20S-Gi
nsenosi
de Rg
3sensi
t
i
zes
Doxor
ubi
ci
n-i
nducedCel
lDeat
h vi
a
Modul
at
i
on ofAut
ophagy i
n Hepat
ocel
l
ul
ar
Car
ci
nomaCel
l
s
by
Dong-Gun Ki
m
Major in Molecular Medicne
Department of Biomedical Sciences
The Graduate School, Ajou University
20S-Gi
nsenosi
de Rg
3sensi
t
i
zesDoxor
ubi
ci
n-i
nduced
Cel
lDeat
h vi
aModul
at
i
on ofAut
ophagy i
n
Hepat
ocel
l
ul
arCar
ci
nomaCel
l
s
by
Dong-Gun Kim
A Dissertation Submitted to The Graduate School of
Ajou University in Partial Fulfillment of the Requirements for
The Degree of Master of Biomedical Sciences
Supervised by
You-Sun Kim, Ph.D.
Major in Molecular Medicine
Department of Biomedical Sciences
This certifies that the dissertation
of Dong-Gun Kim is approved.
SUPERVISORY COMMITTEE
You-Sun Kim Sung-Yong Jeong Chan-Bae ParkThe Graduate School, Ajou University
December, 20th, 2011
- ABSTRACT -
20S-Ginsenoside Rg
3sensitizes Doxorubicin-induced Cell Death via
Modulation of Autophagy in Hepatocellular Carcinoma Cells
Although at present the role of autophagy in cell death is still controversial, there is accumulating evidence demonstrating that autophagy serves as an important cell survival mechanism under various stress conditions, including cancer cells exposing to chemotherapeutic agents. Ginsenoside Rg3 extracted from Panax ginseng C.A. Meyer is
one of the diverse groups of steroidal saponins with high pharmacological activity and it may increase the efficacies of cancer chemotherapy. In this study, we investigated the effect of Rg3 on autophagy, and to evaluate whether such effect is relevant to the
sensitization effect of Rg3 to apoptosis induced by DNA damage agents in
hepatocellular carcinoma cells. We found that that Rg3 is able to markedly increase
autophagy markers in cancer cells, including GFP-LC3 punctuation and LC3 II conversion. However, Rg3 treatment led to accumulation of p62 level and addition of
chloroquine failed to further enhance autophagy markers, indicating that Rg3 is likely to
suppressed autophagic flux at late maturation and degradation stage. Moreover, it was also found that Rg3 exerts synergistic effect on cell death induced by a DNA-damage
agent, doxorubicin in different hepatocellular carcinoma cell lines, suggesting that Rg3
sensitizes doxorubicin-induced cell death via suppression of autophagy. Using the mouse xenograft model, we confirmed that the synergistic effect of Rg3 with
doxorubicin in inhibiting tumor growth. Taken together, our data suggest that the Suppression effect of Rg3 on autophagy is functionally related to its sensitization effect
on doxorubicin-induced cell death which is caspase-independent and such a combination could serve as a novel strategy for chemotherapy for HCC.
TABLE OF CONTENTS
ABSTRACT ··· 1 TABLE OF CONTENTS ··· 2 LIST OF FIGURES ··· 3 I. INTRODUCTION ··· 4 . Ⅱ MATERIALS AND METHODS ··· 6. Ⅲ RESULTS ··· 9
A. Rg3 induces LC3-I to LC3-II conversion in dose- and time- dependent manner in hepatocellular carcinoma cells ··· 9
B. Increased autophagy markers in Rg3 treated cells are unlikely due to enhanced autophagic flux ··· 12
C. Rg3 inhibits autophagy at its late stage of autophagic flux ··· 14
D. Doxorubicin induces autophagy marker in hepatocellular carcinoma cells ··· 17
E. Autophagy induced by doxorubicin serves as a pro-survival mechanism ···· 19
F. Combination of Rg3 and Doxorubicin promotes caspase-independent cell death ··· 22
G. Rg3 sensitizes doxorubicin-induced antitumor activity in vivo mouse model ··· 24
. Ⅳ DISCUSSION ··· 26
REFERENCES ··· 28
LIST OF FIGURES
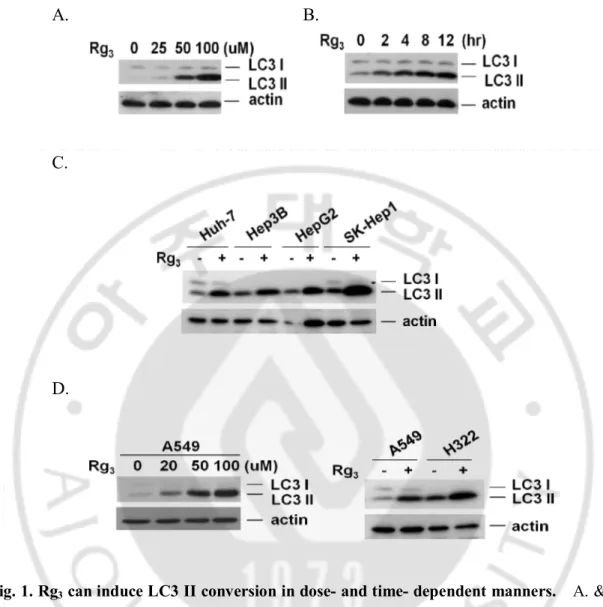
Fig. 1. Rg3 can induce LC3 II conversion in dose- and time- dependent
manners ··· 10
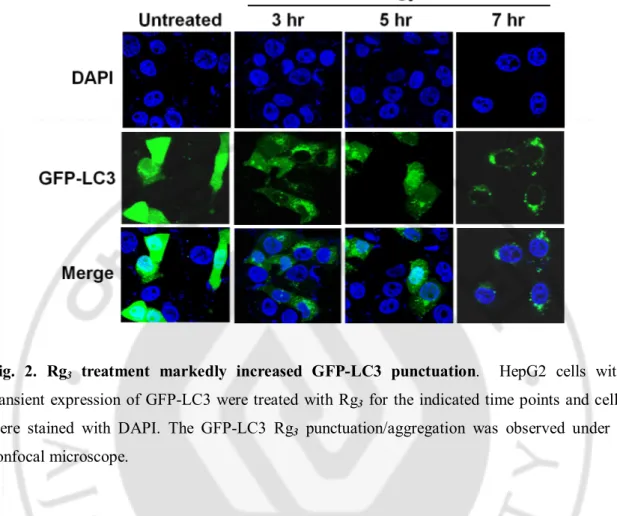
Fig. 2. Rg3 treatment markedly increased GFP-LC3 punctuation ··· 11
Fig. 3. Rg3-induced accumulation of autophagic markers is due to blockage
of lysosomal functions ··· 13
Fig. 4. Effects of Rg3 on autophagosome maturation ··· 15
Fig. 5. LC3-II conversion and punctuation of GFP-LC3 upon Rg3 treatment
are relevant to autophagy ··· 16
Fig. 6. Doxorubicin-induced autophagy served as pro-survival mechanism in human cancer cells ··· 18
Fig. 7. Suppression of autophagy can contribute doxorubicin-induced cancer cell death ··· 20
Fig. 8. Rg3 can sensitize DNA-damage induced cell death ··· 21
Fig. 9. Combination of Rg3 and doxorubicin induced- cell death was caspase-
I. Introduction
Autophagy is a cellular catabolic degradation response to various stress whereby cellular proteins, organelles and cytoplasm are engulfed, digested and recycled to sustain cellular metabolism (Shintani and Klionsky, 2004; Mizushima and Klionsky, 2007; Rubinsztein et al., 2007). In recent years, the importance of autophagy in human diseases, including cancer, neurodegenerative diseases and immunity has been progressively appreciated based on the increasing understanding of the diverse biological function of autophagy (Mizushima et al., 2008). Especially for the cancer, one particular important aspect is its role on cell death and cell survival (Yue et al., 2003; Kuma et al., 2004; Feng et al., 2005). Although autophagy is generally recognized as a defense mechanism in response to a number of cancer chemotherapeutics and therefore, inhibition of autophagy sensitizes cell death (Amaravadi et al., 2007; KO et al., 2009), there is emerging evidence supporting the notion that activation of autophagy sensitizes cancer cell death. Cell death by autophagy, or autophagic cell death