https://doi.org/10.13160/ricns.2020.13.4.170
3D-QSAR, Docking and Molecular Dynamics Simulation Study of
C-Glycosylflavones as GSK-3β Inhibitors
Suparna Ghosh1, Seketoulie Keretsu1, and Seung Joo Cho1,2† Abstract
Abnormal regulation, hyperphosphorylation, and aggregation of the tau protein are the hallmark of several types of dementia, including Alzheimer's Disease. Increased activity of Glycogen Synthase Kinase-3β (GSK-3β) in the Central Nervous System (CNS), increased the tau hyperphosphorylation and caused the neurofibrillary tangles (NFTs) formation in the brain cells. Over the last two decades, numerous adenosine triphosphate (ATP) competitive inhibitors have been discovered that show inhibitory activity against GSK-3β. But these compounds exhibited off-target effects which motivated researchers to find new GSK-3β inhibitors. In the present study, we have collected the dataset of 31 C-Glycosylflavones derivatives that showed inhibitory activity against GSK-3β. Among the dataset, the most active compound was docked with the GSK-3β and molecular dynamics (MD) simulation was performed for 50 ns. Based on the 50 ns MD pose of the most active compound, the other dataset compounds were sketched, minimized, and aligned. The 3D-QSAR based Comparative Molecular Field Analysis (CoMFA) model was developed, which showed a reasonable value of q2=0.664
and r2=0.920. The contour maps generated based on the CoMFA model elaborated on the favorable substitutions at the
R2 position. This study could assist in the future development of new GSK-3β inhibitors.
Keywords: Alzheimer's Disease, Kinase, Molecular Docking, Molecular Dynamics, MM/PBSA, CoMFA
Introduction
Alzheimer’s Disease (AD) is a known neurodegen-erative disorder and common to the aging population. AD patients developed tau lesions in the somas and dendrites due to the aberrant phosphorylation and mis-localization of the tau protein, which is crucial for main-taining the structural integrity of microtubules in the neural cells[1-3]. The formation of β-amyloid (Aβ)
pep-tide with tauopathies was considered as the initiator of AD pathogenesis[2]. Recent studies elucidated that Aβ
deposition triggered the positive regulation of the Gly-cogen synthase kinase-3 (GSK-3 kinase) by preventing its inhibitory phosphorylation[4-6]. The GSK-3 is an
atypical serine/threonine kinase enzyme that has recently been characterized as a regulator of tau
hyper-phosphorylation in AD. There are two different iso-forms of GSK-3 in mammals, namely GSK-3α and GSK-3β, where are highly conserved in their catalytic domain and largely redundant in multiple cell signaling pathways. GSK-3β differs from the GSK-3α at the C-terminus domain and is inhibited upon phosphorylation at residue Ser9 instead of Ser21 in GSK-3α[7,8]. Between
them, GSK-3β was identified as the major dominating kinase for tau hyperphosphorylation in AD patients and transgenic animal models [9,10]. Tau protein
hyperphos-phorylation by hyperactive GSK-3β was found in more than 70% of the AD brain[11,12]. Hence, it is necessary
to design and develop inhibitors against GSK-3β. Sev-eral adenosine triphosphate (ATP) competitive inhibi-tors that target GSK-3β have been developed. However, the off-target selection of these inhibitors remained a significant concern.
In recent studies, natural compounds and their semi-synthetic derivatives have been taken into account for developing selective and irreversible GSK-3β inhibi-tor[13,14]. Liang et al. showed the competitive inhibition
of the 31 natural and semi-synthetic C-Glycosylfavone derivatives against GSK-3β. These compounds exhib-1Department of Biomedical Sciences, College of Medicine, Chosun
University, Gwangju 501-759, Korea
2Department of Cellular Molecular Medicine, College of Medicine,
Chosun University, Gwangju 501-759, Korea
†Corresponding author : [email protected]
(Received : October 2, 2020, Revised : November 11, 2020, Accepted : November 28, 2020)
ited a wide range of activities (i.e., IC50 0.59 ± 0.04 µM
to 5153 ± 31 µM) and were taken for our current study[15]. The compound C30 which showed the highest
activity was docked with the target GSK-3β to elucidate the key molecular interactions within the substrate-bind-ing site. After that, the MD simulation was performed to study the dynamic interactions between C30 and GSK-3β. The CoMFA models were developed to explore the structure-activity relations of the GSK-3β inhibitors.
