충남대학교 바이오응용화학과
(2013년 1월 16일 접수, 2013년 1월 28일 심사, 2013년 2월 12일 채택)
Effect of Boric Acid Treatment on the Electrochemical Properties of the Phenol-Based Activated Carbon
Min-Jung Jung, Hye-Ryeon Yu, Dayoung Lee, and Young-Seak Lee†
Department of Applied Chemistry and Biological Engineering, Chungnam National University, Daejeon 305-764, Korea (Received January 16, 2013; Revised January 28, 2013; Accepted February 12, 2013)
본 연구에서는 전기이중층 커패시터의 전극 활물질로 사용되는 페놀계 활성탄소의 비 정전용량의 증가를 위하여 붕 산을 이용하여 표면처리를 수행하였다. 또한, 붕산 처리가 전기화학 특성에 미치는 영향에 대하여 고찰하였다. 활성 탄소의 붕산 처리는 활성탄소의 표면에 전기화학적 특성 향상에 도움이 되는 퀴논형 관능기(O=C)의 비율을 효과적으로 증가시켰으며, 비표면적과 총 기공 부피 및 미세공 부피를 증가시켰다. 최적의 조건으로 붕산 처리된 활성탄소는 미처리 활성탄소에 비해 비 정전용량이 약 20% 증가하였다. 이러한 결과로부터 활성탄소의 붕산 처리는 활성탄소의 비 정전 용량을 효과적으로 증가시킬 수 있다고 사료된다.
In this study, the surface of a phenol based activated carbon (AC) used as an electrode in an electric double layer capacitor was modified via boric acid treatment for the capacitance investigation. The effect of boric acid treatment on electrochemical performance was also investigated. The AC surface functional groups ratio of quinone-like (O=C) which is electrochemical active functional groups was increased after the boric acid treatment. And, boric acid treated AC showed an increase in the specific surface area, total pore volume, and micropore volume. In case of optimum boric acid treated AC, its specific capaci- tance increased by 20% in comparison to that of untreated AC. These results demonstrate that a boric acid treated carbon surface-based electric double layer capacitor electrode effectively enhances specific capacitance.
Keywords: phenol based activated carbon, surface treatment, boric acid, electric double-layer capacitors
1. 서 론
1)
정보통신 기술의 비약적인 진보에 의해서 스마트폰 등의 모바일 기 기가 급속하게 보급되었고 유비쿼터스 네트워크 시대를 향한 고기능 화가 진행되고 있다. 또한 동시에 지구 온난화로 대표되는 환경문제에 대한 사회적인 연구로서 하이브리드 자동차나 연료전지 자동차의 실 용화 등의 연구도 활발하게 이루어지고 있다. 이러한 세계적인 사회 변화 중에 새로운 기술을 지지하는 전원으로서 전기 이중층 커패시터 (EDLC, electric double layer capacitor)가 주목을 받고 있다[1-4].
EDLC는 전지와 달리 충⋅방전이 활성탄소 표면에 이온들이 물리 적으로 흡⋅탈착하는 원리로 충⋅방전이 일어나는 것으로, 많은 에너 지를 모아두었다가 수초 또는 수십 초 동안에 높은 출력의 에너지를
† Corresponding Author: Chungnam National University Department of Applied Chemistry and Biological Engineering 220 Gung-dong, Yuseong-gu, Daejeon 305-764, Korea Tel: +82-42-821-7007 e-mail: [email protected]
pISSN: 1225-0112 @ 2013 The Korean Society of Industrial and Engineering Chemistry.
All rights reserved.
발산할 수 있다. 또한 짧은 충전시간, 긴 수명 등의 특성을 가지고 있다[5,6]. 그러나 이런 장점에도 불구하고 에너지 밀도가 적어 휴대 통신기기 및 가전전자제품의 메모리 백업용 전원으로의 활용분야가 제한된다[7]. 이러한 문제를 해결하고자 ELDC 전극 물질로 일반적으 로 사용되고 있는 활성탄소보다 더 큰 정전용량을 가지는 소재를 개 발하기 위하여 금속 산화물[8-10]이나 전도성 고분자[11-13]를 이용한 의사커패시터(pseudo-capacitor)용 전극 물질에 대한 연구가 많이 이루 어졌다. 그러나 2000년대에 들어서 다시 탄소재를 이용한 전극 물질 의 연구가 활발히 이루어지고 있는 실정이다[14].