is characterized by (i) independent of apoptosis, (ii) increased autophagic flux in the dying cells, and (iii) pro-death function of autophagy (Shen and Codogno, 2011). The autophagic flux is initiated with formation of phagophore and subsequent complete autophagosomes, and finally is finished by fusion with endosomes-lysosomes to form autolysosomes. Moreover, even there is real increase of autophagic flux in the dying cells, the enhanced autophagy may indicate an attempt by the cell to save from cell death (Levine and Yuan, 2005; Kroemer and Levine, 2008; Scarlatti et al., 2009). Therefore, it is critical to demonstrate the pro-death function of autophagy in defining this form of cell death. Hepatocellular carcinoma (HCC) is one of the most common solid tumor and the third leading cause of cancer death in the worldwide. One of the reasons for the high mortality rate in patients with HCC is the lack of effective treatment, especially for those with advanced disease. To date, systemic therapy with classical cytotoxic chemotherapy has been reported to be poorly tolerated and ineffective. For this reason,
new therapeutic options are eagerly needed for more effective treatment of HCC. To increase anticancer efficacy for HCC, several approaches that combination of autophagy modulation and conventional chemotherapeutics have been developed and suggested as a promising therapeutic strategy in treatment (Takimoto and Awada, 2008; Cabibbo et al., 2009). In these situations, autophagy modulation may be a good therapeutic strategy. However, little is known about autophagy status and corresponding functions in HCCs after use of these therapies.
Ginsenoside Rg3 extracted from Panax ginseng C.A. Meyer is one of the diverse
groups of steroidal saponins with high pharmacological activity. Several findings suggest that Rg3 may increase the efficacies of cancer chemotherapy, and the molecular
mechanisms responsible for such anti-cancer function is related to its inhibitory effects on NF- B and AP-1 activity but it is still unclear that the mechanism of Rg3 in cancer
chemotherapies (Keum et al., 2003). It has been reported that the effects and mechanisms of ginsenoside Rb1 on activation of autophagy in glutamate-injured neurons (Chen et al., 2010) and ginsenoside Rk1 has anti-tumor activity by modulating autophagy (KO et al., 2009). However, the mechanism of autophagy modulation by ginsenoside is unclear and there is no report that effects and mechanisms of ginsenoside Rg3 on activation of autophagy.
In the present study, we investigated the effect of the ginsenoside Rg3 on modulation
of autophagy in hepatocellular carcinoma cell lines and evaluate whether such effect is relevant to the sensitization effect of Rg3 to apoptosis induced by DNA damage agents,
doxorubicin which is commonly used in the treatment of a wide range of cancers. Our data show that Rg3 is capable of repressing autophagic flux via suppressing the late
stage autophagosome maturation and degradation. Importantly, Rg3-induced inhibition
of autophagy contributes to doxorubicin-induced cancer cell death. The synergistic effect of Rg3 on the therapeutic efficacy of doxorubicin was confirmed using the
II. Materials and Methods
A. Rg3 extraction. Sun ginseng (1.6 kg) was extracted with 70% MeOH (1.2 L) under
reflux for 3 hr. The solvent was removed in vacuo to yield 320 g of 70% MeOH extract, which was suspended in water and extracted with n-BuOH. The n-BuOH fraction was concentrated in vacuo to yield 91.5 g of BuOH fraction. 40g of fractions was subjected to silica gel column chromatography. 9 fractions were obtained using stepwise gradient elution (EtOAc : MeOH : H20 =40 : 1 : 1 → 20 : 1 : 1 → 10 : 1 : 1). Fraction 8 was chromatographed over silica gel using CHCl3 : MeOH : H2O = 200 : 20 : 1 → 150 : 20 : 1 solvent. Rg3 rich fraction which contained Rg3 (R) and (S) forms was obtained solvent.
It was further purified over semi-preparative HPLC/ELSD analysis using a reverse-phase column (Phenomenex C18, 250 mm x 10 mm) with 40% ACN to isolate Rg3 (S)
form (10 mg).
B. Reagents. Anti-p62, anti-PARP1 antibodies were purchased from Cell signaling.
Anti-LC3 antibody and Necrostatin-1 were purchased from Sigma. Doxorubicin, Chloroquine diphosphate (CQ), Bafilomycin, 3-methyladenine (3-MA), doxycyclin, and Cycloheximide were purchased from Calbiochem. zVAD was purchased from R&D system.
C. Cell culture. SK-Hep1, HepG2, Hep3B, A549, H322 cells were cultured in
Dulbecco’s modified Eagle’s medium supplemented with 10 % fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. Huh-7 cells were cultured in RPMI 1640 medium supplemented with 10 % fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. The Atg5-/- MEFs and the Tet-off Atg5 MEFs were provided by Dr. N Misushima (Tokyo Medical and Dental University). Normal liver cell line HL-7702 was purchased from Shanghai Institute of
Cell Biology (Shanghai, China) and cells were cultured in RPMI 1640 medium supplemented with 20 % fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin and 100 ug/ml streptomycin.
D. Western blot analysis. Upon treatment, cells were lysed in M2 buffer (20 mM Tris
at pH 7, 0.5 % NP-40, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 2 mM DTT, 0.5 mM PMSF, 20 mM β-glycerol phosphate, 1 mM sodium vanadate, 1 μg/ml leupeptin). Equal amounts of cell extracts were resolved by 12% or 15% SDS-PAGE and analyzed by western blot and visualized by enhanced chemiluminescence (ECL, Amersham).