Methodology
Molecular Docking
A series of 31 GSK-3β inhibitors based on C-Glyco-sylflavones have been chosen for this study.15 The
reported inhibitory concentration (IC50) values were
converted into -logIC50 (pIC50) values. The most active
compound (C30, pIC50= 6.22) was sketched and
mini-mized using tripos forcefield in SYBYL-X 2.1. The crystal structure of GSK-3β (PDB ID: 1PYX)[16] was
collected from the protein databank (www.rcsb.org) and eliminated all the water molecules, ions, and small mol-ecules. The missing residues and loops were modeled using MODELLER[17] and treat as a receptor for the
docking study. Docking between GSK-3β and C30 was performed using AutodockTools[18] described in an
ear-lier study[19]. The grid-box dimension was set to
60×65×65 around the active site of the receptor, whereby the Lamarckian genetic algorithm[20] was
applied as a search method for the ligand-binding inside the pocket. The 100 runs of docking evaluation were carried out, and the binding conformations of the ligand were clustered based on the similarity of docked posi-tion. The cluster-1 conformation of the protein-ligand complex was selected for the molecular dynamics study and MM/PBSA calculation.
Molecular Dynamics (MD) Simulation and Data Analysis
The GROMACS-2020[21] package was used for
molecular dynamics simulation of protein-ligand com-plex applying the CHARMM36[22] forcefield. The
CHARMM general forcefield (CGenFF)[23] was used to
generate the ligand topology file. The C30-GSK-3β complex was then immersed into a cubic box with TIP3P water model. The box padding dimension was
kept to 10 Å from the complex and further neutralized by the addition of 13 Cl- ions. The overall system was
energy minimized by 100 ps steepest descent method, followed by the 100 ps at a constant substance, volume, and temperature (NVT) equilibration and 100 ps at a constant substance, pressure, and temperature (NPT) equilibration by position restraining of the protein. Finally, an unrestrained 50 ns MD run was continued at 300K of temperature and 1 bar of pressure. The MD trajectory was analyzed using the in-built Gromacs packages. Finally, protein-ligand binding energy was calculated by molecular mechanics energies combined with the Poison-Boltzmann and surface area continuum solvation (MM/PBSA) method using the g_mmpbsa package[24] described here in an earlier study[25].
CoMFA
The conformation of C30 from the 50 ns MD trajec-tory was collected to sketch the other compounds of the dataset. All the compounds were charged (Gasteiger charge) and optimized by Tripos forcefield[26] in
SYBYL-X 2.1, described previously[27-29]. The compounds were
aligned based on the common substructure and proceed further for the CoMFA study. The electrostatic and ste-ric potential energy terms were calculated for each model in CoMFA by probing the 3D-grid space around the sp3 hybridized carbon atom (charge +1). Then the
Leave-One-Out (LOO) method was used to cross-vali-date the predictive ability of the 3D-QSAR model. The partial least square analysis (PLS) was calculated between the dependent and independent variables to establish the structure-activity relationship[30,31]. Based
on the optimum number of components (ONC) the non-cross validated analysis was done to generate the final CoMFA model with reasonable q2 and r2 values.
Results and Discussions
Molecular Docking Analysis
Molecular docking result of the C30-GSK-3β com-plex was analyzed by AutoDockTools. Protein-ligand complex from the cluster-1 scored -9.84 Kcal/mol with five hydrogen bond formation and was selected to examine the molecular interaction. In the active site, Ile62, Asn64, Ser66, Val70, Lys85, Asp181, Lys183, Leu188, Cys199, and Asp200 were critical for ligand interactions. The residues Asn64, Ser66, Asp181,
Lys183, and Asp200, formed H-bond interaction with three hydroxyl groups of the tetrahydropyran ring of C30. Besides, residues Ile62, Val70, and Ala83, Cys199 formed π-alkyl and π-σ interaction with the catechol B-ring center of the C-Glycosylflavone. Lys85 formed π-cation interaction with the C-Glycosylflavone core. The details interactions have been summarized in Figure 1. The ligand alignment and interaction with residues were similar to the interactions observed in the crystal struc-tures 1PYX and 6HK3[32]. However, no interactions
were found between the trifluoropropane group of C30 and the surrounding residues.
