that defects in apoptotic signaling pathways contribute to the development of cancer and to therapy resistance in many types of malignant tumors (Roth, 2009). Therefore, cancer cells that have acquired resistance to apoptosis need novel strategies for inducing non-apoptotic cell death. Non-apoptotic cell death may have considerable merit for the treatment of cancer cells which have acquired resistance to apoptosis (Bröker et al., 2005). Paraptosis characteristically lacks the apoptotic features (paraptosis; para = next to or related to, and apoptosis). It typically does not involve the activation of caspases, the formation of apoptotic bodies, or other characteristics of apoptotic morphologies; it is insensitive to apoptotic inhibitors (e.g., caspase inhibitors and Bcl-xL) and requires protein synthesis (Sperandio et al., 2000; Sperandio et al., 2004). Paraptosis is characterized by a requirement for new gene transcription and translation and AIP-1/Alix (a protein that interacts with the cell death-related calcium-binding protein, ALG-2) as an inhibitor of paraptosis (Sperandio et al., 2004; Valamanesh et al., 2007). Recently, paraptosis was shown to be associated with the activation of the mitogen-activated protein kinase pathways as well as generation of reactive oxidant species (ROS) and Ca2+ (Yoon
et al., 2012; Lee et al., 2015; Wang et al., 2012; Kar et al., 2009; Li et al., 2011). However, the underlying mechanism of paraptosis, in particular the signals responsible for triggering mitochondria and ER dilation, have not yet been fully determined.
- 2 -
[ER stress]
The vast majority of proteins that a cell secretes or displays on the cell surface first enter the ER, where they fold and mature. To ascertain fidelity in protein folding, cells regulate the protein folding capacity in the ER according to need (Walter & Ron, 2011). Disruption of balance between protein load and folding capacity induces the ER stress. Upon ER stress, cells activate a series of complementary adaptive mechanisms to cope with protein-folding alteration, which together are known as UPR (Hetz, 2012). The UPR monitors condition in the ER, sensing an insufficiency in the ER’s protein folding capacity and transduces information about the protein-folding status in the ER lumen to the nucleus and cytosol to buffer fluctuation in unfolded protein load (Ron & Walter, 2007; Schröder & Kaufman, 2005; Hetz et al., 2011). Initiation of the UPR is mediated by three ER transmembrane sensors: activating transcription factor 6α (ATF6α), inositol-requiring enzyme 1α (IRE1α), and PKR-like endoplasmic reticulum kinase (PERK) (Ron & Walter, 2007). The UPR results in global changes in gene expression to restore the ER homeostasis (Huber et al., 2013): While ATF6α activates genes to increase the protein-folding capacity through the upregulation of chaperone proteins, IRE1α (via RNase activity) and PERK (via kinase activity) both branches decrease the load of proteins entering the ER through activation of degradation and inhibition of translation. In unstressed ER, Bip/Grp78 binds to the ER luminal domains of these sensor proteins and keeps them inactive (Szegezdi et al., 2006). Upon sensing the accumulation of misfolded protein in the ER lumen, Bip/Grp78 dissociates from its clients and translocates to the ER lumen to help protein folding (Bertolotti et al., 2000; Liu & Kaufman, 2003). Improperly folded proteins in the ER lumen are delivered for proteasomal degradation after retro-translocation into the cytosol, a process called ER-associated degradation (ERAD) (Smith et al., 2011). Therefore, accumulation of misfolded protein is important for the ER stress, and the ERAD pathway is required for maintenance of the ER
- 3 -
homeostasis. But, prolonged activity of the UPR, an indication that ER stress cannot be mitigated and homeostasis cannot be reestablished, correlates with cell death (Walter & Ron, 2011). During the induction of paraptosis, dilation of the ER seems to be mediated by prolonged UPR or the ER stress At least, the ER dilation is accompanied by the increase in the ER stress marker proteins.
[The ubiquitin proteasome system (UPS)]
Like all intracellular components, the proteome is in a dynamic state of biogenesis and proteolysis (Glickman & Ciechanover, 2002). Proteolysis is the enzymatic process that is associated with the breakdown of proteins into their component polypeptide or amino acids. This process is served by a diverse group of proteases, protein hydrolysis enzymes, involving the proteasome, which is multifunctional proteolytic complex. Proteasome is the 26S proteasome complex, macromolecular machinery, which consists of two 19S regulatory subunits and one 20S catalytic subunit (Gallastegui et al., 2010). Proteasome differs from typical proteases in many characteristics, including ATP dependency, systematic process, and unique recognizing-molecule, etc. (Kisselev & Goldberg, 2001). In contrast to non-specific degradation by lysosomes, proteasomes are highly selective and destroy only the proteins that are covalently labelled with small proteins, called ubiquitins (Grigoreva et al., 2015). Because studies on the proteasome inhibitors have shown that the bulk of cellular proteins, 80–90%, are degraded by the proteasome, ubiquitin-proteasome system (UPS) is important for the maintenance of protein homeostasis via protein degradation (Voges et al., 1999; Rock et al., 1994; Craiu et al., 1997; Crawford & Irvine, 2013). The UPS is tightly regulated to prevent the intracellular environment from accumulation of poly-ubiquitinated proteins, involving abnormal and damaged proteins (Ciechanover, 2005). UPS is accomplished in multi-steps protein degradation: 1) poly-ubiquitination of proteins, 2) recognition through poly-ubiquitin chains by the 19S proteasome, and 3)
- 4 -
degradation within 20S proteasome (Ciechanover, 2005). For the poly-ubiquitination, three different enzymes are involved in the cascade reaction for covalent conjugation of ubiquitin protein to the target proteins (Hershko & Ciechanover 1998): Ub-activating enzymes (E1), Ub-conjugating or -carrier enzymes (E2), and Ub-protein ligases (E3). In next step, poly-ubiquitin chain plays as a signal peptide to target the substrate to the proteasome for degradation. Poly-ubiquitin chain is recognized through several subunits in 19S proteasome prior to translocation into 20S core complex (Finley, 2009; Hao et al., 2013). In the last steps, translocated protein is cleaved by several proteolytic subunits in 20S proteasome. 20S proteasome has barrel-like shape and is comprised of two outer layers containing seven α-subunits (α1- α7 subunits) and two inner rings made up seven β-subunits (β1- β7 subunits) (Lander et al., 2012). Some of them, β1, β2, and β5 subunits have a proteolytic activity: β1 is related with a caspase-like activity, β2 with trypsin-like activity, and β5 with chymotrypsin-like activity (Grigoreva et al., 2015; Groll et al., 1999; Kisselev et al., 2003; Britton et al., 2009). UPS plays a critical role in the fate of proteins that are involved in major cellular processes, including signal transduction, gene expression, cell cycle, replication, differentiation, immune response, cellular response to stress, etc. (Voges et al., 1999; Grigoreva et al., 2015; D'Arcy et al., 2015). Importantly, many diseases, including neurodegenerative diseases and cancers, are intimately connected to the activity of proteasomes making them an important pharmacological target (Grigoreva et al., 2015; D'Arcy et al., 2015).