EDLC는 분리막, 양극, 음극, 집전체, 전해질로 구성되어 있다. 이 중에서 EDLC의 핵심을 이루는 기술은 분극성 물질을 사용하는 전극 분야이다. 전기이중층 커패시터의 정전용량을 증가시키기 위해서 가 장 핵심이 되는 부분은 전극 활물질의 개선이다. 전극 활물질은 최소 의 전압강하가 이루도록 전기전도성이 크고, 비표면적이 크며 일정전 위에서 산화⋅환원 반응이 일어나지 않는 전기 화학적으로 안정한 물 질이어야 한다[15]. 이러한 기준으로 볼 때 탄소재료가 가장 적합한 재료로 여겨지고 있다. 이는 활성탄소가 비표면적이 높고 전기전도도 가 크며 전기화학적으로 안정하기 때문이다[16-18]. 따라서 활성탄소
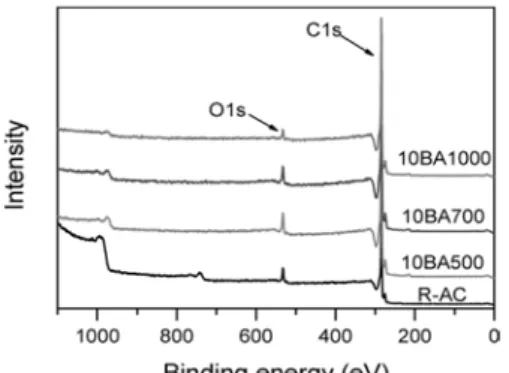
Figure 1. XPS spectra of untreated and boric acid treated activated carbon.
Table 1. XPS Surface Elemental-analysis Parameters for Untreated and Boric Acid Treated Activated Carbon
Sample
Elemental contents
(Atomic percent) O/C (%)
B/C
C O B (%)
R-AC 92.1 7.9 - 8.58 -
10BA500 94.7 5.3 - 5.60 -
10BA700 95.9 4.1 - 4.28 -
10BA1000 97.4 2.6 - 2.67 -
5BA700 96.7 3.3 - 3.41 -
20BA700 97.6 2.4 - 2.46 -
50BA700 94.2 4.5 1.3 4.78 1.38
의 구조 및 물성이 EDLC에 적합하도록 제조하는 것은 매우 중요한 일이며 이를 위한 다양한 방법들이 고안되고 있다. 위에서 설명하였 듯이, 전기이중층 커패시터는 전해질 내부의 이온이 전극 표면에 물 리적으로 흡⋅탈착되는 장치이므로 활물질 표면의 표면특성이 매우 중요하다[19-21]. 따라서 표면개질을 통한 탄소재 활물질의 표면에 관 능기를 도입하는 것은 활성탄소의 물성 변화로 인해 탄소재료의 전기 화학적 특성을 변화시켜 전기이중층 커패시터에 적합한 재료로 제조 하는 것이 가능하므로 이와 연관된 연구가 활발히 진행되고 있다 [22,23].
따라서 본 연구에서는 활물질인 활성탄소를 붕산으로 표면처리하고, 반응 온도 및 붕산과 활성탄소의 비율 변화가 활성탄소의 표면화학 특성 및 기공 특성 변화에 어떠한 영향을 주는지에 대하여 고찰하고자 한다. 또한 붕산 처리된 활성탄소의 전기화학적 특성변화에 대해 고찰 하고, EDLC 전극재료로서의 이용가능성에 대하여 평가하고자 한다.
2. 실 험
2.1. 실험 재료
본 실험에서는 활성탄소에 붕산 처리를 위하여 페놀계 활성탄소 (MSP-20, Kansai Coke & Chemicals Co.), 붕산(Samchun Chemicals, 99.5%), 메탄올(Samchun Chemicals)을 사용하였다. 또한 전극 제조를 위하여 활물질로는 미처리 및 붕산 처리된 페놀계 활성탄소를, 도전 제로 카본블랙(Super P, Timcal Ltd.), 결합제로 Polyvinylidene fluoride (PVDF, Aldrich)를 사용하였다.