E. Transfection. Transfection experiments in hepatoma cells were performed with
Lipofectamine PLUS reagent by following the instruction provided by the manufacturer (GIBCO/BRL). Cells were transfected with the GFP-LC3 construct. The mRFP-GFP tandem fluorescent-tagged LC3 construct (tfLC3) was provided by Dr. T Yoshimori (Osaka University) (Kimura et al., 2007).
F. Confocal microscopy. Cells were seeded to a coverglass slide chamber (Lab-Tek®,
NUNC), after designated treatments, the cells were examined under a confocal microscope (Carl Zeiss LSM710). The GFP-LC3 puncta were examined under a confocal microscope and tfLC3 were observed for the change of both green and red fluorescence using a confocal microscope. The data presented were from one representative experiment of at least 3 independent repeats.
G. Cytotoxicity assay. Cell death was determined using tetrazolium dye colorimetric
test (MTT test). The MTT absorbance was then read at 570 nm. Representative images were taken by a phase-contrast microscope.
H. Reverse Transcription-PCR. RNA was extracted with the RNeasy Kit (Qiagen). 1
ug of total RNA from each sample was used as a template for cDNA synthesis with a reverse transcriptase kit (Invitrogen). An equal amounts of cDNA product was used in the PCR performed using the Taq DNA polymerase (Takara). PCR amplification was performed using the following primers: human p62 gene sense (5'-AATGTGATCTGCGATGGCTG-3'), p62 antisense (5'-CCAAGGCGATCTTCCTCATC T-3') and β-actin gene sense (5′-CAGGTCATCACCATTGGCAATGAGC-3′), β-actin gene antisense (5′-GATGTCCACGTCACACTTCATGA-3′ ). The final PCR products were resolved in 1.5 % agarose gel and stained with ethidium bromide.
I. Tumor xenograft study. Male nude mice were obtained from Central Lab. Animal
Inc. (Seoul, Korea). The animals were fed standard rat chow and tap water ad libitum, and were maintained under 12 h dark/light cycle at 21 ºC. Male, 6-week-old nude mice were randomly divided into four groups (control, Rg3, Doxorubicin, Rg3+Doxorubicin,
n=8/group). Huh-7 cells were harvested and mixed with PBS (200 μl/mouse) and then inoculated into one flank of each nude mouse (5x106 Huh-7 cells). When the tumors had reached a volume of about 50-70 mm3, mice were given a daily oral dose of 20 mg/kg
Rg3 or the vehicle (200 μl PBS, control group), and i.p three times/week at dose of 1
mg/kg Doxorubicin, for 21 days, respectively. The tumor dimensions were measured twice a week using a digital caliper and the tumor volume was calculated using the formula: V = length x width2 x 0.5. At the end of the experiment, the mice were killed and the tumors were excised and weighed. Histopathological analysis for tumors was carried out by using hematoxylin and eosin (H&E) staining.
III. Results
A. Rg3 induces LC3-I to LC3-II conversion in dose- and time- dependent manner
in hepatocellular carcinoma cells.
Several earlier studies have suggested that Rg3 may have anti-tumor functions but the
mechanism is still unclear. We first examined the effect of Rg3 on autophagy by
measuring Rg3-induced LC3 I to LC3 II conversion. As shown in Fig. 1.A and B, Rg3
can induce LC3 II conversion in a dose- and time-dependent manner in SK-Hep1 hepatocellular carcinoma cells. In order to confirm the above results of LC3 II conversion by Rg3 in not cell line specific, we next tested the effect of Rg3 on 4
different hepatocellular carcinoma cells (Fig. 1.C) and two different lung cancer cell lines of A549 and H322 cells (Fig. 1.D). All cells tested show similar LC3 II conversion following Rg3 treatment, suggesting that the effect of Rg3 on autophagy is not cell type
specific. To further confirm the effect of Rg3 on autophagy, we next tested the changes
of GFP-LC3 puncta formation in HepG2 cells with transient expression of the GFP-LC3. As shown in Fig. 2, Rg3 treatment markedly increased GFP-LC3 punctuation. Taken
together, these results demonstrate that Rg3 treatment is capable of enhancing the
A. B.
C.
D.
Fig. 1. Rg3 can induce LC3 II conversion in dose- and time- dependent manners. A. & B. Rg3 can induce LC3 II conversion. SK-Hep1 cells were treated for 12 hours in different
concentration of Rg3 and cell lysates were analyzed by western blot using anti-LC3 antibody
(A), and cells were treated for indicated time point in 100 uM of Rg3 and cell lysates were
analyzed by western blot (B). C. Rg3-induced LC3 II conversion in different Hepatoma cell
lines. Four different hepatocellular carcinoma cells were treated with Rg3 for 12 hours at 100
uM concentration and cells lysates were analyzed by western blot. D. Rg3-induced LC3 II
conversion in different type of cancer cell lines. A549 and H322 lung cancer cells were treated with Rg3 for 12 hour and cell lysates were analyzed by western blot.
Fig. 2. Rg3 treatment markedly increased GFP-LC3 punctuation. HepG2 cells with
transient expression of GFP-LC3 were treated with Rg3 for the indicated time points and cells
were stained with DAPI. The GFP-LC3 Rg3 punctuation/aggregation was observed under a
B. Increased autophagy markers in Rg3 treated cells are unlikely due to enhanced
autophagic flux.
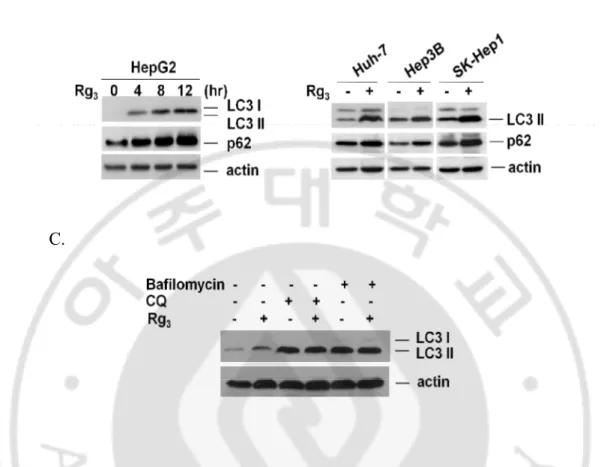
One important note in evaluation of autophagy is that the increase of autophagic markers may represent either the increased generation of autophagosomes and/or a blockage in autophagosomal maturation and degradation (Mizushima, 2004; Mizushima and Yoshimori, 2007; Klionsky et al., 2008; Wu et al., 2008). p62/SQSTM1 is a multifunctional protein that binds to LC3 and can be destructed within the autolysosome (Pankiv et al., 2007). Thus, reduction of p62 has been regarded as a marker for increase of autophagic flux (Bjørkøy et al., 2005; Mizushima and Yoshimori, 2007). To examine the changes of autophagic flux by Rg3, we checked p62 protein level in Rg3-treated cells.