[Bortezomib]
Proteasome specific inhibitors have been regarded to be positive clinical benefits for cancer therapy. Bortezomib (PS341, Velcade), the first FDA-approved proteasome inhibitors (PIs), is now used in the clinic for the treatment of newly diagnosed and relapsed multiple myeloma and mantle cell lymphoma (Kane et al.,
- 5 -
2006). Bortezomib inhibits mainly β5 and partially β1 of proteasome complex (Crawford et al. 2006, Chen et al., 2011) and results in the disruption of protein homeostasis and induction of ER stress, contributing to its cytotoxicity (Obeng et al., 2006). Since proteasome activity is required for the retrotranslocation of misfolded proteins across the ER membrane into the cytoplasm and subsequent degradation of these proteins, proteasome inhibitors induce accumulation of misfolded protein in the ER lumen, followed by the stress to ER (Kostova Z & Wolf, 2003; Obeng et al., 2006). Under this stress condition, cells utilize the unfolded protein response (UPR) signaling for recovery. Prolonged ER stress induces the terminal UPR associated with ER-stress-mediated cell death, although transient UPR initially increases the machinery for resolving these problems (Hetz et al. 2015; Schröder & Kaufman, 2005). However, recent studies have shown that when bortezomib was used as a single agent in newly diagnosed multiple myeloma patients, approximately 50% did not achieve a partial response or better (Ruschak et al., 2011; Dispenzieri et al., 2010). Moreover, the clinical response to bortezomib in other hematologic malignancies and solid tumors remain unsatisfactory (Cortes J et al., 2004; Kale AJ & Moore BS, 2012). Therefore, there is a growing challenge for overcoming the resistance to bortezomib and for the improvement of the survival of patients.
[Nutlin-3]
Nutlin-3 is a small molecule inhibitor of the MDM2/p53 interaction, which leads to the non-genotoxic p53 stabilization, activation of cell cycle arrest and apoptosis pathways (Secchiero et al., 2011). MDM2 protein is an ubiquitin-E3 ligase to be able to degrade p53 (Chène, 2003). But, recently several research groups have shown that the nutlin-3 has another function in the cells, independently of p53 (Kurokawa et al., 2013; Valentine et al., 2011).
- 6 -
Here, we show that the nutlin-3 effectively overcomes the resistance of various cancer cells with defective p53 to bortezomib, and thus through p53-independent mechanism. Interestingly, combined treatment with subtoxic doses of bortezomib and nutlin-3 induced severe vacuolization, which was derived of the dilation of both the ER and mitochondria, prior to cell death. In this process, accumulation of poly-ubiquitinated proteins and the proteins associated with ER stress, including CHOP, as well as mitochondria Ca2+ influx was observed. We found that CHOP induction plays a critical role in the ER dilation and mitochondrial Ca2+ influx contributes to the cell death induced by the combined treatment with bortezomib and nultlin-3. Combined regimen of bortezomib and nultin-3 may offer an effective therapeutic strategy to overcome the resistance of cancer cells to bortezomib. In this study, we found that nutlin-3 effectively overcomes the resistance of various cancer cells to bortezomib, independently of p53, via induction of paraptosis-like cell death and explored its underlying mechanisms.
- 7 -
II. MATERIALS AND METHODS
A. Chemicals and antibodies
Chemical against 3-methyladenine (3-MA), bafilomycin A1, necrostatin-1, N-acetylcysteine (NAC), reduced glutathione (GSH), cycloheximide (CHX), 1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid acetoxymethyl ester (BAPTA-AM), ruthenium red and crystal violet were (Sigma Chemical Corp); z-VAD-fmk (R&D systems); nutlin-3 (TOCRIS); Rhod-2-AM, MitoTracker-Red (MTR), MitoTracker-Green (MTG), Lysotracker-red, calcein- acetoxymethyl ester (calcein-AM), ethidium homodimer-1 (EthD-1), and 4',6-diamidino-2-phenylindole (DAPI) (Molecular Probes); Ru360, 2-Aminoethoxydiphenyl borate (2-APB), MnTBAP (Mn(III)tetralis benxoic acid porphyrin), SB203580, PD98059, U0126, and SP600125 (Calbiochem); Bortezomib (Selleckchem).
Antibodies against β-actin and Mfn1 (Abcam); caspase-3, caspase-4 (Stressgen); ubiquitin, ATF4, MCU, MDM2, p53, Mfn2, and Mcl-1 (Santa Cruz); PARP (Epitomics Inc.); ERK1/2, ERK1/2, phospho-p38, p38, phosphor-JNK,JNK, CHOP/GADD153, GRP78, Grp75, and AIP/Alix (Cell Signaling); Fis1 (Origene) Noxa, Bim, p62, OPA1, and Drp1 (BD biosciences pharmingen); rabbit IgG HRP, mouse IgG HRP, and goat IgG HRP (Molecular probes)
B. Cell Culture
The MDA-MB 435 human breast cancer cell lines, DLD-1human colon cancer cell lines, HeLa human ovarian cancer cell lines, T98G human glioblastoma cancer cell lines, and HCT116 1human colon cancer cell lines were purchased from American Type Culture Collection. Cells were cultured in DMEM supplemented with 10%
- 8 -
fetal bovine serum (FBS) and antibiotics (GIBCO-BRL) and incubated in 5% CO2
at 37℃.
C. Examination of the stable cell lines expressing the fluorescence specifically in mitochondria or endoplasmic reticulum.
To establish the stable cell lines expressing the fluorescence specifically in mitochondria or endoplasmic reticulum (ER), MDA-MB 435 cells were transfected with the pEYFP-Mito or pEYFP-ER vector (Clontech, Mountain, CA). Stable cell lines expressing pEYFP-Mito or pEYFP-ER (YFP-Mito or YFP-ER) were selected with fresh medium containing 500 µg/mL G418 (Calbiochem). To quantitatively measure the dilation of the ER and mitochondria induced by treatment with bortzomib and/or nutlin-3, we analyzed the average width of the vacuoles originated from mitochondria and the ER in YFP-ER cells and YFP-Mito cells using AxioVision Rel. 4.8 software (Zeiss). More than 200 clearly identifiable vacuoles derived from the ER in 50 YFP-ER cells and more than 200 clearly identifiable vacuoles derived from mitochondria in 50 YFP-Mito cells per experiment, randomly selected, were measured in three independent experiments.
D. Measurement of cellular viability
Cell viability was assessed by double labeling of cells with 2 µM calcein-AM and 4 µM EthD-1. The calcein-positive live cells and EthD-1-positive dead cells were visualized using the fluorescence microscope (Axiovert 200M; Carl Zeiss) equipped with Zeiss filter set #10 (excitation band pass, 450-490 nm; emission band pass, 565 nm) and #20 (excitation band pass, 546 nm; emission band pass, 640 nm) and counted.
- 9 -
E. MTT assay
MTT assay was performed according to the manufacturer’s protocol (Sigma). Absorption at 570 nm was normalized to that of untreated control (100%), and the results were expressed as Viability of control.
F. Western blotting
Cells were washed in PBS and lysed in boiling sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer (62.5 mM Tris [pH 6.8], 1% SDS, 10% glycerol, and 5% b-mercaptoethanol). The lysates were boiled for 5 min, separated by SDS–PAGE, and transferred to an Immobilon membrane (Millipore, Bredford, MA, USA). After blocking nonspecific binding sites for 1 h using 5% skim milk, membranes were incubated for 2 h with specific Abs. Membranes were then washed three times with TBST and incubated further for 1 h with horseradish peroxidase- conjugated anti-rabbit, -mouse or -goat antibody. Visualization of protein bands was accomplished using ECL (Amersham Life Science, Buckinghamshire, UK).
G. Immunocytochemistry
After treatments, cells were fixed with acetone/methanol (1:1) for 5 min at -20℃ and blocking in 5% BSA in PBS for 30 min. Fixed cells were incubated overnight at 4℃ with primary antibody [anti-COX II (1:500, mouse, Invitrogen), anti-PDI (1:500, rabbit, Stressgen), anti-CHOP (1:200, mouse, Cell Signaling Technology), anti-MCU (1:200, goat, Santa Cruz Biochemistry), or anti-COX IV (1:500, rabbit, Gentex)] diluted in PBS and then washed three times in PBS and incubated for 1 h at room temperature with anti-rabbit or anti-mouse Alexa Fluor 488 or 594 (1:500,
- 10 -
Molecular Probes). Slides were mounted with ProLong Gold antifade mounting reagent (Molecular probes) and cell staining was visualized with a fluorescence microscope using Zeiss filter sets #46 and #64HE (excitation band pass, 598/25 nm; emission band pass, 647/70 nm).