2.2. 페놀계 활성탄소의 붕산 처리
붕산을 이용한 페놀계 활성탄소 표면 처리를 위하여 활성탄소와 붕 산, 메탄올을 상온에서 1 h 동안 교반하였다. 이때 붕산의 양은 활성 탄소 대비 각각 5, 10, 20, 50 wt%로 하였고, 메탄올은 활성탄소 1 g 당 10 g으로 고정하였다. 교반 후 100 ℃에서 24 h 동안 건조하고, 건 조된 활성탄소는 불활성 분위기에서 5 ℃/min으로 승온하여 각각 500, 700, 1000 ℃의 온도에서 1 h 동안 열처리를 하였다. 처리하지 않은 활성탄소는 R-AC로, 붕산 처리된 활성탄소는 XBAY의 형식으로 명 명하였는데 여기서 X는 활성탄소 대비 붕산의 비율을 의미하고 Y는 반응 온도를 의미한다.
2.3. 특성평가
붕산 처리에 따른 페놀계 활성탄소의 표면 관능기 변화를 확인하기 위하여 X-ray photoelectron spectroscopy (XPS, MultiLab 2000, Thermo) 분석과 Energy dispersive spectroscopy mapping (EDX mapping, S-4700,
Hitachi) 분석을 수행하였다. 또한, 붕산 처리에 따른 페놀계 활성탄소 의 기공특성 변화를 확인하기 위하여 액체질소 온도(-196 ℃) 하에서 질소기체 흡ㆍ탈착 분석을 실시하였다[24]. 비표면적 및 미세기공 부 피는 Brunauer-Emmett-Teller (BET), t-plot, density functional theory (DFT) 등의 방법을 통하여 계산하여 평가하였다.
2.4. 전기화학 특성분석
붕산 처리된 페놀계 활성탄소의 전기화학 특성 변화를 알아보기 위하여 다음과 같이 전극을 제조하였다. 활물질인 활성탄소와 도전 제인 카본블랙, 결합제 PVDF를 각각 80, 10, 10 wt% 비율로 혼합 하여 슬러리를 제조하였다. 이렇게 제조한 슬러리를 티타늄 판에 바 (bar) 코팅방법을 이용하여 약 100 µm 두께로 코팅한 후 120 ℃에서 24 h 동안 건조하였다. 그 후 고온고압 성형기(Hot press)를 이용하여 150 ℃에서 200 kgf/cm2의 입력으로 압착하였다. 이렇게 제조된 전극 을 computer-controlled porentiostat/galcanostat (Ivium Technologies, Netherlands)를 이용하여 삼전극 방법으로 cyclic voltammetry 측정을 실시하였다. 작업전극으로는 상기 방법에 의해 제조된 활성탄소 전극, 기준전극으로 Ag/AgCl, 상대전극으로는 백금 전극을 사용하였다.
Cyclic voltammetry (CV) 측정은 1 M H2SO4전해질을 사용하여 0∼1 V 범위에서 5와 50 mV/s의 전압주입속도로 실시되었다.
3. 결과 및 고찰
3.1. 붕산 처리에 따른 페놀계 활성탄소의 표면 화학적 특성 붕산 처리 전후 페놀계 활성탄소의 표면 변화를 살펴보기 위하여 XPS 스펙트럼과 표면화학 조성을 Figure 1과 Table 1에 나타내었다.
(a) (b)
(c) (d)
Figure 2. SEM and EDX boron (B) mapping images of (a) 10BA700, (b) 10BA1000, (c) 20BA700, and (d) 50BA700.