MD Simulation and MM/PBSA Analysis
MD simulation of the C30-GSK-3β complex was
performed for 50 ns. Multiple ligand RMSDs of C30 based on the structures of the ligand at 0 ns, 10 ns, and 30 ns of the MD trajectory as references are shown in Figure 2(C). The RMSD plots suggested the ligand underwent a major conformational change during the first 2 ns. The RMSD plots plateaued after the initial 2 ns of the production run suggesting stabilization of the ligand position. Comparison of the ligand position at 0 ns and 50 ns trajectories Figure 2(A) and 2(B) showed that the -CF3 moiety shifted from its initial position and formed new interactions with Tyr221 and Tyr222. The -CF3 moiety was stabilized at the binding site by form-ing π-anion interaction with Tyr221. At 50 ns, com-pound C30 formed the hydrogen bond interaction with the residue Gln185, Asn186, and Lys188 and the hydro-Fig. 1. (A) The molecular structure of the most active compound C30. (B) C30 docked pose in the GSK-3β active site cavity. (C) Molecular interaction analysis after docking with C30 and GSK-3β, and (D) 2D ligand interaction map.
phobic interaction with the residue Val70, Leu188, Thr138, and Tyr140.
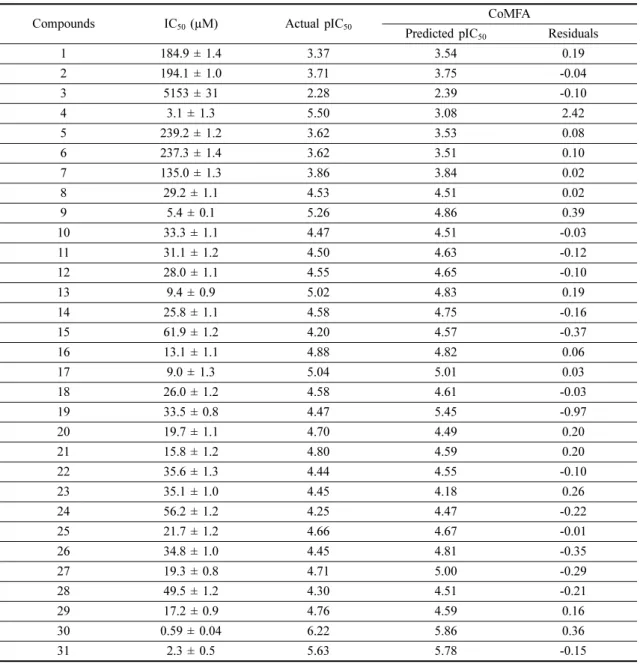
The final 3 ns of the MD trajectory were collected for binding energy (BE) calculation. BE calculation was done using the g_mmpbsa package and summarized in Table 2. The van der Waals interactions and electro-static interactions played a significant role in total BE
energy, which was found to be -54.60 kJ/mol. The res-idues, Val70, Thr138, Arg144, Gln185, Leu188, were the major BE contributing residues with BE values of -4.28 kJ/mol, -3.63 kJ/mol, -3.50 kJ/mol, -4.80 kJ/mol, and -7.24 kJ/mol respectively. The detailed analysis was depicted in Figure 2(D).
Fig. 2. (A) Ligand position at 0 ns and 50 ns of MD simulation. (B) Molecular interaction of C30 with the active site residues at 50 ns. (C) RMS D analysis of C30, measured based on the position taken at 0 ns (C30_0ns_ref), 10 ns (C30_10ns_ref) and 30 ns (C30_30ns_ref).