H. Measurement of cytosolic and mitochondrial Ca²⁺ levels
To measure cytosolic [Ca2+]c levels, treated cells were incubated with 2.5 µM
Fluo-3-AM at 37℃ for 20 min, washed with HBSS (without Ca2+ or Mg2+), and
analyzed immediately by flow cytometry. To measure mitochondrial [Ca2+]mt levels,
treated cells were incubated with 2.5 µM Rhod-2-AM at 4 ℃ for 30 min, washed with HBSS (without Ca2+ or Mg2+), further incubated with HBSS at 37 ℃ for 20 min, and then analyzed by flow cytometry. To confirm the mitochondrial localization of the Rhod-2 probe, cells were loaded with 2.5 µM Rhod-2-AM in HBSS (without Ca2+ or Mg2+) for 30 min at 4 ℃. The cells were then washed with HBSS, loaded with 100 nM MitoTracker-Green for 10 min in HBSS, and visualized by the fluorescence microscopy using Zeiss filter sets #10 and #20.
I. Small interfering RNAs
The small interfering RNA (siRNA) duplexes used in this study were purchased from Invitrogen and gave the following sequences: Negative Universal ControlTM (Invitrogen) was used as the control. After annealing of the pairs of siRNA oligos, cells were transfected with siRNA oligonucleotides. To confirm successful siRNA-mediated knockdown, we performed western blotting of the proteins of interest.
J. shRNA-mediated knockdown of proteins
Knockdown of the CHOP protein in breast cancer cells was achieved by lentiviral infection of viral vectors that express different shRNA directed to CHOP mRNA and were purchased from Sigma-Aldrich (ShRNA MISSION). Viruses were
- 11 -
generated by transfection of HEK 293T cells with MISSION shRNA vectors and DNRF vectors encoding for gag- pol, and CMV-VSVG encoding for envelop glycoprotein of vesicular stomatitis virus. The medium of transfected 293T cells containing lentiviruses was used to infect myoblasts that were further selected with puromycin (3 mg/ml). Knockdown efficiency was analyzed by western blotting. Viral particles that caused maximal repression of CHOP expression relative to control particles were chosen for the knockdown experiments.
K. Transmission electron microscopy
Cells were prefixed in Karnovsky’s solution (1% paraformaldehyde, 2% glutaraldehyde, 2 mM calcium chloride, 0.1 M cacodylate buffer, pH 7.4) for 2 h and washed with cacodylate buffer. Post-fixing was carried out in 1% osmium tetroxide and 1.5% potassium ferrocyanide for 1 h. After dehydration with 50-100% alcohol, the cells were embedded in Poly/Bed 812 resin (Pelco, Redding, CA), polymerized, and observed under electron microscope (EM 902A, Zeiss).
L. Statistical analysis
All data were presented as mean ± S.D. (standard deviation) from at least three separate experiments. Student’s t test was applied to evaluate the differences between treated and control groups with cell viability. Data from multiple groups were analyzed by one-way ANOVA, followed by Bonferroni multiple comparison test. For all the tests, the level of significance was values of P < 0.05.
M. Isobologram analysis
To determine the effect of combination of bortezomib and nutlin-3 on MDA-MB 435S, DLD-1, HeLa, and T98G cells, dose-dependent effects were determined for each compound and for one compound with fixed concentrations of another. The interaction of bortezomib and nutlin-3 was quantified by determining the
- 12 -
combination index (CI), in accordance with the following classic isobologram (Chou TC & Talalay P., 1984). The equation for the isobologram is shown as CI = (D)1/(Dx)1 + (D)2/(Dx)2, where (Dx)1 and (Dx)2 indicate the individual dose of
bortezomib and nutlin-3 required to produce an effect, and (D)1 and (D)2 are the
doses of bortezomib and nutlin-3, respectively, in combination that produce the same effect. From this analysis, the combined effects of the two drugs can be summarized as follows: CI < 1 indicates synergism; CI = 1 indicates summation (additive and zero interaction); and CI > 1 indicates antagonism.
- 13 -
III. RESULTS
1. Bortezomib and nutlin-3 synergistically induce cell death in various cancer cell lines with defective p53
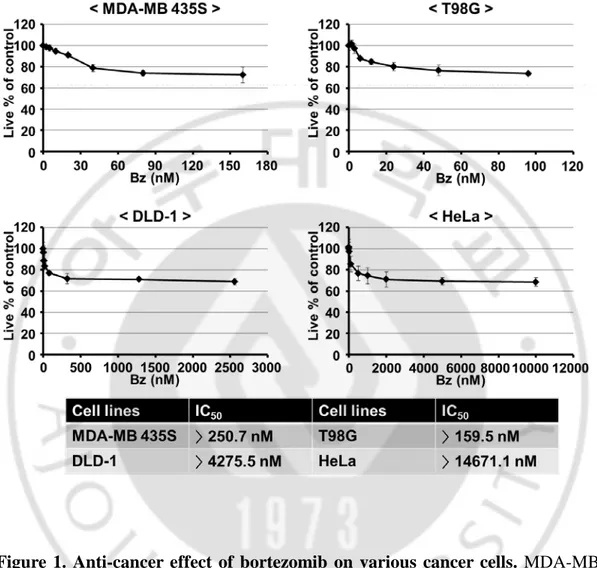
First, we investigated whether nutlin-3 could overcome the resistance of various cancer cell lines with defective p53 to bortezomib. For this purpose, we used MDA-MB 435S (breast cancer cells, G266E mutant p53, Gartel et al., 2003), T98G cells (glioma cells, M237I mutant p53, Villalonga-Planells et al., 2011), DLD-1 (colon cancer cells, S241P mutant p53, Li et al., 2005), and HeLa (cervix cancer cells, non-functional p53 due to E6 expression, Hoppe-Seyler & Butz, 1993)). We treated these cancer cells with various doses of bortezomib and/or nutlin-3 for 24 h and performed cell viability assay using calcein-AM and EthD-1, to detect live and dead cells, respectively. When we examined the effect of bortezomib on these cancer cells, bortezomib treatment did not show dose-dependent cytotoxicity at higher doses exceeding the certain concentrations and more than 70% cells treated with bortezomib of different doses were viable (Figure 1). When we assessed IC50s
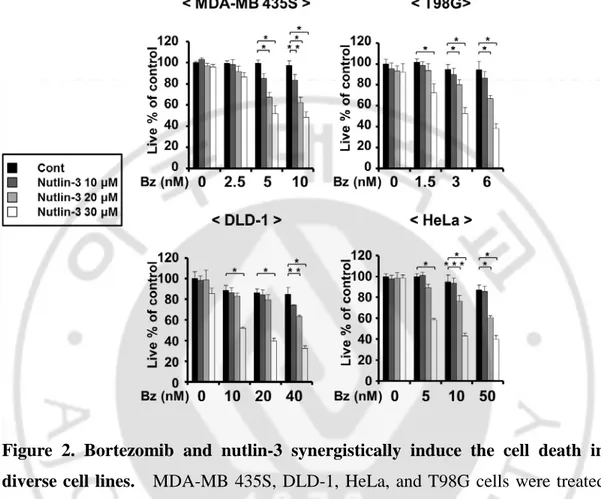
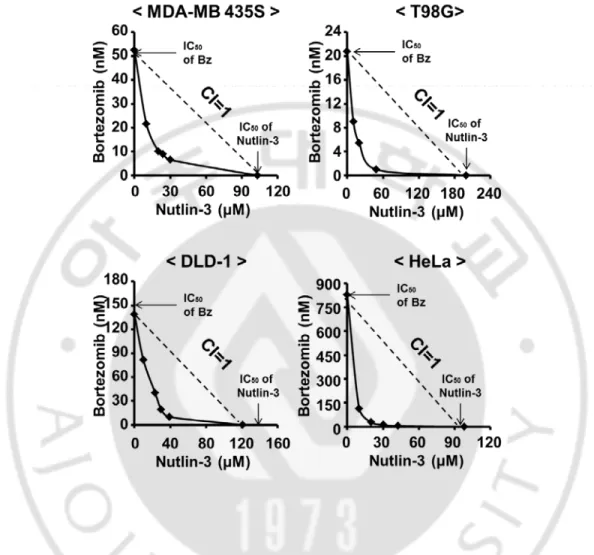
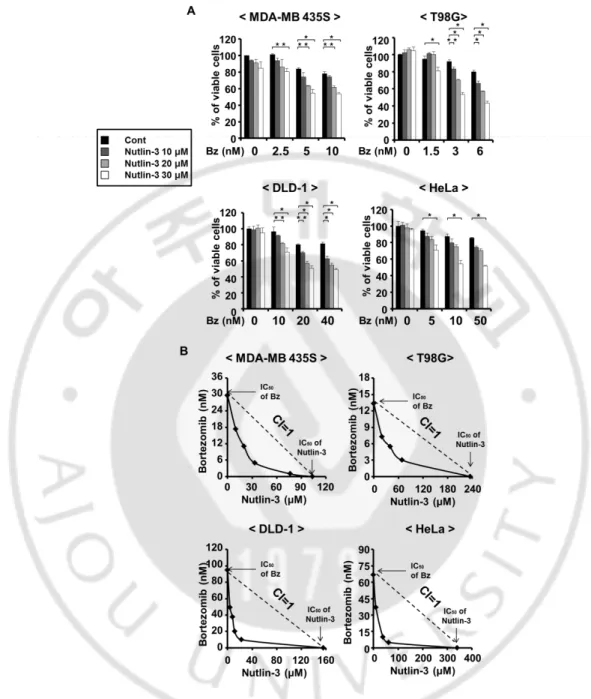