As shown in Fig. 3. A, Rg3 treatment led to evident increase of p62 protein level as
similar pattern as LC3 II conversion, being inconsistent with the common understanding that effect in autophagy could result in decrease of p62. Rg3 was able to significantly
enhance the p62 protein level in all three hepatocellular carcinoma cell lines tested (Fig. 3. B). Moreover, Rg3 could not to further enhance the LC3 II conversion in cells treated
with chloroquine (CQ), an agent capable of impairing lysosomal acidification and suppressing the protease activity or bafilomycin and the level of LC3 II conversion by Rg3 alone was much lesser than that of CQ or bafilomycin alone (Fig. 3. C). Taken
together, our data suggest that the increased autophagy markers (LC3 II conversion and GFP-LC3 punctuation) in Rg3-treated cells are unlikely due to enhanced autophagic flux,
but rather by suppression of the late maturation and degradation stage.
A. B.
C.
Fig. 3. Rg3-induced accumulation of autophagic markers is due to blockage of lysosomal functions. A. Rg3 up-regulates autophagic marker p62/SQSTM1. HepG2 cells were treated
with Rg3 for indicated time periods and cell lysates were collected and subject to western blot.
B. Accumulation of p62 in Rg3 treatment. Three different hepatocellular carcinoma cells were
treated with Rg3 and cell lysates were collected and subject to western blot. C. Rg3 does not
promote autophagic flux. HepG2 cells were pretreated with chloroquine (CQ) or bafilomycin for 30 min, followed by treatment with Rg3 for another 12 hours and cell lysates were collected
C. Rg3 inhibits autophagy at its late stage of autophagic flux.
In order to further understanding the inhibitory effect of Rg3 on autophagy, here we
set to examine whether the increased autophagy markers as observed above in cells treated with Rg3 is due to suppressed autophagic flux. The tfLC3 has been reported to
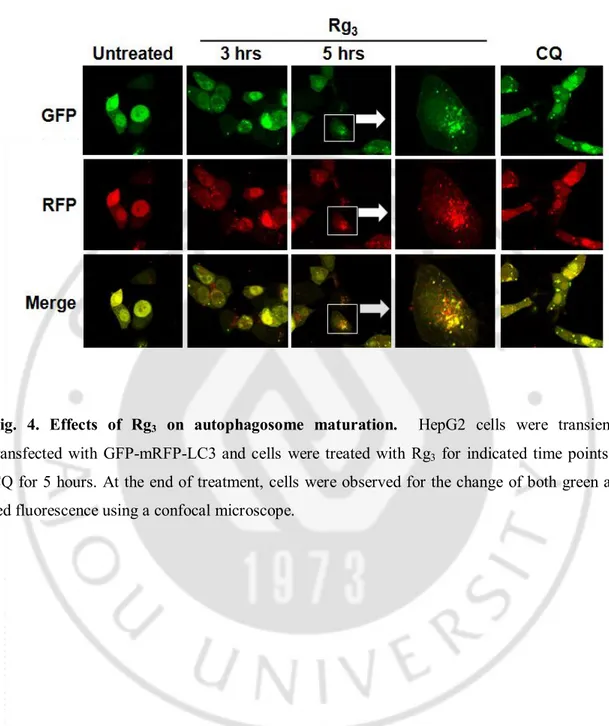
be a useful tool for examining autophagosome maturation and autolysosome formation (Katayama et al., 2007; Kimura et al., 2007). We used the HepG2 cells with transient transfection of tfLC3 to analyze the effect of Rg3 on this process. As shown in Fig. 4,
treatment with Rg3 led to enhance both GFP and mRFP punctuation and co-localization
to appear as yellow suggesting that the autophagosome does not fuse with lysosome or the lysosome function is impaired. More importantly, CQ also led to be similar to that of Rg3, although to a more extent suggesting the possibility that the Rg3 may inhibit
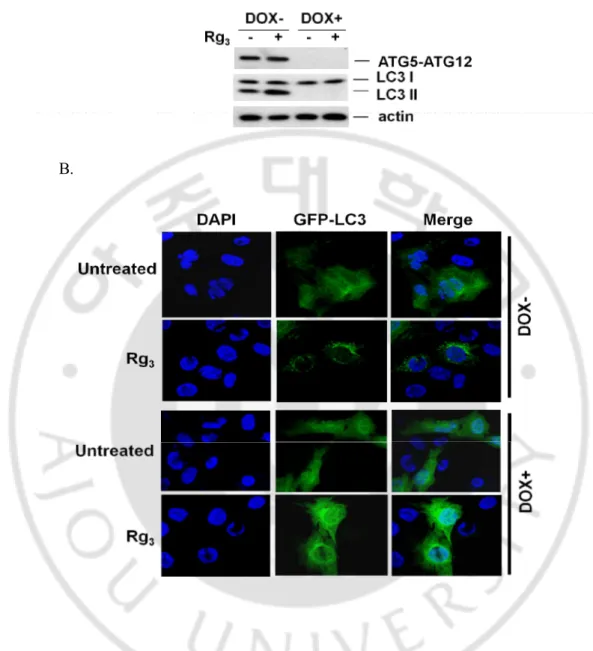
autolysosome formation by interfering the fusion of autophagosome with lysosome. We next further tested whether the induction of LC3 II conversion and punctuation of GFP-LC3 upon Rg3 treatment are relevant to autophagy by using Tet-off Atg5 MEF cells in
which the Atg5 and the GFP-LC3 were stably expressed (Hosokawa et al., 2006). In this system, the Atg5 gene expression was abolished following treatment with doxycyclin (DOX). As shown in Fig. 5. A , Rg3 induced LC3 II conversion in MEF cells with the
presence of Atg5 (without treatment of DOX), but not in cells with the absence of Atg5 (pretreated with DOX). Consistent with LC3 II conversion, the punctuation/aggregation of GFP-LC3 was only induced in cells expressing Atg5, but not in cells without Atg5 (Fig. 5. B). Taken together, data from this part of our study present clear evidence that suggests Rg3 enhances the level of autophagy markers by suppressing autophagic flux.
Fig. 4. Effects of Rg3 on autophagosome maturation. HepG2 cells were transiently transfected with GFP-mRFP-LC3 and cells were treated with Rg3 for indicated time points or
CQ for 5 hours. At the end of treatment, cells were observed for the change of both green and red fluorescence using a confocal microscope.
A.
B.
Fig. 5. LC3-II conversion and punctuation of GFP-LC3 upon Rg3 treatment are relevant to autophagy. A. Rg3-induced LC3 I to II conversion is ATG5-dependent. Tet-off Atg5 MEFs
with stable expression of GFP-LC3 were pretreated with or without doxycyclin (DOX) for 4 days, then cells were treated with Rg3 for 12 hrs, and cell lysate was subject to western blotting.