CoMFA Model Study
Based on the conformation of C30 at 50 ns, the rest of the compounds were drawn, applied Gasteiger charges, and minimized in SYBYL-X 2.1. All the com-pounds were aligned by taking C30 as a template. The aligned compounds were shown in Figure 3(A). Chem-ical structures and their respective pIC50 values were
given in Table 1. The generated CoMFA model showed the q2 and r2 values of 0.664 and 0.920 respectively, at
the ONC of 4. The detailed statical analysis was pro-vided in Table 3. The steric and electrostatic field con-tribution was found to be 81% and 19%, respectively. The statistical results indicated a reasonable predictive ability of the CoMFA model. The actual vs. predicted pIC50 values of the compounds and their residuals were
tabulated in Table 4. The scatterplot between the pre-dicted and actual pIC50 was shown in Figure 3(B).
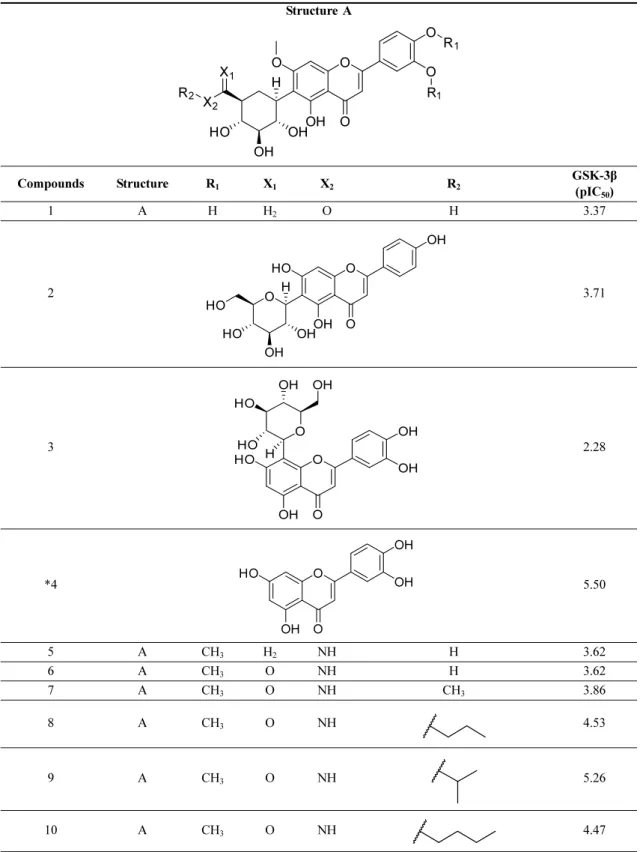
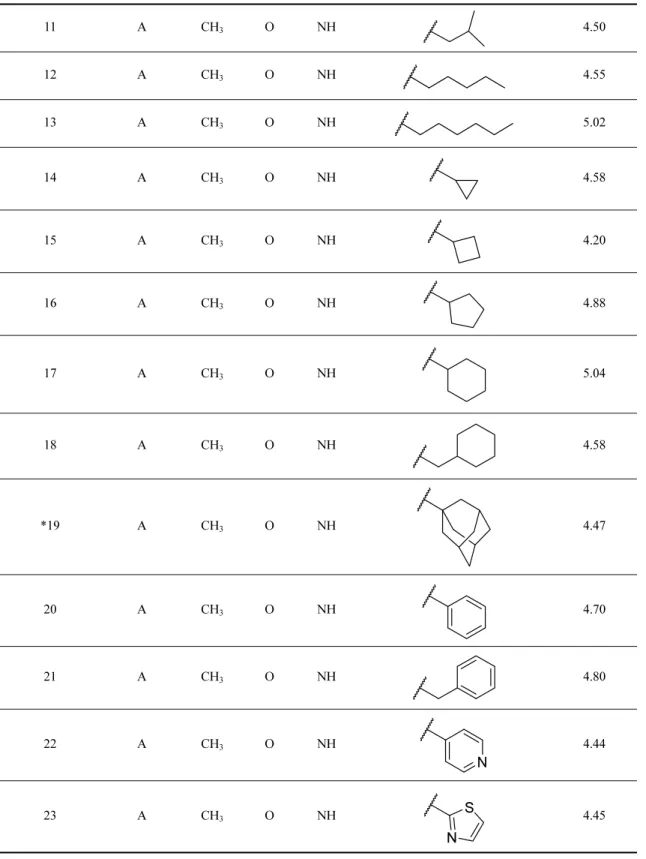
Compound C4 and C19 were set as outliers (assigned by *) due to their high residual values.