of bortezomib in these cancer cells, they were higher than 14671.1 nM, 4275.5 nM, 250.7 nM, and 159.5 nM in DLD-1, MDA-MB 435S, HeLa, and T98G cells, respectively (Figure 1). While treatment with nultlin-3 up to 30 µM was not cytotoxic to all the tested cancer cells, co-treatment with bortezomib and nultlin-3 significantly enhanced cell death in all the tested cancer cells, compared to the treatment with bortezomib alone (Figure 2), in spite of the differences in the relative sensitivity to bortezomib (Figure 1). Isobologram analysis showed that bortezomib and nutlin-3 synergistically induces cell death in these cancer cells (Figure 3). MTT assay also showed that bortezomib and nutlin-3 synergistically induced cancer cell death (Figure 4).
- 14 -
Since nutlin-3 is known to be an MDM2 inhibitor, that can lead to stabilization of p53 (Jones, 2011), we investigated whether the sensitizing effect of nutlin-3 on bortezomib-mediated cell death is associated with MDM2 and/or p53. MDM2, an ubiquitin E3 ligase, is known to be upregulated by p53-mediated transcriptional activation (Miyachi et al., 2009) or proteasome inhibition (Hideshima et al., 2003). When we first examined the expression of MDM2 and p53 in MDA-MB 435S cells, we found that the protein levels of MDM2 were not affected by nutlin-3, but markedly increased by bortezomib (Figure 5A). p53 proteins were highly expressed in untreated MDA-MB 435S cells and their expression was not altered by treatment with bortezomib and/or nutlin-3, possibly due to the fact that this cancer cell line harbors the mutation in p53 (Gartel et al., 2003). In addition, pifithrin-α, an inhibitor of p53-dependent transcriptional activity (Dagher, 2004) did not block the cell death by bortezomib plus nutlin-3 in MDA-MB 435S cells (Figure 5B). Therefore, these results suggest that upregulation of MDM2 by bortezomib is independent of p53 and the sensitizing effect of nutlin-3 on bortezomib-mediated cell death may not be associated with its activity as an MDM2 inhibitor to indirectly upregulate p53. In addition, when we performed immunocytochemistry of p53 in MDA-MB 435S cells, high expression of p53 in the nuclei was not further accumulated by bortezomib and/or nultlin-3 (Figure 6). To further confirm whether nutlin-3 could sensitize cancer cells to bortezomib-mediated cell death via p53-independent mechanism, we employed two HCT116 isogenic cell lines differing in the p53 status, HCT116 wild-type (WT) cells and HCT116 p53-null (p53-/-) cells. Ten µM bortezomib was toxic neither to HCT116 WT cells nor to HCT116 p53-/- cells (Figure 7). Interestingly, treatment with nutlin-3 alone reduced the cell viability in HCT116 WT cells in a dose-dependent manner and this nutlin-3-induced cytotoxicity was very effectively inhibited by pifithrin-α pretreatment (Figure 7). In addition, co-treatment with bortezomib enhanced nutlin-3-induced cell death and pifithrin-α pretreatment significantly but partially
- 15 -
inhibited the cell death by bortezomib plus nutlin-3. These results suggest that p53 may be critically involved not only in nutlin-3-induced cell death but also in the cell death by the combined treatment with bortezomib and nutlin-3 in HCT116 WT cells. In contrast, treatment with nutlin-3 up to 20 µM was not cytotoxic to HCT116 p53-/- cells, but nutlin-3 treatment dose-dependently recovered the cellular sensitivity to bortezomib (Figure 7). However, pifithrin-α pretreatment did not attenuate the cell death by bortezomib plus nutlin-3 in these cells, suggesting that nutlin-3-stimulated bortezomib-mediated cell death is independent of p53. When we performed western blotting, not only p53 protein levels but also the protein levels of MDM2 and p21, downstream targets of p53, were markedly increased by nutlin-3 (Figure 7). The protein levels of p53, MDM2, and p21 were also increased by bortezomib and they were further increased by the combined treatment with bortezomib and nutlin-3, suggesting the possible involvement of p53 in the cellular response to nutlin-3 and/or bortezomib in HCT116 WT cells. In contrast, the protein levels of MDM2 and p53 were not altered by nutlin-3, but by bortezomib, in HCT116 p53-/- cells and they were rather slightly decreased by bortezomib plus nutlin-3. Taken together, these results suggest that since bortezomib plus bortezomib can induce cell death independently of p53, this combined treatment may provide a way to overcome the resistance of bortezomib in cancer cells harboring defective p53.
To understand the underlying mechanism by which nutlin-3 sensitizes bortezomib-mediated cell death, independently of p53, we first observed the cancer cell morphologies following treatment with bortezomib and/or nultlin-3. We found that treatment with bortezomib or nutlin-3 alone did not noticeably alter the cellular morphologies in MDA-MB 435S, T98G, DLD-1 and HeLa cells, because their subtoxic does were used in this experiment (Figure 8). However interestingly, combination of bortezomib and nutlin-3 at the same concentrations induced extensive cytoplasmic vacuolation prior to cell death in all the tested cancer cells.
- 16 -
To investigate the cell death mode induced by the combination of bortezomib and nutlin-3, we tested the effects of various inhibitors, including z-VAD-fmk to block caspase-mediated apoptosis, necrostatin-1 to block necroptosis, and 3-MA or bafilomycin A to block autophagy. However, neither the cell death nor cytoplasmic vacuolation induced by bortezomib plus nultlin-3 was inhibited by any tested inhibitors in these cancer cells (Figure 9, 10), suggesting that apoptosis, necrosis, or autophagy is not critically involved in the cell death by bortezomib plus nultlin-3. Collectively, these results suggest that nutlin-3 overcomes the resistance of these several cells to bortezomib through induction of a novel cell death mode.