B. Tet-off Atg5 MEFs were prepared as described in (A), and treated with Rg3 for 3 hrs. Cells
D. Doxorubicin induces autophagy marker in hepatocellular carcinoma cells.
Several studies have suggested that modulation of autophagy can sensitize cancer cell death induced by DNA damage agents (Lambert et al., 2008; Li et al., 2010). Since our data suggest that Rg3 treatment can suppress autophagic flux, it will be of interest to
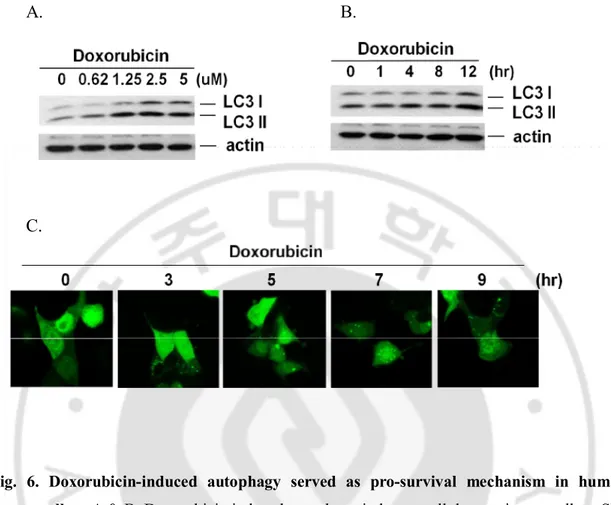
investigate whether such effect is related to cancer cell death induced by DNA damage. Doxorubicin is a commonly used anticancer drug which causes DNA damage and kills cancer cells mainly by apoptosis (Lee et al., 2002). Several reports suggest that doxorubicin-induced cardiotoxicity is associated with the depletion of a variety of long- and short-lived proteins (Kim et al., 2003; Aries et al., 2004; Lim et al., 2004; Olson et al., 2005; Li et al., 2006; Oliveira and Wallace, 2006), indicating an activation of protein degradation systems including the ubiquitin proteasome system (Kumarapeli et al., 2005) and autophagy (Semenov et al., 2001). As shown in Fig. 6. A and B, doxorubicin can induce LC3 II conversion in a dose- and time-dependent manner in SK-Hep1 cells. Moreover, we observed similar effects by doxorubicin on autophagy by showing the GFP-LC3 punctuation (Fig. 6. C), indicating the possibility that induction of autophagy by doxorubicin in hepatocullular carcinoma cells.
A. B.
C.
Fig. 6. Doxorubicin-induced autophagy served as pro-survival mechanism in human cancer cells. A & B. Doxorubicin induced autophagy in hepatocellular carcinoma cells.
SK-Hep1 cells were treated with Doxorubicin for indicated concentrations for 12 hour (A) or indicated time periods with 2.5 uM concentration (B) and cell lysates were subject to western blotting. C. Doxorubicin induces autophagy in hepatocellular carcinoma cells. HepG2 cells were treated with doxorubicin for indicated time points and cells were examined with a confocal microscope for GFP-LC3 punctuation/aggregation.
E. Autophagy induced by doxorubicin serves as a pro-survival mechanism.
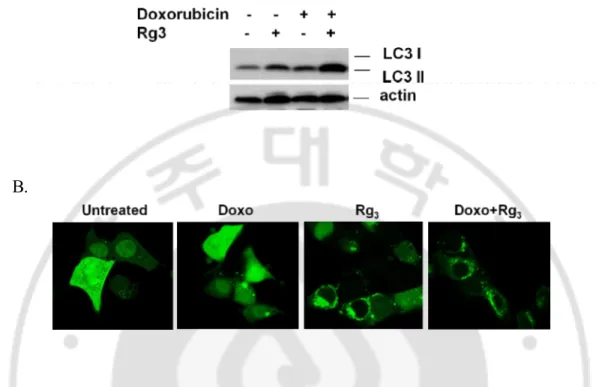
Here we attempted to determine whether the suppression of autophagic flux induced by Rg3 serves as pro-death mechanism in hepatocellular carcinoma cells. We first tested
whether Rg3 is able to suppress autophagy by doxorubicin in hepatocellular carcinoma
cells. As shown in Fig. 7. A, Rg3 enhanced doxorubicin-induced LC3 II conversion,
confirming the blockage of autophagy by Rg3 treatment. Similar with this data,
doxorubicin was found to induce the GFP-LC3 punctuation which was further increased by Rg3 (Fig. 7. B).
Since our data suggest that autophagy plays a pro-survival role in doxorubicin-induced cell death, whereas doxorubicin suppresses autophagy at its late stage of autophagic flux, here we examined the effect of Rg3 on doxorubicin-induced cell death.
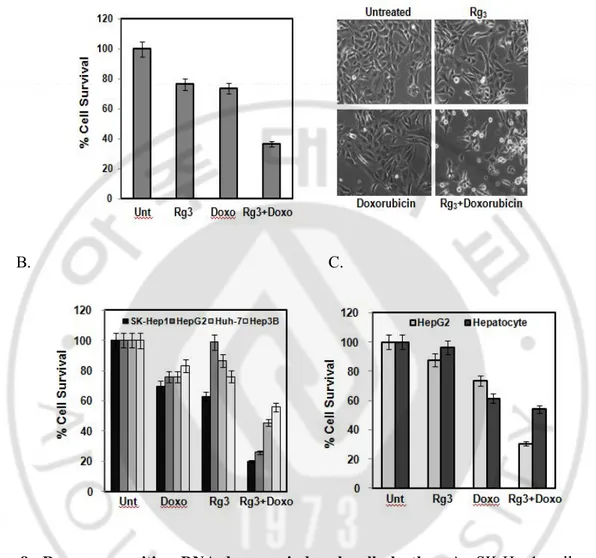
When cells were pretreated with Rg3 for 30 min followed by a low cytotoxic
concentration of doxorubicin for 24 hr, cells underwent dramatic cell death as evidenced by MTT assay and morphological changes (Fig. 8. A). The sensitizing effect of Rg3 and
doxorubicin was observed in all the hepatocellular carcinoma cell lines tested (Fig. 8. B), but not in the normal human liver cells, HL 7702, suggesting that Rg3 plus
A.
B.
Fig. 7. Suppression of autophagy can contribute doxorubicin-induced cancer cell death.
A. LC3 turnover assay in Rg3 induced autophagy. HepG2 cells were pretreated with Rg3 for 30
min, followed by Doxorubicin for another 12 hours, cell lysates were collected and subject to western blot. B. Combination of Rg3 and Doxorubicin markedly increased GFP-LC3
punctuation. Cells were transfected with GFP-LC3 and treated with Rg3, Doxorubicin, or Rg3 +
Doxorubicin for indicated time points. Cells were examined with a confocal microscope for GFP-LC3 punctuation/aggregation.
A.