Steric and Electrostatic contour maps from the CoMFA analysis were shown in Figure 3(C) and (D) by taking the most active compound C30 as a reference. The green contour signifies favorable bulky substitu-tion, whereas the yellow contour indicates an unfavor-able one. Similarly, the blue contour favored the positive charge substitution, whereas the red contour favored negative charge substitution. A green and red contour map was found near R2 position, indicating the
bulky and electronegative group is favorable at this site. Most active compound C30 and second most active compound C31 poses (S)-CF3 and (R)-CF3 substitution
in their R2 position. But, C30 has more negatively
charged -CF3 towards the Tyr221 (S-configuration) and showed higher activity compared to C31. Compounds Fig. 3. (A) Data set alignment by making C30 as a template. (B) Scatterplot diagram of the CoMFA model to show the actual vs. predicted pIC50. (C) Steric and (D) Electrostatic contour map from the CoMFA analysis.
Table 1. Structure of the C-Glycosylflavones and their pIC50 values for GSK-3β
Structure A
Compounds Structure R1 X1 X2 R2 GSK-3β (pIC
50) 1 A H H2 O H 3.37 2 3.71 3 2.28 *4 5.50 5 A CH3 H2 NH H 3.62 6 A CH3 O NH H 3.62 7 A CH3 O NH CH3 3.86 8 A CH3 O NH 4.53 9 A CH3 O NH 5.26 10 A CH3 O NH 4.47 O O OH O O O R1 R1 X1 X2 R2 OH HO OH H O O OH OH HO O OH OH HO HO H O O OH OH HO O OH HO H OH HO OH O O OH OH HO OH
Table 1. Continued 11 A CH3 O NH 4.50 12 A CH3 O NH 4.55 13 A CH3 O NH 5.02 14 A CH3 O NH 4.58 15 A CH3 O NH 4.20 16 A CH3 O NH 4.88 17 A CH3 O NH 5.04 18 A CH3 O NH 4.58 *19 A CH3 O NH 4.47 20 A CH3 O NH 4.70 21 A CH3 O NH 4.80 22 A CH3 O NH 4.44 23 A CH3 O NH 4.45 N N S
C1, C2, C3, C5, and C7 were not bearing any bulky substitution and displayed lower activity values than the compounds C16, C17, C18, C21, C22, and C23, which bore bulky substitutions. Compound C27 and C29 have the electronegative -CF3 towards the Tyr221
and Tyr220, compared to C26 and C28, having only single -F. This substitution could be the reason behind the higher activity values of C27 and C29 compared to C26 and C28 against GSK-3β inhibition. The con-tour map analysis suggested that compounds C30, C31, C16, and C17 expressed higher activity values among the dataset compounds due to the favorable substituents. Table 1. Continued 24 A CH3 O NH 4.25 25 A CH3 O NH 4.66 26 A CH3 O NH 4.45 27 A CH3 O NH 4.71 28 A CH3 O NH 4.30 29 A CH3 O NH 4.76 30 A CH3 O NH 6.22 31 A CH3 O NH 5.63 *Outliers
Table 2. Summary of MM/PBSA binding energy evaluation
Energy term Binding Energy contribution
Van der Waal energy -192.12 ± 19.82 kJ/mol
Electrostatic energy -48.67 ± 15.54 kJ/mol
Polar solvation energy 210.40 ± 28.47 kJ/mol
SASA energy -24.21 ± 1.75 kJ/mol
Total Binding energy -54.60 ± 11.99 kJ/mol
F F F F F CF3 CF3 (S) CF3 (R)
Table 3. Statistical summary of the CoMFA model
Parameters CoMFA MODEL
q2 0.664 ONC 4 SEP 0.443 r2 0.920 SEE 0.217 F value 68.858 Steric contribution 81% Electrostatic contribution 19%
q2: cross-validated correlation coefficient; ONC: optimum
number of components; SEP: standard error of prediction; r2 : non-cross-validated correlation coefficient; value:
Conclusion
We have performed the molecular modeling study on the C-Glycosylflavone derivatives. C-Glycosylflavones are an interesting candidate over the existing inhibitors due to their ease of chemical modification and higher selectivity for the GSK-3β. In this study, we have per-formed docking and MD simulation, which showed the critical interactions for binding with GSK-3β. MM/
PBSA evaluation showed the important residues that contributed to binding energy for ligand binding. The 3D-QSAR based CoMFA model was developed to study the structure-activity relationship. The contour map analysis from CoMFA showed favorable substitu-tion in the R2 position for steric and electrostatic
inter-actions. This study could help in developing new C-Glycosylflavon based derivatives to inhibit the GSK-3β for the treatment of AD patients.