- 17 -
Figure 1. Anti-cancer effect of bortezomib on various cancer cells. MDA-MB
435S, DLD-1, HeLa, and T98G cells were treated with various concentrations of bortezomib for 24 h and then the viability was assessed using calcein-AM and EthD-1 (Live/Dead assay). The values of IC50s of bortezomib were assessed in the
- 18 -
Figure 2. Bortezomib and nutlin-3 synergistically induce the cell death in diverse cell lines. MDA-MB 435S, DLD-1, HeLa, and T98G cells were treated
with the indicated concentrations of bortezomib and/or nutlin-3 for 24 h and then the viability was assessed using calcein-AM and EthD-1(Live/Dead assay). *P < 0.05; **P < 0.01; ***P < 0.005 versus bortezomib alone treated groups.
- 19 -
Figure 3. Synergistic induction of cell death by bortezomib and nutlin-3. The
classic Isobologram at IC₅₀ (the concentration of each drug that is required to reduce the viability of treated cells for 24 h to 50%). Diverse cancer cells were treated with bortezomib and nutlin-3 for 24 h. Isoboles for the combination of bortezomib and nutlin-3 that were isoeffective (IC₅₀) for inhibition of cell viability are shown.
- 20 -
Figure 4. Bortezomib and nutlin-3 synergistically induce cell death in various cancer cell lines. (A) Cells were treated with the indicated concentrations of
bortezomib and/or nutlin-3 for 24 h and viability was measured by MTT assay. *P < 0.05; **P < 0.01; ***P < 0.005 versus bortezomib alone treatment groups. (B) The classic Isobologram at IC₅₀.
- 21 -
Figure 5. Combined treatment with bortezomib and nutlin-3 induces the cell death independent of p53. (A) MDA-MB 435S cells are treated with combination
of bortezomib and/or nutlin-3 at the indicated concentrations for 16 h and then western blotting of MDM2, p53, and β-actin was performed. (B) Cells were untreated or pretreated with indicated concentrations of pifithrin-α and further treated with bortezomib plus nutlin-3 with indicated concentration for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. *P < 0.005 versus non treated groups.
- 22 -
Figure 6. High expression of p53 in the nuclei is not further accumulated by bortezomib and/or nultlin-3. MDA-MB 435S cells treated as indicated were fixed
- 23 -
Figure 7. Cell death induced by bortezomib plus nutlin-3 is not associated with pro-cell death activity of p53. Cells were untreated or pretreated with indicated
concentrations of pifithrin-α and further treated with bortezomib and/or nutlin-3 with indicated concentration for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. *P < 0.005 versus DMSE treated groups; **P < 0.005 versus bortezomib plus nutlin-3 treated groups; ***P < 0.005 versus pifithrin-α alone treated groups. #P < 0.005 versus 20 μM nutlin-3 treated groups. The presence/absence of p53 and its transcription activity were confirmed by western blotting of p53 and p53-related proteins following treatment with indicated concentrations of bortezomib and/or nutlin-3 for 12 h. β-actin was used as a loading control in western blots.
- 24 -
Figure 8. Combined treatment with bortezomib and nutlin-3 induces cytoplasmic vacuolation followed by cell death in all the tested cancer cells.
Cells were treated with indicated concentrations of bortezomib and/or nutlin-3 for 24 h. And the cells were observed under the phase contrast microscopy. Bars, 10 μm
- 25 -
Figure 9. Combination treatment with bortezomib and nutlin-3 induces alternative cell death in all the tested cells. Cells were untreated or pretreated
with indicated concentrations of the respective inhibitors, including z-VAD-fmk, necrostatin-1, 3-MA, and bafilomycin A, and further treated with bortezomib plus nutlin-3 with indicated concentration for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. *P < 0.005 versus non treated groups.
- 26 -
Figure 10. Each inhibitor did not alter the cytoplasmic vacuolation induced by bortezomib plus nutlin-3 in all the tested cells. Cells were untreated or pretreated
with indicated concentrations of each inhibitor, including z-VAD-fmk, necrostatin-1, 3-MA, and bafilomycin A, and further treated with bortezomib plus nutlin-3 with indicated concentration for 24 h. And the cells were observed under the phase contrast microscopy. Bars, 10 μm
- 27 -
2. Vacuolation induced by bortezomib plus nutlin-3 is derived from the dilation of both the ER and mitochondria
Recently, we have shown that curcumin or celastrol induces paraptosis, a cell death mode which is accompanied by the dilation of the ER and mitochondria, and proteasome inhibition plays a critical role in this cell death (Yoon et al., 2010; Yoon et al., 2014). Since combination of bortezomib and nutlin-3 induced non-apoptotic cell death accompanied by extensive vacuolation, we next examined whether the cell death by bortezomib plus nutlin-3 is associated with paraptosis. For this purpose, we employed the MDA-MB 435S sublines transfected with the YFP-ER plasmid and YFP-Mito plasmid for the labeling of the ER and mitochondria (YFP-ER cells and YFP-Mito cells). When we first performed the fluorescence microscopy in YFP-ER cells, the ER structures of reticular shapes were not noticeably affected by treatment with bortezomib or nultlin-3 alone (Figure 11A). In contrast, at 8 h of the combined treatment with bortezomib and nutlin-3, numerous fluorescent ER-derived vacuoles were generated. And with the increased incubation time, the sizes of the vacuoles were increased, whereas their numbers were reduced, suggesting that the fusion among the swollen ER might be progressed (Figure 11B and 11C). We further examined the changes in mitochondrial structures using the YFP-Mito cells. As shown in Figure 12, while mitochondria in non-treated YFP-Mito cells exhibited filamentous and elongated morphology, combined treatment with bortezomib and nutlin-3 induced mitochondrial dilation around the nuclei at 8 h. The intensity of mitochondrial fluorescence was somewhat weakened at 24 h, when cellular spaces were almost occupied with a few giant ER-derived vacuoles. In contrast, treatment with bortezomib alone induced mitochondrial fragmentation, rather than dilation. Interestingly, treatment with nultlin-3 alone induced dilation of mitochondria at 8 h, although the sizes of mitochondria-derived vacuoles were smaller than those
- 28 -
induced by bortezomib plus nutlin-3 at the same time. However, at 24 h of nultin-3 treatment, the sizes of mitochondria-derived vacuoles were noticeably reduced, suggesting that mitochondrial structures might be recovered from dilation, possibly via mitochondrial fission. These results suggest that combination of bortezomib and nultlin-3 induces paraptosis-like morphologies and nultlin-3 may contribute to mitochondrial dilation during this process. Dilation of the ER and mitochondria was further confirmed by the fluorescence microscopy after staining of YFP-ER cells treated with bortezomib and/or nutlin-3 for 8 h with MitoTracker Red (MTR) (Figure 13). To confirm whether dilation of the ER and mitochondria by the combination of bortezomib and nutlin-3 is not restricted to MDA-MB 435S cells, we performed immunocytochemistry using the specific antibodies against protein disulfide isomerase (PDI), an ER-resident protein, and cytochrome oxidase subunit II (COXII), a protein localized in the inner mitochondrial membrane in four different cancer cells, including MDA-M 435S, T98G, DLD-1, and HeLa cells. We found the expression of COXII with the small ring shape in the perinuclear area and PDI expression with larger ring shapes in all the tested cancer cells treated with bortezomib and nutlin-3 for 16 h (Figure 14). These results suggest that combination of bortezomib and nutlin-3 commonly induces paraptosis-like morphologies via dilation of the ER and mitochondria in many cancer cells with defective p53.
- 29 -
Figure 11. Vacuolation induced by bortezomib plus nutlin-3 is mainly derived from dilation of the ER. (A) MDA-MB 435S sublines (YFP-ER/435S) expressing
the fluorescence selectively in the ER were treated with bortezomib and/or nutlin-3 at the indicated concentrations and for the indicated time points and observed under the fluorescent and phase contrast microscope. (B) The average widths of the vacuoles originated from the ER were measured in YFP-ER cells treated with combination of bortezomib and/or nutlin-3 using AxioVision Rel. 4.8 software (Zeiss). (C) The average numbers of the vacuoles per cell were assessed in YFP-ER cells treated with bortezomib and/or nutlin-3. Bars, 20 μm.
