Dongsoo Koh
Department of Applied Chemistry, Dongduk Women's University, Seoul 136-714, Korea Received March 8, 2004; Accepted June 3, 2004
An enantiomeric pair of C2-symmetric (R, R)- and (S, S)-enonates (6,12) was synthesized from D-(-)- and L-(+)-arabinose respectively as optically active starting materials. Chain extended sugar enonate 3 was obtained by Wittig reaction of aldehyde group of D-(-)-arabinose without protection. After Wittig reaction, tetraol of sugar moiety was protected with dimethoxypropane. Selectively partial deprotection at terminal hydroxyl group was made with Dowex resin. Diol cleavage followed by Wittig reaction gave chiral C2-symmetric (R, R)-enonate 6. The same (S, S)-enonate (12) with opposite configurations was prepared from L-(+)-arabinose.
Key words: C2-symmetric, enonate, enantiomer, Wittig reaction, arabinose, ylide.
C2-symmetric compounds have attracted much attention because of their potential ability for excellent stereoregulation.
The optically active C2-symmetric compounds make highly advantageous chiral environmental in a variety of stoichiometric and catalytic asymmetric syntheses1) and have emerged as versatile auxiliaries in many asymmetric transformations.2,3) In order to obtain optically active C2- symmetric compounds, naturally occurring chiral pools Chirons have been widely applied due to the reliability on their stereogenic centers.4) Different starting materials, however, have been applied to obtain enantiomeric C2- symmetric compounds with opposite configurations at stereocenters each other. As part of a program aimed at investigating whether the chirality at γ-position of α, β- unsaturated esters can control the diastereofacial selectivity of asymmetric Diels-Alder reaction,5-7) convenient syntheses of C2-symmetric chiral ,-unsaturated esters(6, 12) from the same starting material, both naturally occurring D- and L-arabinose, are reported here (Fig. 1).
Materials and Methods
General methods. T.l.c. was performed on precoated glass plates of Silica Gel 60F-254 (E. Merck), and compounds on the plate were detected by spraying with 10% aq H2SO4
solution with subsequent heating or irradiation of UV-light.
Flash-column chromatography was performed on 230-300 mesh silica gel (Merck) as described in the literature.8) Melting points were determined using a Thomas-Hoover Unimelt
apparatus. Optical rotations were measured with a Perkin- Elmer model 141 polarimeter at 25oC unless otherwise noted.
1H-NMR and 13C-NMR spectra were recorded using a Bruker Avance 400 spectrometer system (9.4 T) at a temperature of 298K in CDCl3 with TMS as an internal reference, unless otherwise specified. Splitting patterns are designated as: s, singlet; d, doublet; dd, double doublet; t, triplet; m, multiplet.
Methyl (Triphenylphosphoranylidene)acetate (2). To a clear solution of methyl bromoacetate (51.84 g, 339 mmol) in diethyl ether (800 ml) was added triphenylphospine (90.5 g, 345 mmol) portionwise and stirred for 24 h at room temperature. The precipitation formed was filtered and washed with additional ether (200 ml) to give 113.8 g of phosphonium salt. The solid was dissolved in chloroform (400 ml). To the above clear solution was slowly added aqueous NaOH solution (52 g/450 ml H2O) with vigorously stirring for 30 min. The aqueous layer was wash twice with chloroform (2
×50 ml) and combined organic layer was dried over anhydrous MgSO4. Filtration and evaporation gave ylide 2, (97.5 g, 86%, for two step reaction). m.p.; 168-169oC, lit9) m.p.; 169-171oC. The phosphonium salt intermediate does not need to be isolated for better yield and more convenience.
Therefore, modification was made as follow. Methyl bromoacetate (3.7 g, 24.2 mmol) in chloroform (50 ml) was treated with triphenylphospine (6.46 g, 24.6 mmol) at room temperature overnight. To the solution was added aqueous NaOH solution (4 g/50 ml H2O) and stirred for 20 min. The aqueous layer was wash chloroform (20 ml) and combined organic layer was dried over anhydrous MgSO4. Filtration and evaporation gave ylide 2, (7.76 g, 96%).
