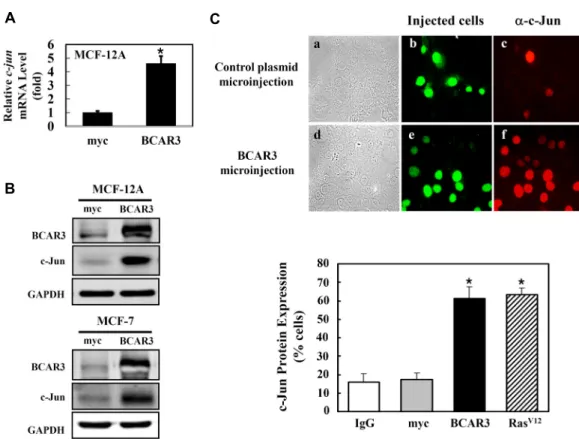
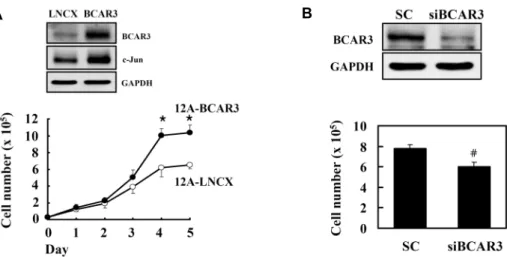
Induction of c-Jun Expression by Breast Cancer Anti-estrogen Resistance-3 (BCAR3) in Human Breast MCF-12A Cells
Myung-Ju Oh
2, Ji-Hyun Kim
1and Byung Hak Jhun
1*
1
College of Nanoscience and Nanotechnology, Pusan National University, Miryang, Geongnam 727-706, Korea
2