- 30 -
Figure 12. Combined treatment with bortezomib and nutlin-3 also induces the formation of mitochondria-derived vacuoles. MDA-MB 435S sublines
(YFP-Mito/435S) expressing the fluorescence selectively in mitochondria were treated with combination of bortezomib and/or nutlin-3 for the indicated concentrations and time points and observed under the fluorescent and phase contrast microscope. And morphologic difference of mitochondrial was quantified. Bars, 20 μm
- 31 -
Figure 13. Cytoplasmic vacuolation is derived from dilation of both the ER and mitochondria. YFP-ER cells treated with 5 nM bortezomib and/or 30 μM
nutlin-3 for 8 h were stained with 100 nM MitoTracker-red (MTR) and observed under the phase contrast and fluorescence microscopy. Bars, 20 μm
- 32 -
Figure 14. Dilation of the ER and mitochondria is induced by bortezomib plus nutlin-3 in various cancer cells with defective p53. All the tested cells were
treated with the indicated concentrations of bortezomib and/or nutlin-3 for 16 h. Immunocytochemistry using anti-PDI and anti-COX II was performed and the representative images of cells are shown. Bars, 20 μm
- 33 -
3. Protein synthesis is required for vacuolation and subsequent cell death by bortezomib plus nutlin-3
Since paraptosis is known to require protein synthesis (Sperandio et al., 2010), we next examined whether the cell death induced by bortezomib plus nutlin-3 is blocked by the protein synthesis inhibitor, cyclohexamide (CHX). We found that CHX pretreatment very effectively and commonly blocked the cell death induced by bortezomib plus nutlin-3 in MDA-MB 435S, T98G, DLD-1, and HeLa cells. In addition, CHX pretreatment almost completely blocked the dilation of both the ER and mitochondria in MDA-MB 435S cells treated with bortezomib and nutlin-3 (Figure 15, 16). These results suggest that the combined treatment with bortezomib and nutlin-3 may induce paraptosis-associated cell death.
- 34 -
Figure 15. CHX pretreatment effectively blocks the cell death by bortezomib plus nutlin-3. Cells were pre-treated with or without CHX for the indicated
concentrations and further treated with bortezomib plus nutlin-3 of indicated concentrations for 24 h. Cell viability was assessed using the Live/Dead assay. *P < 0.05 versus non-treat control group; #P < 0.005 versus bortezomib plus nutlin-3 group.
- 35 -
Figure 16. CHX pretreatment effectively blocks the dilation of both the ER and mitochondria induced by bortezomib plus nutlin-3. YFP-ER cells were
pre-treated with or without 2 μM CHX and further pre-treated with 5 nM bortezomib and 30 μM nutlin-3 for 8 h. Treated cells were stained with 100 nM MitoTracker-red and observed under the phase contrast and fluorescence microscopy. Bars, 20 μm
- 36 -
4. CHOP induction critically contributes to the ER dilation and the cell death by bortezomib plus nultin-3
In an attempt to understand the underlying mechanisms by which combined treatment with bortezomib and nutlin-3 induces paraptosis-associated cell death, we first examined whether combined treatment induces ER stress in cancer cells, as reflected by extensive ER-derived dilation. We found that the protein levels of GRP78 and GRP94 were not noticeably affected by bortezomib and/or nutlin-3 in all the tested cancer cells (Figure 17). In contrast, while treatment with bortezomib or nutlin-3 alone slightly increased the protein levels of ATF4 and CHOP, combined treatment markedly much further increased their protein levels, suggesting that enhanced ER stress may be associated with this cell death by bortezomib plus nultlin-3 (Figure 17). We next examined whether bortezomib-induced proteasome inhibition is affected by co-treatment with nutlin-3. We found that while polyubiquitinated proteins were progressively accumulated by bortezomib treatment alone, combined treatment markedly increased their accumulation, suggesting that bortezomib-mediated impairment may be aggravated by co-treatment with nultlin-3, contributing to the enhanced ER stress. Since we previously showed that CHOP plays a critical role in ER-originated vacuolation and consequently paraptotic cell death induced by curcumin and dimethoxycurcumin (Yoon et al., 2014a), we tested the effect of CHOP knockdown on vacuolation and cell death by bortezomib plus nutlin-3. When we treated MDA-MB 435S cells with the lentivirus containing non-targeting shRNA (shNT) or CHOP-targeting shRNA (shCHOP) and further treated with bortezomib plus nutlin-3, the cell death by bortezomib plus nutlin-3 was markedly attenuated by CHOP knockdown (Figure 18A). Vacuolation induced by the combined treatment was markedly but not completely inhibited by CHOP knockdown (Figure 18B). When we further examined the effect of CHOP knockdown on the dilation of the ER and mitochondria by the immunocytochemistry of PDI and COXII, we found
- 37 -
that CHOP knockdown remarkably inhibited the dilation of the ER but not mitochondrial dilation (Figure 19). Taken together, these results show that increased expression of CHOP may critically contribute to ER dilation and subsequent cell death induced by the combined treatment with bortezomib and nutlin-3 in cancer cells.
- 38 -
Figure 17. Nutlin-3 enhances bortezomib-induced accumulation of ubiquitin conjugated proteins and ER stress. Cells were treated with bortezomib and/or
nutlin-3 at the indicated concentrations and for the indicated time points, and then western blotting of the proteins associated with ER stress was performed. β-actin was used as a loading control in western blots.