Methyl (trans)-2,3-dideoxy-D-arabino-hept-2-enonate (3).
To a suspension of unprotected D-(-)-arabinose 1 (9 g, 60 mmol) in THF (80 ml) was added ylide 2 (23.4 g, 70 mmol) and catalytic amounts (40 mg) of benzoic acid. The mixture was refluxed for 1 h when it turned into clear solution. After
*Corresponding author
Phone: +82-2-940-4512; Fax: +82-2-940-4193 E-mail: [email protected]
Abbreviations: T.l.c., thin layer chromatography; THF, Tetrahydro- furane; DMSO, Dimethylsulfoxide; NMR, nuclear magnetic resonance
additional 3 h refluxing, the mixture was cooled at room temperature. The solvent was evaporated to give a white solid, which was partitioned between H2O (200 ml) and chloroform (300 ml). Desired product sugar enonate was dissolved into water layer, and by-product triphenylphospine oxide was soluble in organic layer. Water layer was washed with chloroform (100 ml) and evaporated to give a white solid.
Recrystallization from ethanol gave the pure trans-enonate 3, (10.25 g, 83%). [α]D; +8.7o, (c 1.3, H2O), m.p.; 161oC, 1H- NMR (400 MHz, DMSO-d6); δ6.94 (dd, 1H, J3,4 4.2 Hz, H-3), 5.98 (dd, 1H, J2,3 15.7 Hz, J2,4 1.9 Hz, H-2), 5.16 (d, 1H, J6.4 Hz, OH), 4.93 (d, 1H, J 5.5 Hz, OH), 4.82 (d, 1H, J 7,1 Hz, OH), 4.77 (t, 1H, J 5.6 Hz, OH), 4.40 (m, 1H, H-4), 3.61 (s, 3H, OCH3), 3.58 (m, 1H), 3.49 (m, 1H), 3.32-3.41 (m, 2H),
13C-NMR (100 MHz, DMSO-d6); δ165.7, 151.7, 119.9, 73.4, 71.7, 70,3, 63.7, 51.7. The enantiomer 9, derived from L-(+)- arabinose, showed the same spectral data except opposite optical rotation [α]D; −8.2o, (c 1.1, H2O)
Methyl (trans)-4,5;6,7-di-O-isopropylidene-2,3-dideoxy- D-arabino-hept-2-enonate (4). To a mixture of ester 3 (1.44 g, 7 mmol), Drierite (3.5 g) in THF (40 ml) was added catalytic amounts of p-toluenesulfonic acid (50 mg) and stirred 30 min at room temperature. 2,2-Dimethoxypropane (4.37 g, 21 mmol) was added to the mixture and stirring was continued overnight. Then 0.15 g of NaHCO3 was added and mixture was filtered and washed with methanol (50 ml).
Concentration of filtrates afforded a residue, which was passed short column of silica gel to give diisopropylidene enonate 4 (1.86 g, 90%). [α]D; −1.7o, (c 1.2, CHCl3), 1H-NMR (400 MHz); δ6.97 (dd, 1H, J2,4 1.7 Hz, H-3), 6.13 (dd, 1H, J2,3
15.7 Hz, H-2), 4.49 (ddd, 1H, J3,4 4.5 Hz, H-4), 4.02-4.11 (m, 2H, H-7,7), 3.90 (m, 1H, J5,6 3.6 Hz, H-6), 3.70 (s, 3H, OCH3), 3.64 (t, 1H, J4,5 7.7 Hz, H-5), 1.30-1.37 (4s, 12H, 4CH3), 13C- NMR (100 MHz); δ166.5, 145.3, 120.9, 110.2, 109.8, 81.8, 76.9, 78.9, 67.4, 51.5, 26.9,26.7, 26.0, 25.3. The enantiomer 10 had [α]D; +1.5o, (c 1.1, CHCl3).