B. C.
Fig. 8. Rg3 can sensitize DNA-damage induced cell death. A. SK-Hep1 cells were pretreated with Rg3 for 30 min, followed by Doxorubicin for another 18 hours and cells viability
was an analyzed by MTT assay (Left panel) and representative images were taken by a phase-contrast microscope (Right panel). Results are averages +/- SEM. B. Four different hepatocellular carcinoma cells were treated with combination of Rg3 and Doxorubicin for 18
hours and cells viability was an analyzed by MTT assay. Results are averages +/- SEM. C. Rg3
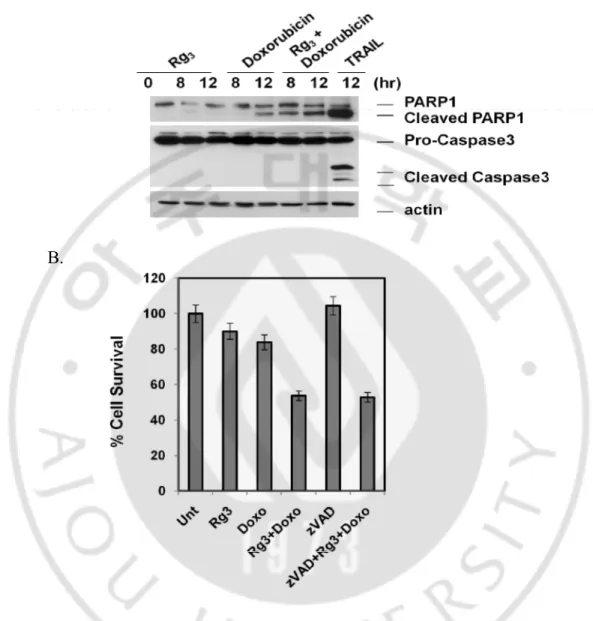
F. Combination of Rg3 and Doxorubicin promotes caspase-independent cell death.
Functional significance of autophagy on cell death has to be determined individually within the specific context under study. One important aspect is that autophagy has been associated with apoptosis and autophagy and apoptosis can act as partners to coordinately induce cell death (Kobayashi et al., 2010). In this study, we checked the combination of Rg3 and doxorubicin on the form of cell death. To do so, we pretreated
with Rg3 for 30 min followed by doxorubicin treatment for indicated time points.
Importantly, TRAIL, which is known as a chemotherapeutic agent to induce apoptotic cell death in cancer cells, can induce PARP and caspase 3 cleavage, but pretreatment with Rg3 significantly augmented doxorubicin-induced cleavage of one of the caspase-3
substrates PARP, without significant cleavage/activation of caspase 3 (Fig. 9. A). Therefore, the above data thus indicate that the form of cell death induced by combination of Rg3 and doxorubicin is likely to be caspase-independent cell death. To
further support this results, we treated cell with a general caspase inhibitor (Z-VAD-FMK) which is able to prevent caspase-dependent cell death. As consistent with western blot data, zVAD failed to block the cell death induced by the combination of Rg3 and
doxorubicin (Fig. 9. B), indicating that this cell death is caspase-independent non-apoptotic. Moreover, necrostatin-1 was unable to block the cell death by combination of Rg3 and doxorubicin (data not shown).
A.
B.
Fig. 9. Combination of Rg3 and doxorubicin induced- cell death was caspase-independent. A. Combination of Rg3 and doxorubicin does not induced caspase 3 cleavage. SK-Hep1 cells
were treated with Rg3, Doxorubicin, or Rg3 + Doxorubicin for indicated time periods and cell
were also treated with GST-TRAIL for 12 hour and cell lysates were subject to western blotting to showing cleavage of PARP1 and caspase 3. B. Caspase inhibitor, zVAD had no effect to
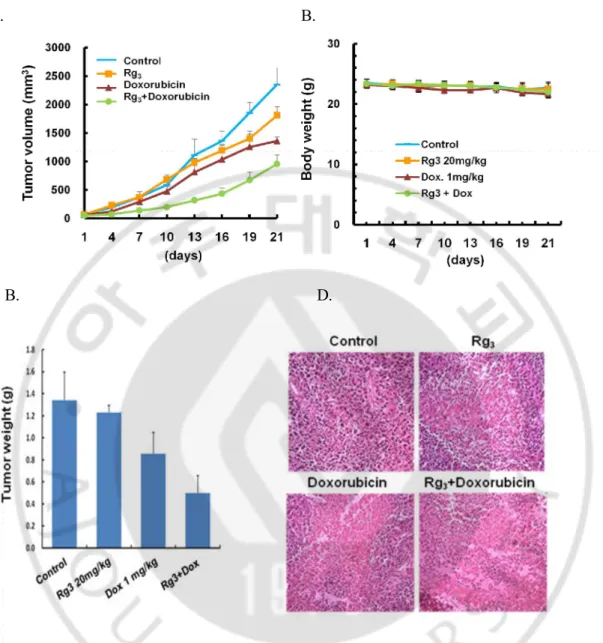
G. Rg3 sensitizes doxorubicin-induced antitumor activity in vivo mouse model.
Since our data suggest that promotion of autophagy by Rg3 can sensitize
doxorubicin-induced cell death, we carried the subsequent study on the therapeutic efficacy of combined treatment of Rg3 and using a xenograft mouse model. When the tumors were
measureable, mice were matched for tumor volumes and assigned to control, Rg3,
doxorubicin, or combination of Rg3 and doxorubicin. As shown in Fig. 10. A, Rg3 or
doxorubicin treatment alone had minimal effect on the growth of tumors, with the tumor size similar to the control mice whereas combined treatment with Rg3 and doxorubicin
led to significant reduction of the tumor volume. Furthermore, no significant weight loss was observed during these periods of combination treatments (Fig. 10. B). Consistent with the data of tumor volume, the tumor weight was also significantly decreased in combination of Rg3 and doxorubicin (Fig. 10. C). Taken together, these findings suggest
that Rg3 potently enhances the therapeutic activity of doxorubicin c in vivo. We next
investigate the effect of the drug combination on in vivo using H&E staining of paraffin-embedded sections of xenografted tumors. As shown in Fig. 10. D, Cell density of the combination of Rg3 and Doxorubicin group was greatly lower than Rg3 or