- 39 -
Figure 18. Knockdown of CHOP attenuates cytoplasmic vacuolation and subsequent cell death induced by bortezomib plus nutlin-3. MDA-MB 435S
cells were infected with the lentivirus containing non-targeting (NT) shRNA or a CHOP-targeting shRNA (CHOP shRNA) for 24 h. Infected MDA-MB 435S cells were treated with 5 nM bortezomib plus 30 μM nutlin-3 for 24 h. Cell viability was assessed using the Live/Dead assay and observed under a phase contrast microscope. *P < 0.005 versus treat control group; #P < 0.005 versus non-target shRNA group treated with bortezomib plus nutlin-3. The knockdown of CHOP was confirmed by its western blotting. β-actin was used as a loading control in western blots. Bars, 10 μm
- 40 -
Figure 19. Knockdown of CHOP inhibits ER-derived vacuolation, but not mitochondria-derived vacuolation. MDA-MB 435S cells were infected with the
lentivirus containing non-targeting (NT) shRNA or a CHOP-targeting shRNA (CHOP shRNA) for 24 h. Infected MDA-MB 435S cells were treated with 5 nM bortezomib plus 30 μM nutlin-3 for 16 h, fixed, and subjected for immunocytochemistry of COX II and PDI. Bars, 20 μm
- 41 -
5. Mitochondrial Ca2+ uniporter-mediated mitochondrial Ca²⁺ overload is important for the cell death induced by bortezomib plus nutlin-3
Previously, we have shown that mitochondria calcium overload plays a crucial role in paraptosis induced by curcumin or celastrol (Yoon et al., 2012; Yoon et al., 2014a). Therefore, we next tested whether combined treatment also increases mitochondrial Ca2+ levels, if so it is important for paraptosis-associated cell death induced by bortezomib plus nutlin-3. We measured the changes in the mitochondrial Ca2+ levels by flow cytometry using Rhod-2 (a cell permeable mitochondria calcium-indicator dye). We found that treatment of MDA-MB 435S cells with bortezomib plus nutlin-3 effectively increased the mitochondria calcium levels, which peaked at 16 h post treatment (Figure 20). The increase in mitochondrial Ca2+ levels by bortezomib plus nultin-3 was further confirmed by fluorescence microscopy by staining with Rhod-2 and MitoTracker Green (MTG), showing that enhanced Rhod-2 fluorescence was co-localized with mitochondria. Calcium reportedly enters mitochondria though the mitochondrial Ca2+ uniporter (MCU) (De Stefani & Rizzuto, 2014) and MCU-mediated mitochondrial Ca2+ uptake is known to be inhibited by Ru360 (García-Rivas Gde et al., 2006). We found that enhanced Rhod-2 fluorescence in mitochondria of MDA-MB 435S cells treated with bortezomib plus nutlin-3 was diminished by Ru360 pretreatment (Figure 21). When we investigated the effect of Ru360 on the cell death by bortezomib plus nutlin-3, Ru360 pretreatment dose-dependently and significantly inhibited the cell death by the combined treatment (Figure 22A). These results suggest that mitochondrial Ca2+ overload may critically contribute to the cell death
by bortezomib plus nutlin-3. However, interestingly, phase contrast microscopy showed that cellular vacuolation induced by bortezomib plus nutlin-3 was not affected by Ru360 pretreatment, whereas cellular detachment by death was markedly reduced by it (Figure 22B). In order to further examine the effect of
- 42 -
Ru360 on the dilation of the mitochondria and the ER induced by bortezomib and nutlin-3, we treated YFP-ER cells with bortezomib plus nutlin-3 for 8 h in the presence and absence of RU360, and further stained with MTR. We found that dilation of not only mitochondria but also the ER induced by the combined treatment was not greatly affected by RU360 pretreatment (Figure 23). Next, we investigated whether MCU knockdown demonstrates a similar effect of RU360 on the cell death and vacuolation induced by bortezomib and nutlin-3. Similar to the effect of Ru360 pretreatment, MCU knockdown slightly but significantly attenuated the cell death induced by bortezomib plus nutlin-3 (Figure 24). In addition, phase contrast microscopy showed that MCU knockdown did not inhibit vacuolation induced by bortezomib plus nutlin-3, but alleviated the cell death (Figure 25). Furthermore, fluorescence microscopy using the specific antibodies against COXII and PDI showed that dilation of both mitochondria and the ER induced by bortezomib plus nutlin-3 was not affected by MCU knockdown (Figure 26). Collectively, these results suggest that MCU-mediated mitochondrial Ca²⁺ overload may critically contribute to cell death induced by the combination of bortezomib and nutlin-3 in breast cancer cells, although it may not be a direct cause of vacuolation.
- 43 -
Figure 20. Combination treatment with bortezomib and nutlin-3 increases the mitochondrial calcium uptake. MDA-MB 435S cells treated with combination of
5 nM bortezomib and 30 μM nutlin-3 for the indicated time points were stained with 2.5 μM Rhod-2, and processed for FACS analysis. Rhod-2 fluorescence intensities (FI) in cells treated with 5 nM bortezomib plus 30 μM nutlin-3 were compared with that of untreated cells and denoted in the graph.
- 44 -
Figure 21. Ru360 pre-treatment effectively attenuates the mitochondrial calcium uptake induced by bortezomib plus nutlin-3. MDA-MB 435S cells
pre-treated with or without 20 μM Ru360 and further pre-treated with combination of 5 nM bortezomib and 30 μM nutlin-3 for 8 h were co-stained with 2.5 μM Rhod-2 and 100 nM MitoTracker-red and observed under the phase contrast and fluorescence microscopy. Bars, 10 μm
- 45 -
Figure 22. Ru360 pretreatment effectively blocks the cell death by bortezomib plus nutlin-3. (A) MDA-MB 435S cells were pre-treated with or without Ru360 at
the indicated concentrations and further treated with 5 nM bortezomib plus 30 μM nutlin-3 indicated concentrations for 24 h. Cell viability was assessed using the Live/Dead assay. *P < 0.05 versus non-treat control group; #P < 0.01, ##P < 0.005 versus bortezomib plus nutlin-3 group. (B) And the cells were observed under the phase contrast microscopy. Bars, 10 μm
- 46 -
Figure 23. Mitochondria calcium uptake dose not contribute to the dilation of mitochondria and the ER. YFP-ER cells pre-treated with or without 20 μM
Ru360 and further treated with 5 nM bortezomib plus 30 μM nutlin-3 for 8 h. Treated cells were stained with 100 nM MitoTracker-red and observed under the phase contrast and fluorescence microscopy. Bars, 10 μm
- 47 -
Figure 24. MCU-mediated mitochondrial Ca²⁺ overload is critical for the cell death induced by bortezomib plus nutlin-3. MDA-MB 435S cells were
transfected with the fluorescent oligonucleotide (F.O) or a MCU-targeting siRNA (siMCU) for 24 h. Transfected MDA-MB 435S cells were treated with 5 nM bortezomib plus 30 μM nutlin-3 for 24 h. Cell viability was assessed using the Live/Dead assay. *P < 0.005 versus treat control group; #P < 0.005 versus non-target F.O group treated bortezomib plus nutlin-3. Their knockdown was confirmed by western blotting of MCU. β-actin was used as a loading control in western blots.
- 48 -
Figure 25. MCU-mediated mitochondrial Ca²⁺ overload is critical for the cell death by bortezomib plus nutlin-3, but not for cytoplasmic vacuolation.
MDA-MB 435S cells were transfected with the fluorescent oligomer (F.O) or a MCU-targeting siRNA (siMCU) for 24 h. transfected MDA-MB 435S cells were treated with 5 nM bortezomib plus 30 μM nutlin-3 for 24 h. Treated cells were observed under a phase contrast microscope. Bars, 10 μm
- 49 -
Figure 26. Knockdown of MCU inhibits the cell death induced by bortezomib plus nutlin-3, but not dilation of the ER and mitochondria. MDA-MB 435S
cells were transfected with the fluorescent oligomer (F.O) or a MCU-targeting siRNA (siMCU) for 24 h. Transfected MDA-MB 435S cells were treated with 5 nM bortezomib plus 30 μM nutlin-3 for 16 h, fixed, and subjected for immunocytochemistry of COX II and PDI. Bars, 20 μm
- 50 -
IV. DISCUSSION
[Summary]
Although bortezomib has demonstrated a promising anti-cancer effect on multiple myeloma and mantle cell lymphoma (Kane et al., 2006), bortezomib-treated patients tend to acquire the resistance to bortezomib, resulting in the recurrence of cancer and failure in further cancer therapy. In addition, the therapeutic effect of bortezomib in solid tumors remains has not been satisfactory (Cortes et al., 2004; Kale & Moore, 2012; Chen et al., 2011). Therefore, the strategies to sensitize resistant cancer cells to the cytotoxic effect of bortezomib should be developed. Previously, nutlin-3, a specific inhibitor of MDM-2, has been shown to sensitize several types of cancer cells, including multiple myeloma and lung cancer cells, to bortezomib mainly via p53-dependent mechanism (Saha et al., 2010; Ooi et al., 2009; Tabe et al., 2009). In the present study, we show for the first time that nutlin-3 effectively overcomes the resistance of various tumor cells with defective p53 via induction of paraptosis-like cell death.
In the present study, combined treatment with bortezomib and nutlin-3 induced the formation of megamitochondria and expanded ER dilation, although the targets of nutlin-3 for the sensitization of cytotoxicity of bortezomib are not clear. Interestingly, treatment with nutlin-3 alone reversibly induced mitochondrial dilation with a peak at 8 h in terms of the diameter of dilated mitochondria (Figure 12). These results suggest that treatment with nutlin-3 alone may induce mitochondrial fusion, which may be recovered via mitochondrial fission. But when nutlin-3 was combined with bortezomib, both the ER and mitochondria progressively and irreversibly were dilated, leading to paraptosis-associated cell death (Figure 12). Bortezomib-induced ER stress and accumulation of poly-ubiquitinated proteins were further aggravated by co-treatment with nutlin-3. In addition, combined treatment induced mitochondrial Ca2+ overload, contributing to
- 51 -
cell death. Taken together, combined treatment with bortezomib and nutlin-3 may induce paraptosis-like cell death via disruption of cellular proteostasis and Ca2+ homeostasis.