Methyl (trans)-4,5-O-isopropylidene-2,3-dideoxy-D- arabino-hept-2-enonate (5). The diisopropylidenated enonate 4 (3.31 g, 1.15 mmol) was dissolved in aqueous methanol (H2O : MeOH = 1 : 9, 100 ml). To the above clear solution was added Dowex 50W-X8 resin (4 g) and resultant suspension was stirred 22 h at room temperature. T.l.c.
showed no starting material and one major product (Rf 0.8, 3 : 2 Ethyl acetate- Hexane) and one minor product (Rf 0.05, 3 : 2 Ethyl acetate- Hexane). The reaction mixture was filtered and filtrate was evaporated to give a syrup, which was dissolved in
methylene chloride and water (CHCl2, 60 ml - H2O, 40 ml).
The major product was dissolved into organic layer and the minor one dissolved into water layer. The organic layer was dried over anhydrous MgSO4. Filtration and concentration of filtrates afforded syrup of a monoisopropylidene enonate 5 (2.21 g, 76%). 1H-NMR (400 MHz); δ7.03 (dd, 1H, J3,4 4.6 Hz, H-3), 6.19 (dd, 1H, J2,3 15.7 Hz, J2,4 1.7 Hz, H-2), 4.62 (ddd, 1H, H-4), 3.77-3.80 (m, 3H), 3.75 (s, 3H, OCH3), 3.62- 3.66 (m, 1H), 1.43 (s, 3H, CH3), 1.40 (s, 3H, CH3), 13C-NMR (100 MHz); δ167.50, 146.46, 121.32, 110.49, 80.66, 78.52, 73.36, 64.09, 52.13, 27.25, 26.95
Methyl-(trans, trans)-4S,5S-α, β-unsaturated diester (6).
The monoisopropylidene enonate 5 (492 mg, 2 mmol) was dissolved in methanol (40 ml), and aqueous solution of sodium periodate (470 mg, 2.2 mmol in 10 ml H2O) was added. After 2 h stirring at room temperature, insoluble solids were removed. The filtrate was concentrated under reduced pressure. The residue was triturated with chloroform (40 ml), and insoluble solids were removed. The residue was dissolved THF (50 ml), and ylide 2 (835 mg, 2.5 mmol) was added. The mixture was refluxed for 3 h and solvent was removed under reduced pressure. Diethyl ether was added to the residue, and insoluble by-product triphenylphospine oxide was filtered.
Filtration and concentration of filtrates afforded syrup, which was purified by chromatography to give dienonate 6 (362 mg, 71%). [α]D; +67.6o, (c 1.8, CHCl3), 1H-NMR (400 MHz);
δ6.79 (m, 2H), 6.08 (dd, 2H, J2,3 15.8 Hz, J2,4 1.5 Hz), 4.28 (m, 2H), 3.72 (s, 6H), 1.42 (s, 6H), 13C-NMR (100 MHz); δ166.1, 142.2 122.4, 110.3, 80.2, 52.2 26.7. The enantiomer 12 had [α]D; −68.4o, (c 1.4, CHCl3).
(4R, 5R)-4,5-(Isopropylidene)dioxy-1,8-octandiol. A suspension of dienonate 6 (306 g, 1.2 mmol) and 10%
palladium-charcoal (30 mg) in methanol (30 ml) was stirred under H2 atmosphere for 10 h. Removal of catalyst by filtration through Celite. Filtrate was concentrated under reduced pressure to give crude diester, which was used for the next step without further purification. To a solution of diester in ether (30 ml) was added LiAlH4 (76 mg, 2 mmol) in ether (10 ml) under ice-bath. After stirring 10 h at room temperature, the mixture was quenched with H2O (0.5 ml).
Insoluble solids were removed and washed with ether. The combined filtrate and washings were concentrated in vacuo.