Table 4. Actual vs predicted pIC50 values and their residuals generated from the CoMFA analysis
Compounds IC50 (µM) Actual pIC50 CoMFA
Predicted pIC50 Residuals
1 184.9 ± 1.4 3.37 3.54 0.19 2 194.1 ± 1.0 3.71 3.75 -0.04 3 5153 ± 31 2.28 2.39 -0.10 4 3.1 ± 1.3 5.50 3.08 2.42 5 239.2 ± 1.2 3.62 3.53 0.08 6 237.3 ± 1.4 3.62 3.51 0.10 7 135.0 ± 1.3 3.86 3.84 0.02 8 29.2 ± 1.1 4.53 4.51 0.02 9 5.4 ± 0.1 5.26 4.86 0.39 10 33.3 ± 1.1 4.47 4.51 -0.03 11 31.1 ± 1.2 4.50 4.63 -0.12 12 28.0 ± 1.1 4.55 4.65 -0.10 13 9.4 ± 0.9 5.02 4.83 0.19 14 25.8 ± 1.1 4.58 4.75 -0.16 15 61.9 ± 1.2 4.20 4.57 -0.37 16 13.1 ± 1.1 4.88 4.82 0.06 17 9.0 ± 1.3 5.04 5.01 0.03 18 26.0 ± 1.2 4.58 4.61 -0.03 19 33.5 ± 0.8 4.47 5.45 -0.97 20 19.7 ± 1.1 4.70 4.49 0.20 21 15.8 ± 1.2 4.80 4.59 0.20 22 35.6 ± 1.3 4.44 4.55 -0.10 23 35.1 ± 1.0 4.45 4.18 0.26 24 56.2 ± 1.2 4.25 4.47 -0.22 25 21.7 ± 1.2 4.66 4.67 -0.01 26 34.8 ± 1.0 4.45 4.81 -0.35 27 19.3 ± 0.8 4.71 5.00 -0.29 28 49.5 ± 1.2 4.30 4.51 -0.21 29 17.2 ± 0.9 4.76 4.59 0.16 30 0.59 ± 0.04 6.22 5.86 0.36 31 2.3 ± 0.5 5.63 5.78 -0.15
Acknowledgment
This study was supported by research funds from Chosun University 2020.
References
[1] Hoover, B. R.; Reed, M. N.; Su, J.; Penrod, R. D.; Kotilinek, L. A.; Grant, M. K.; Pitstick, R.; Carlson, G. A.; Lanier, L. M.; Yuan, L.-L., Tau Mislocaliza-tion to Dendritic Spines Mediates Synaptic Dys-function Independently of Neurodegeneration. Neuron 2010, 68, 1067-1081.
[2] Miller, E. C.; Teravskis, P. J.; Dummer, B. W.; Zhao, X.; Huganir, R. L.; Liao, D., Tau Phosphor-ylation and Tau Mislocalization Mediate Soluble Aβ Oligomer‐Induced Ampa Glutamate Receptor Sig-naling Deficits. European Journal of Neuroscience 2014, 39, 1214-1224.