In our study, cellular morphologies following treatment with bortezomib plus nutlin-3 are similar to paraptosis, the cell death mode accompanied by cytoplasmic vacuolation driven by the dilation of the ER and/or mitochondria (Sperandio et al., 2000). Although the underlying mechanisms of paraptosis are not still clearly understood, paraptosis is known to require protein synthesis (Sperandio et al., 2000; Sperandio et al., 2004). In the present study, both the cellular vacuolation and subsequent cell death were very effectively blocked by CHX pretreatment. In addition, Yoon et al. (2014a) showed that CHOP protein is important for curcumin- or dimethoxycurcumin-induced paraptosis through the blockage of ER dilation. Similar to these results, we found that CHOP knockdown effectively attenuated the cell death induced by bortezomib plus nutlin-3, inhibiting the dilation of the ER, but not dilation of mitochondria. Mitochondrial calcium overload has been shown to be important for paraptosis, in particular for mitochondrial dilation (Yoon et al., 2010; Yoon et al., 2012; Yoon et al., 2014a; Zhang et al., 2009). In the present study, mitochondrial Ca2+ overload also critically contributes to the cell death induced by bortezomib plus nutlin-3. Inhibition of mitochondrial Ca2+ uptake employing RU360 or MCU knockdown significantly attenuated the cell death by bortezomib plus nutlin-3. However, this inhibition of mitochondrial Ca2+ uptake did not affect the dilation of the ER and mitochondria. These results suggest that mitochondrial Ca2+ overload may not be the direct cause of mitochondrial dilation, but it can contribute to shift the cellular balance toward cell death. Previously, MAPKs have been implicated as the signals mediating paraptosis (Yoon et al., 2010; Yoon et al., 2012; Yoon et al., 2014a; Yoon et al., 2014b; Kar et al., 2009; Zhang et al., 2009; Korsnes et al., 2011). However, since the specific inhibitors of the respective MAP kinases had no effect on the cell death by bortezomib plus nutlin-3, MAP kinases
- 52 -
may not be critically involved in this cell death (Figure 27). In addition, although ROS are reported to act as key signals for the paraptosis induced by curcumin or dimethoxycurcumin, in the present study, any anti-oxidant agents including NAC, GSH, MnTBAP did not affect the dilation of the ER or mitochondria as well as the cell death induced by bortezomib plus nutlin-3. Collectively, these results indicate that paraptosis-associated cell death induced by bortezomib plus nutlin-3 share some signals associated with paraptosis, but not all. Mitochondria is known to communicate with the ER to execute cell death (Marchi et al., 2014), although its underlying mechanisms are not clearly understood. Therefore, we cannot exclude the possibility that nutlin-3-mediated mitochondrial dilation may somehow negatively affects the adaptive response induced by bortezomib, such as the ER-associated with degradation (ERAD) and ER-ER-associated autophagy (ERAA), and thus further aggravating the accumulation of misfolded proteins within the ER (ER stress) and subsequent dilation of the ER.
[Mitochondria and cell death]
Mitochondria are key organelles which are associated with cellular metabolism, energy conversion, and Ca2+ homeostasis (Dimmer & Scorrano, 2006). At the same manner, mitochondria localize where high ATP amounts are required, or where Ca2+ signaling needs to be regulated, as if they are movable tuners of signaling events (Scorrano, 2013; Hoth et al., 1997). They also integrate and amplify various cell deaths, accompanied by the morphological alteration of mitochondria (Cogliati et al., 2013). Changes in mitochondrial shapes influence crucial cellular function, from Ca2+ signaling to ROS generation (Scorrano, 2013). The loss of Ca2+ homeostasis within intracellular compartments can lead to cell death (Zhivotovsky & Orrenius, 2011), and excessive ROS also can damage cellular components such as DNA, protein, and lipid, resulting in oxidative stress and consequent cell death
- 53 -
(Zhang et al., 2015). Therefore, it is reasonable scenario that confusion of Ca2+ signaling or ROS can alter the mitochondria morphology and, reversely, that change of mitochondria morphology by intrinsic or extrinsic factors affects to disruption of Ca2+ homeostasis or generation of ROS followed by cell death (Scorrano, 2013). Therefore, we tested the effects of various antioxidant agents and Ca2+ antagonists on the dilation of mitochondria and the ER and subsequent cell death. Among many tested inhibitors, Ru360, an inhibitor of mitochondrial Ca2+ uptake, effectively attenuated the cell death by bortezomib plus nutlin-3 (Figure 28), but many antioxidants did not (Figure 29). In addition, both Ru360 and NAC had no effect on the dilation of the ER or mitochondria induced by bortezomib plus nutlin-3 (Figure 23, 30). Interestingly, CHX pre-treatment blocked all the changes by bortezomib plus nutlin-3, including the dilation of the ER and mitochondria, ER stress, and mitochondrial calcium uptake (Figure 31). These results suggest that blocking of protein synthesis by CHX may potently inhibit paraptosis-like cell death by reducing the burden on the homeostatic protein-folding mechanisms and inhibiting the perturbation of Ca2+ homeostasis.
[ER stress and calcium]
In our results, mitochondrial calcium influx may be directly or indirectly associated with the ER dilation. The ER is a major organelle that is responsible for Ca2+ storage and signaling as well as the folding, modification, and sorting of newly synthesized proteins in mammalian cells. Disruption of any of these functions can induce the ER stress, and prolonged ER stress can result in the disruption in Ca2+ homeostasis as well as protein homeostasis (Zhang et al., 2015). In those cases, Ca2+ is released from the ER to cytosol or to other cytosolic organelles, and excessive calcium may affect almost all the organelles, contributing to cell death. Especially, mitochondria are first barrier to reduce the increased
- 54 -
cytosolic calcium levels, called “calcium buffering” (Scorrano, 2013). Upon the transient ER stress, calcium homeostasis is properly regulated by mitochondria, but prolonged ER stress excessively disrupts calcium homeostasis, and overwhelming the capacity of mitochondria to calcium buffering which consequently induce the cell death. Indeed, interaction between mitochondria and the ER was found to be essential for the transfer of Ca2+ between the two organelles (Rizzuto et al., 1998; Rizzuto et al., 1992), and the determination of the cell death by various stimuli (Alirol et al., 2006). Several reports indicate that the changes in mitochondrial shape and ultrastructure are very important marker determining the cell fates under various stimuli (Scorrano, 2013). In the present study, the simultaneous dilation of the ER and mitochondria may disrupt the homeostatic mechanisms of both organelles for the cell survival.
[Mitochondrial swelling and mitochondrial UPR]
In this study, we further attempted to investigate the underlying mechanism involved in nutlin-3-mediated the dilation of mitochondria. Mitochondria shape is known to be affected by diverse stimuli, including the activity changes of the proteins related to mitochondrial dynamics, Ca2+ homeostasis via mitochondria-ER tethering, and ROS (Teranishi et al., 1999; von Ahsen et al., 2000; Seo et al., 2010; Gdynia et al., 2010; Ferreirinha et al., 2004; Arbustini et al., 1998). When we tested the knockdown effect of various proteins associated with mitochondrial dynamics, including Drp1, Fis1, OPA1, and MFN1, no one demonstrated the blocking effect on cytoplasmic vacuolation and cell death induced by bortezomib plus nutlin-3 (Figure 32). As we mentioned above, various antioxidants or mitochondrial calcium uptake inhibitors did not affect cytoplasmic vacuolation or mitochondrial swelling by bortezomib plus nutlin-3. Therefore, we attempted to investigate another pathway that may affect mitochondrial dilation.