A. B.
B. D.
Fig. 10. Combination of Rg3 plus Doxorubicin inhibits hepatoma growth in mouse xenograft model. A & B. Effect of Rg3 plus Doxorubicin on Huh-7 hepatocellular carcinoma
cell xenograft in athymic BALB/c nude mice. Huh-7 tumors were established subcutaneously and treated with Rg3, Doxorubicin or Rg3 plus Doxorubicin for 21 days. Tumor growth (tumor
volume in A and body weight in B) was monitored. C. Tumor weight was also measured after 21 days treatment. At the end of treatment mice were killed and measured tumor weight and
IV. Discussion
At present, it has been well established that autophagy possesses important biological functions and has been also implicated closely in oncogenesis and cancer development (Mathew et al., 2007; Levine and Kroemer, 2008; Apel et al., 2009; Chen and Debnath, 2010). One particularly important and yet highly controversial issue in autophagy study is the role of autophagy in cell death (Gozuacik and Kimchi, 2007; Kroemer et al., 2008; Scarlatti et al., 2008). In general, autophagy serves as pro-survival function under starvation and several other cell stress condition and suppression of autophagy has been accepted as a novel therapeutic strategy in combination cancer therapy (Kourtis and Tavernarakis, 2008; Dalby et al., 2010). It is believed that the exact function of autophagy on cancer is depending on the various factors, including cell type, the subcellular environment, the nature and intensity of the stimulus, and the levels of autophagy induced (Levine, 2007; Brech et al., 2009; White and DiPaola, 2009). Ginsenosides are a class of steroid glycosides, and triterpene saponins, have been the target of research, as they are viewed as the active compounds behind the claims of ginseng's efficacy. Because ginsenosides appear to affect multiple signaling pathways, their effects are complex and it is unclear their action of mechanisms. In the present study, we provide convincing evidence suggesting that suppression effect of Rg3 on
autophagy is functionally related to its sensitization effect on a DNA-damage agent, doxorubicin and such a combination could serve as a novel strategy to improve cancer therapeutic efficiency, based on the following observation: First, we found that that Rg3
enhances autophagy markers by suppressing autophagic flux. Suppressing autophagic flux was analyzed by the tfLC3. Essentially, this method is based on the difference in the nature of the two fluorescent proteins (GFP and mRFP), i.e. mRFP is much more resistant to lysosomal quenching than GFP in the acidic environment. Therefore, if the autolysosome maturation proceeds normally, it would give rise to more red-only puncta. Reversely, if the autophagosome does not fuse with lysosome or the lysosome function
is blocked, most of the puncta should exhibit both red and green signals and appear to be yellow (Kimura et al., 2007). Based on this, our results provided evidence that Rg3 is
likely to inhibit autophagosome maturation and autolysosome formation. Although the underlying molecular mechanisms for Rg3’s effects on inhibition of late stage of
autophagic flux remain to be further investigated.
Secondly, Rg3 suppressed autophagy can contribute synergic effects on
doxorubicin-induced cell death. DNA damage induces autophagy and autophagy delays apoptotic cell death induced by the DNA-damaging agents. Doxorubicin has been recently shown to be paralleled by autophagy (Kobayashi et al., 2010) and low doses of doxorubicin induced autophagy and apoptosis simultaneously. For instance, in multiple myeloma cell lines, pharmacologic or genetic inhibition by knockdown of Beclin 1 or Atg5, markedly augmented doxorubicin-induced cell death (Pan et al., 2011). Consistent with this report, our results revealed that inhibition of autophagy by Rg3 treatment markedly
augmented doxorubicin-induced cell death in hepatocellular carcinoma cells in vitro, which was further evidenced by using mouse xenograft model. Therefore, suppression of autophagy becomes a potent strategy in combination therapy to overcome the resistance and enhance the chemotherapeutic efficacy. Although the role of autophagy in cancer cell fate is still controversial, there is growing evidence that autophagy plays a pro-survival mechanism in cancer cells and there is an urgent need to develop specific autophagy inhibitors that are clinically viable. At present, few inhibitors of autophagy are used in autophagy research and CQ is the only autophagy inhibitor in clinical trial (Rubinsztein et al., 2007). Based on the above, Rg3 could be further developed as
References
1. Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB: Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 117: 326-336, 2007
2. Apel A, Zentgraf H, Buchler MW, Herr I: Autophagy-A double-edged sword in oncology. Int J Cancer 125: 991-995, 2009
3. Aries A, Paradis P, Lefebvre C, Schwartz RJ, Nemer M: Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proceedings of the National
Academy of Sciences of the United States of America 101: 6975, 2004
4. Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A, Stenmark H, Johansen T: p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. The Journal of Cell
Biology 171: 603, 2005
5. Brech A, Ahlquist T, Lothe R, Stenmark H: Autophagy in tumour suppression and promotion. Molecular Oncology 3: 366-375, 2009
6. Cabibbo G, Latteri F, Antonucci M, Craxì A: Multimodal approaches to the treatment of hepatocellular carcinoma. Nature Clinical Practice Gastroenterology & Hepatology 6: 159-169, 2009
7. Chen N, Debnath J: Autophagy and tumorigenesis. FEBS Lett 584: 1427-1435, 2010
8. Chen Z, Lu T, Yue X, Wei N, Jiang Y, Chen M, Ni G, Liu X, Xu G: Neuroprotective effect of ginsenoside Rb1 on glutamate-induced neurotoxicity: With emphasis on autophagy. Neuroscience letters 482: 264-268, 2010
9. Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B: Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer.
10. Feng Z, Zhang H, Levine AJ, Jin S: The coordinate regulation of the p53 and mTOR pathways in cells. Proceedings of the National Academy of Sciences of
the United States of America 102: 8204, 2005
11. Gozuacik D, Kimchi A: Autophagy and cell death. Current topics in
developmental biology 78: 217-245, 2007
12. Hosokawa N, Hara Y, Mizushima N: Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS
letters 580: 2623-2629, 2006
13. Katayama H, Yamamoto A, Mizushima N, Yoshimori T, Miyawaki A: GFP-like proteins stably accumulate in lysosomes. Cell structure and function: 802040006, 2007
14. Keum YS, Han SS, Chun KS, Park KK, Park JH, Lee SK, Surh YJ: Inhibitory effects of the ginsenoside Rg3 on phorbol ester-induced cyclooxygenase-2 expression, NF-kappaB activation and tumor promotion. Mutat Res 523-524: 75-85, 2003
15. Kim Y, Ma AG, Kitta K, Fitch SN, Ikeda T, Ihara Y, Simon AR, Evans T, Suzuki YJ: Anthracycline-induced suppression of GATA-4 transcription factor: implication in the regulation of cardiac myocyte apoptosis. Molecular
pharmacology 63: 368-377, 2003
16. Kimura S, Noda T, Yoshimori T: Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3: 452, 2007
17. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A: Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes.