The residue was chromatographed on silica gel to give diol 7 as an oil (230 g, 87%). [α]D; +28.2o, (c 1.1, CHCl3), lit10) [α]D; +29o, (c 0.51, CHCl3), 1H-NMR (400 MHz); δ3.62-3.67 (m, 6H), 2.34 (bs, 4H), 1.48-1.85 (m, 8H), 1.41 (s, 6H), 13C-NMR Fig. 1. Structures of enantiomeric pair of C2-symmetric (R, R)- and (S, S)-enonates 6 and 12.
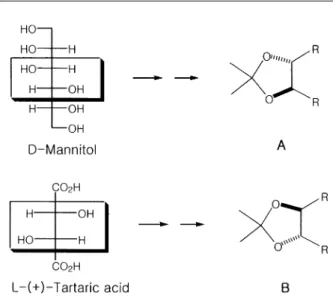
As shown in Fig. 2, different starting materials are usually required to produce enantiomeric pair of C2-symmetric molecules A and B. (Fig. 2). D-Mannitol, obtainable from naturally occurring D-mannose by one step reduction, is usually applied to produce building block A. Naturally abundant L-(+)-tartaric acid, however, is transformed to its enantiomer B. If different starting material is applied, all chemical procedures must be altered. Therefore it is desirable to obtain a pair of enantiomeric building blocks from the same starting material with opposite optical rotation. Arabinose, inexpensively commecial available in both D-(-) and L-(+) form, was the choice of starting material.
D-(-)-Arabinose (1), without any protection, was elongated Fig. 2. Structures of naturally abundant Chirons D-manni-
tol and L-tartaric acid and their use in chiral C2-symmetric compounds as enantiomeric pairs.
Fig. 3. Synthetic methods for C2-symmetric (R, R)-enonates 6 from D-(-)-arabinose.
by Wittig reaction. (Fig. 3).
It was treated with methyl (triphenylphosphoranylidene) acetate (2) in refluxing THF with catalytic amounts of benzoic acid.11) Trans enonate 3 with D-arabino configuration was obtained in 92% crude yield. After recrystallization in ethanol, analytically pure product was made in 83% yield. The ylide 2 was conveniently prepared from the reaction between methyl bromoacetate and triphenylphosphine, followed by treatmentment with aqueous NaOH solution, with slight modification of literature method.9) Protection of tetraol in 3 was performed by diisopropylidenation with 2,2- dimethoxypropane. Fully protected diisopropylidene enonate 4 was converted to monoisopropylidene enonate 5 by selective deprotection. Among the partial, selective deprotecting methods,12) Dowex resin method13) was good for enonate 4. It provided simple and selective deprotection of diol at the position including primary hydroxyl group. Monoisopropylidene enonate 5 was transformed to desired C2-symmetric dienonate by diol-cleavage and subsequent Wittig chain elongation.
Conventional diol cleavage with lead tetraacetate14,15) was not
good for diol 5, affording poor yield of aldehyde. Instead, reaction of diol with sodium periodate16,17) in aqueous methanol afforded improved yield of aldehyde, which was sequentially treated with ylide 5 in refluxing THF to produce C2-symmetric enonate. The resultant products showed trans configuration at newly formed C=C double bond as a major, contaminated with cis minor by NMR.18) In order to confirm the optical purity, the C2-symmetric enonate 6 was transformed to known diol 7. Hydrogenation of C=C double bond in 6 and followed by reduction with LiAlH4 gave corresponding diol in good yield. Optical rotation of diol 7, [α]D; +28.2o, (c 1.1, CHCl3), showed good agreement with literature10) [α]D; +29o, (c 0.51, CHCl3).