[3] Probst, A.; Tolnay, M.; Langui, D.; G oedert, M.; Spillantini, M., Pick’s Disease: Hyperphosphory-lated Tau Protein Segregates to the Somatoaxonal Compartment. Acta neuropathologica 1996, 92, 588-596.
[4] Qu, Z.-S.; Li, L.; Sun, X.-J.; Zhao, Y.-W.; Zhang, J.; Geng, Z.; Fu, J.-L.; Ren, Q.-G., Glycogen Syn-thase Kinase-3 Regulates Production of Amyloid-<I>Β</I> Peptides and Tau Phosphorylation in Dia-betic Rat Brain. The Scientific World Journal 2014, 2014, 878123.
[5] Reddy, P. H., Amyloid Beta-Induced Glycogen Syn-thase Kinase 3β Phosphorylated Vdac1 in Alzhei-mer’s Disease: Implications for Synaptic Dysfunction and Neuronal Damage. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2013, 1832, 1913-1921.
[6] Sofola, O.; Kerr, F.; Rogers, I.; Killick, R.; Augus-tin, H.; Gandy, C.; Allen, M. J.; Hardy, J.; Love-stone, S.; Partridge, L., Inhibition of Gsk-3 Ameliorates Aβ Pathology in an Adult-Onset Dro-sophila Model of Alzheimer's Disease. PLoS Genet 2010, 6, e1001087.
[7] Beurel, E.; Grieco, S. F.; Jope, R. S., Glycogen Syn-thase Kinase-3 (Gsk3): Regulation, Actions, and Diseases. Pharmacology & therapeutics 2015, 148, 114-131.
[8] Hernandez, F.; Lucas, J. J.; Avila, J., Gsk3 and Tau: Two Convergence Points in Alzheimer's Disease. Journal of Alzheimer's disease 2013, 33, S141-S144.
[9] Medina, M.; Garrido, J. J.; Wandosell, F. G., Mod-ulation of Gsk-3 as a Therapeutic Strategy on Tau Pathologies. Frontiers in molecular neuroscience 2011, 4, 24.
[10] Toral-Rios, D.; Pichardo-Rojas, P. S.; Alonso-Vane-gas, M.; Campos-Peña, V., Gsk3β and Tau Protein in Alzheimer’s Disease and Epilepsy. Frontiers in Cellular Neuroscience 2020, 14.
[11] Boutajangout, A.; M Sigurdsson, E.; K Krishnamur-thy, P., Tau as a Therapeutic Target for Alzheimer's Disease. Current Alzheimer Research 2011, 8, 666-677.
[12] Ko, H.-J.; Chiou, S.-J.; Wong, Y.-H.; Wang, Y.-H.; Lai, Y.-L.; Chou, C.-H.; Wang, C.; Loh, J.-K.; Lieu, A.-S.; Cheng, J.-T., Gskip-Mediated Anchoring Increases Phosphorylation of Tau by Pka but Not by Gsk3beta Via Camp/Pka/Gskip/Gsk3/Tau Axis Sig-naling in Cerebrospinal Fluid and Ips Cells in Alz-heimer Disease. Journal of clinical medicine 2019, 8, 1751.
[13] Eldar-Finkelman, H.; Martinez, A., Gsk-3 Inhibi-tors: Preclinical and Clinical Focus on Cns. Fron-tiers in molecular neuroscience 2011, 4, 32. [14] Khan, I.; Tantray, M. A.; Alam, M. S.; Hamid, H.,
Natural and Synthetic Bioactive Inhibitors of Gly-cogen Synthase Kinase. European journal of medic-inal chemistry 2017, 125, 464-477.
[15] Liang, Z.; Li, Q. X., Discovery of Selective, Sub-strate-Competitive, and Passive Membrane Perme-able Glycogen Synthase Kinase-3β Inhibitors: Synthesis, Biological Evaluation, and Molecular Modeling of New C-Glycosylflavones. ACS chem-ical neuroscience 2018, 9, 1166-1183.
[16] Bertrand, J.; Thieffine, S.; Vulpetti, A.; Cristiani, C.; Valsasina, B.; Knapp, S.; Kalisz, H.; Flocco, M., Structural Characterization of the Gsk-3β Active Site Using Selective and Non-Selective Atp-Mimetic Inhibitors. Journal of molecular biology 2003, 333, 393-407.