19. Kobayashi S, Volden P, Timm D, Mao K, Xu X, Liang Q: Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death.
Journal of Biological Chemistry 285: 793, 2010
20. Kourtis N, Tavernarakis N: Autophagy and cell death in model organisms. Cell
Death & Differentiation 16: 21-30, 2008
21. Kroemer G, Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC: To die or not to die: that is the autophagic question. Current molecular medicine 8: 78-91, 2008
22. Kroemer G, Levine B: Autophagic cell death: the story of a misnomer. Nat Rev
Mol Cell Biol 9: 1004-1010, 2008
23. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N: The role of autophagy during the early neonatal starvation period. Nature 432: 1032-1036, 2004
24. Kumarapeli ARK, Horak KM, Glasford JW, Li J, Chen Q, Liu J, Zheng H, Wang X: A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. The FASEB journal 19: 2051-2053, 2005
25. Lambert LA, Qiao N, Hunt KK, Lambert DH, Mills GB, Meijer L, Keyomarsi K: Autophagy: a novel mechanism of synergistic cytotoxicity between doxorubicin and roscovitine in a sarcoma model. Cancer research 68: 7966, 2008
26. Lee T, Lau T, Ng I: Doxorubicin-induced apoptosis and chemosensitivity in hepatoma cell lines. Cancer chemotherapy and pharmacology 49: 78-86, 2002 27. Levine B: Cell biology: autophagy and cancer. Nature 446: 745-747, 2007 28. Levine B, Kroemer G: Autophagy in the pathogenesis of disease. Cell 132:
27-42, 2008
29. Levine B, Yuan J: Autophagy in cell death: an innocent convict? J Clin Invest 115: 2679-2688, 2005
30. Li J, Hou N, Faried A, Tsutsumi S, Kuwano H: Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in
vivo model. European Journal of Cancer 46: 1900-1909, 2010
31. Li L, Takemura G, Li Y, Miyata S, Esaki M, Okada H, Kanamori H, Khai NC, Maruyama R, Ogino A: Preventive effect of erythropoietin on cardiac dysfunction in doxorubicin-induced cardiomyopathy. Circulation 113: 535-543, 2006
32. Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, Suter TM, Liao R, Sawyer DB: Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. Journal of Biological Chemistry 279: 8290, 2004
33. Mathew R, Karantza-Wadsworth V, White E: Role of autophagy in cancer. Nat
Rev Cancer 7: 961-967, 2007
34. Mizushima N: Methods for monitoring autophagy. The international journal of
biochemistry & cell biology 36: 2491-2502, 2004
35. Mizushima N, Klionsky DJ: Protein Turnover Via Autophagy: Implications for Metabolism*. Annu. Rev. Nutr. 27: 19-40, 2007
36. Mizushima N, Levine B, Cuervo A, Klionsky D: Autophagy fights disease through cellular self-digestion. Nature 451: 1069, 2008
37. Mizushima N, Yoshimori T: How to interpret LC3 immunoblotting. Autophagy 3: 542, 2007
38. Oliveira PJ, Wallace KB: Depletion of adenine nucleotide translocator protein in heart mitochondria from doxorubicin-treated rats--relevance for mitochondrial dysfunction. Toxicology 220: 160-168, 2006
39. Olson RD, Gambliel HA, Vestal RE, Shadle SE, Charlier HA, Cusack BJ: Doxorubicin cardiac dysfunction. Cardiovascular Toxicology 5: 269-283, 2005 40. Pan Y, Gao Y, Chen L, Gao G, Dong H, Yang Y, Dong B, Chen X: Targeting
DNA-degradation of ubiquitinated protein aggregates by autophagy. Journal of
Biological Chemistry 282: 24131, 2007
42. Rubinsztein D, Gestwicki J, Murphy L, Klionsky D: Potential therapeutic applications of autophagy. Nature Reviews Drug Discovery 6: 304-312, 2007 43. Scarlatti F, Granata R, Meijer A, Codogno P: Does autophagy have a license to
kill mammalian cells? Cell Death & Differ 16: 12-20, 2008
44. Scarlatti F, Granata R, Meijer AJ, Codogno P: Does autophagy have a license to kill mammalian cells? Cell Death Differ 16: 12-20, 2009
45. Semenov D, Lushnikova E, Nepomnyashchikh L: Anthracycline-induced cardiomyopathy is manifested in decreased protein synthesis, impaired intracellular regeneration, and non-necrotic death of cardiomyocytes. Bulletin of
experimental biology and medicine 131: 505-510, 2001
46. Shen HM, Codogno P: Autophagic cell death: Loch Ness monster or endangered species? Autophagy 7, 2011
47. Shintani T, Klionsky DJ: Autophagy in health and disease: a double-edged sword. Science 306: 990, 2004
48. Takimoto CH, Awada A: Safety and anti-tumor activity of sorafenib (Nexavar®) in combination with other anti-cancer agents: a review of clinical trials. Cancer
chemotherapy and pharmacology 61: 535-548, 2008
49. White E, DiPaola RS: The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 15: 5308-5316, 2009
50. Wu Y, Tan H, Huang Q, Kim Y, Pan N, Ong W, Liu Z, Ong C, Shen H: Autophagy plays a protective role during zVAD-induced necrotic cell death.
Autophagy 4: 457, 2008
51. Yue Z, Jin S, Yang C, Levine AJ, Heintz N: Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proceedings of the National Academy of Sciences 100: 15077, 2003
- 국문요약 -
간암세포에서
Rg
3에
의한 자가포식작용 조절을 통한
DNA-손상 약물 매개 세포사의 감작
아주대학교 대학원 의생명과학과 김 동 건 (지도교수 : 김 유 선) Autophagy 는 세포사멸에 있어서 그 역할이 상반되는 의견이 제시되고 있지만 최근 많은 연구에 의하면 암세포에 있어서 다양한 스트레스 조건, 즉 항암제 처리와 같은 세포사멸 조건에서는 세포의 생존 전략으로 이해되고 있다. Ginsenoside Rg3 는 홍삼에서 추출한 높은 약리활성을 가진 사포닌 계열로 항암화학물의 효율을 증가시키는 능력이 있음이 알려져 있다. 본 연구를 통해, Rg3 의 autophagy 조절 능력을 확인하였고 이러한 Rg3 가 간암 세포에서 DNA 손상 약물로 인한 세포 사멸을 촉진시킨다는 것을 확인하였다. Rg3는 autophagy 마커인 LC3 II 변환과 GFP-LC3 punctuation 을 증가시켰으며 p62 단백질의 축적을 일으키는 것을 확인하였고, 이러한 autophagy marker 의 증가는 autophagy 억제제인 chloroquine 에 의해 변화가 없는 것으로 확인되어 Rg3 는 autophagosome 의 숙성과 분해를 저해하여 autophagicflux 를 억제함으로써 autophagy markers 의 증가를 보인 것으로 확인되었다. 그리고 Rg3 는 DNA-손상 약물인 doxorubicin 에 의한 세포 사멸이 다양한 간암 세포주에서
증가됨이 관찰되어 Rg3는 암세포 내에서 auotphagy 를 억제하여 doxorubicin 에 의한
세포 사멸을 촉진시킨다는 사실을 발견하였고 mouse xenograft model 에서도 Rg3 와
doxorubicin 의 병합에 의한 종양성장억제가 확인되었다. 이상의 연구를 바탕으로 Rg3 에 의한 autophagy 억제 현상은 doxorubicin 과 병합을 통해 caspase-비의존적
세포사멸을 촉진시키며 이러한 병합은 간암의 약물치료에 새로운 전략으로 사용될 가능성을 제시하고 있다.
핵심어 : Autophagy, Hepatocellular carcinoma, Ginsenoside Rg3, DNA damage agent