The same procedures were used as described for D- arabinose, but starting from L-arabinose (Fig.4). The desired , -unsaturated esters (12) with opposite configuration of stereocenters was obtained in the same manner. Throughout the chemical transformations, the configurations of stereocenters at 2,3-position of arabinose (see box in Fig. 3 and 4) were maintained, providing C2-symmetric compounds Fig. 4. Synthetic methods for C2-symmetric (S, S)-enonates 12 from L-(+)-arabinose.
tion. Chem. Rev. 89, 1581-1590.
2. Blaser, H. U. (1992) The chiral pool as a source of enanti- oselective catalysts and auxiliaries. Chem. Rev. 92, 935-952.
3. Koh, D. and Moon, H. (1998) Synthesis and determination of optical purity of C2-symmetric pyrrolidine amides as chiral auxiliaries. J. of Korean Ind. & Eng. Chemistry 9, 914-919.
4. Henessian, S. (1983) Total synthesis of natural products:
The Chiron approach, Pergamon Press.
5. Horton, D., Koh, D., Takagi, Y. and Usui, T. (1992) Stereo- control in Diels-Alder cycloaddition to unsaturated sugars as a function of structure and stereochemistry. American Chemical society Symposium Series 494, 66-80.
6. Horton, D. and Koh, D. (1993) Generalized approach from sugars to enantiomerically pure tetra-C-substituted carbocy- cles. Tetrahedron Lett. 34, 2283-2287.
7. Horton, D., Koh, D. and Takagi, Y. (1993) Stereocontrol in Diels-Alder cycloaddition to unsaturated sugars: Reactivi- ties of cis dienophiles with cyclopentadiene. Carbohydr.
Res. 250, 261-274.
8. Still, W. C., Kahn. M. and Mitra, A. (1978) Rapid chro- matographic technique for preparative separation with mod- erate resolution. J. Org. Chem. 43, 2923-2925.
9. Railton, C. J. and Clive, D. L. J. (1996) Wittig chain exten- sion of unprotected carbohydrates: formation of carbohy- drate-derived , -unsaturated esters. Carbohydr. Res. 281, 69- 77.
10. Ruan, Z. and Mootoo, D. R. (1999) A novel desymmetriza- tion reaction of an acetogenin precursor: A formal synthe-
group, including 1,2- and 1,3-diols (3rd ed.) Wiley Inter- science.
13. Park, K. H., Yoon, Y. J. and Lee, S. G. (1994) Efficient cleavage of terminal acetonide group: Chirospecific synthe- sis of 2,4-dideoxy-2,5-imino-d-mannitol. Tetrahedron Lett.
35, 9737-9790.
14. Pianetti, P., Rollin, P. and Pougny, J. R. (1986) Optically active propargylic alcohols from D-xylose useful precursors for LTB4 synthesis Tetrahedron Lett. 27, 5853-5856.
15. Chen, S.-H., Horvath, R., Joglar, J, Fisher, M. and Dan- ishefsky, D. (1991) Application of the Ibuka-Yamamoto reaction to a problem in stereochemocal communication : A strategy for the stereospecific synthesis and stabilization of the triene substructure of rapamycin through sulfone substi- tution. J. Org. Chem. 56, 5834-5845.
16. Schmid, C. R., Bryant, J. D., Dowlatzedah, M., Phillips, J.
L., Prather, D. E., Schantz, R. D., Sear, N. L. and Vianco, C. S. (1991) Synthesis of 2,3-O-isopropylidene-D-glyceral- dehyde in high chemical and optical purity: Observations on the development of a practical bulk process. J. Org. Chem.
27, 4056-4-58.
17. Audia, J. E., Boisvert, L., Patten, A. D., Villalobos, A. and Danishefsky, D. (1989) Synthesis of two useful, enantiomer- ically pure derivatives of (S)-4-hydroxy-2-cyclohexenone. J.
Org. Chem. 54, 3738-3740.
18. Harcken, C. and Martin, S. F. (2001) Improved E-selectiv- ity in the Wittig reaction of stabilized ylides with α-alkoxy- aldehydes and sugar lactols. Org. Lett. 3, 3591-3593.