[17] Webb, B.; Sali, A., Comparative Protein Structure Modeling Using Modeller. Current protocols in bio-informatics 2016, 54, 5.6. 1-5.6. 37.
[18] Huey, R.; Morris, G. M., Using Autodock 4 with Autodocktools: A Tutorial. The Scripps Research Institute, USA 2008, 54-56.
[19] Keretsu, S.; Bhujbal, S. P.; Cho, S. J., Docking and 3d-Qsar Studies of Hydrazone and Triazole Deriv-atives for Selective Inhibition of Grk2 over Rock2. Letters in Drug Design & Discovery 2020, 17, 618-632.
Huey, R.; Hart, W. E.; Belew, R. K.; Olson, A. J., Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. Journal of computational chemistry 1998, 19, 1639-1662.
[21] Lindahl, E.; Abraham, M.; Hess, B.; van der Spoel, D., Gromacs 2020 Manual. Version: 2020. [22] Huang, J.; MacKerell Jr, A. D., Charmm36
All‐Atom Additive Protein Force Field: Validation Based on Comparison to Nmr Data. Journal of com-putational chemistry 2013, 34, 2135-2145. [23] Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.;
Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I., Charmm Gen-eral Force Field: A Force Field for Drug‐Like Mol-ecules Compatible with the Charmm All‐Atom Additive Biological Force Fields. Journal of com-putational chemistry 2010, 31, 671-690.
[24] Kumari, R.; Kumar, R.; Consortium, O. S. D. D.; Lynn, A., G_Mmpbsa a Gromacs Tool for High-Throughput Mm-Pbsa Calculations. Journal of chemical information and modeling 2014, 54, 1951-1962.
[25] Keretsu, S.; Bhujbal, S. P.; Cho, S. J., Computa-tional Study of Paroxetine-Like Inhibitors Reveals New Molecular Insight to Inhibit Grk2 with Selec-tivity over Rock1. Scientific reports 2019, 9, 1-14. [26] Clark, M.; Cramer III, R. D.; Van Opdenbosch, N., Validation of the General Purpose Tripos 5.2 Force Field. Journal of Computational Chemistry 1989, 10, 982-1012.
[27] Gadhe, C. G.; Kothandan, G.; Cho, S. J., Large Vari-ation in Electrostatic Contours Upon Addition of Steric Parameters and the Effect of Charge Calcu-lation Schemes in Comfa on Mutagenicity of Mx Analogues. Molecular Simulation 2012, 38, 861-871.
[28] Gadhe, C. G.; Madhavan, T.; Kothandan, G.; Cho, S. J., In Silico Quantitative Structure-Activity Rela-tionship Studies on P-Gp Modulators of Tetrahy-droisoquinoline-Ethyl-Phenylamine Series. BMC structural biology 2011, 11, 5.
[29] San Juan, A. A.; Cho, S. J., 3d-Qsar Study of Mic-rosomal Prostaglandin E 2 Synthase (Mpges-1) Inhibitors. Journal of Molecular Modeling 2007, 13, 601-610.
[30] Bang, S. J.; Cho, S. J., Comparative Molecular Field Analysis (Comfa) and Comparative Molecular Similarity Index Analysis (Comsia) Study of Muta-gen X. BULLETIN-KOREAN CHEMICAL SOCI-ETY 2004, 25, 1525-1530.
[31] Pasha, F.; Cho, S. J.; Beg, Y.; Tripathi, Y., Quantum Chemical Qsar Study of Flavones and Their Radi-cal-Scavenging Activity. Medicinal Chemistry Research 2007, 16, 408-417.
[32] Gobbo, D.; Piretti, V.; Di Martino, R. M. C.; Trip-athi, S. K.; Giabbai, B.; Storici, P.; Demitri, N.; Girotto, S.; Decherchi, S.; Cavalli, A., Investigating Drug–Target Residence Time in Kinases through Enhanced Sampling Simulations. Journal of Chem-ical Theory and Computation 2019, 15, 4646-4659.