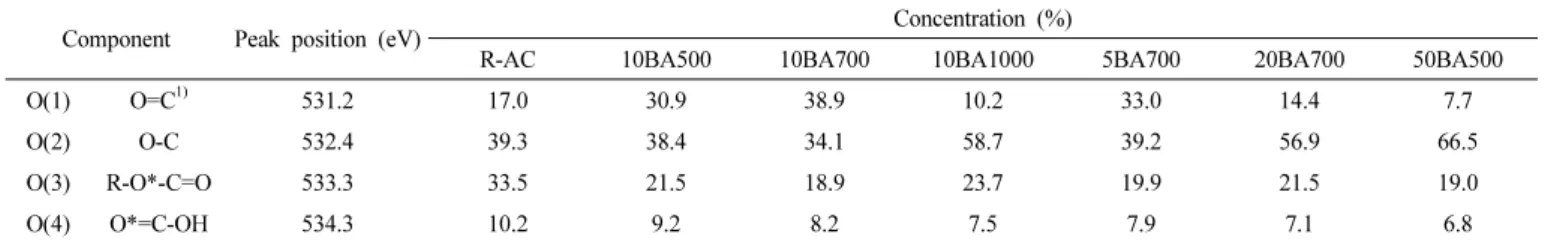
Table 2. O1s Peak Parameters of Untreated and Boric Acid Treated Activated Carbon
Component Peak position (eV) Concentration (%)
R-AC 10BA500 10BA700 10BA1000 5BA700 20BA700 50BA500
O(1) O=C1) 531.2 17.0 30.9 38.9 10.2 33.0 14.4 7.7
O(2) O-C 532.4 39.3 38.4 34.1 58.7 39.2 56.9 66.5
O(3) R-O*-C=O 533.3 33.5 21.5 18.9 23.7 19.9 21.5 19.0
O(4) O*=C-OH 534.3 10.2 9.2 8.2 7.5 7.9 7.1 6.8
1) Quinone-like functional groups
미처리 및 붕산 처리된 활성탄소에서 결합에너지 284.5 eV 부근에서 탄소 피크, 532 eV 부근에서 산소 피크가 나타나는 것을 확인할 수 있었으며, 50BA700의 경우에서만 192 eV 부근에서 붕소 피크가 새롭 게 나타나는 것을 확인할 수 있었다. Table 1의 표면화학 조성 결과를 살펴보면, 붕산 처리 시 반응 온도가 증가할수록 활성탄소 표면의 산소 함량이 감소하는 것을 확인할 수 있었다. 그러나 붕산 처리 시 활성탄 소와 붕산의 비율은 활성탄소 표면의 산소 함량 변화에 큰 영향을 미 치지 않는 것으로 사료된다. 붕산 처리된 활성탄소라도 50BA700을 제외하고는 표면에서 붕소가 검출되지 않았는데, 이것은 이 실험분석 에서 수행된 XPS 분석에서는 표면에서의 원소 비율이 1.0 at% 이내인 경우 검출되었다고 평가하기에 어렵기 때문인 것으로 여겨진다[25].
그래서 EDX 분석을 통하여 50BA700을 제외한 다른 붕소 처리된 활 성탄소 표면에 붕소가 도입되었는지에 대한 여부를 확인하고자 하였고, 이를 Figure 2에 나타내었다.
Figure 2의 SEM 사진을 보면 페놀계 활성탄소는 파쇄된 형태의 입 자로 대략 10 µm 정도의 입자 크기를 가지며, 파쇄된 미분말이 존재 하고 있음을 알 수 있다. 또한 활성탄소에 붕산 처리 시, 입자 크기나 형태에 큰 영향을 주지 않는 것으로 보인다. Figure 2의 EDX mapping 결과로부터 알 수 있듯이, 비록 XPS 분석에서는 너무 적은 양이기에 검출되지 않았지만 붕산 처리된 활성탄소 표면에 붕소가 도입된 것을 확인할 수 있었다. EDX을 통해 얻어진 표면 원소 비율 결과를 통해서 10BA700은 12.66%, 10BA1000은 13.18%, 20BA700은 13.64%, 50BA700 은 13.70%의 붕산이 검출되었음을 알 수 있었다. 붕산 처리 시 반응온 도나 활성탄소와 붕산의 비율의 증가가 활성탄소 표면의 붕산 도입을
증가시키기는 하지만 그 증가의 폭이 미미함을 볼 수 있다. 일반적으로 이는 붕소-탄소 결합을 갖는 물질을 제조하기 위해서는 1000 ℃ 이상의 고온이 필요하기 때문에[26], 이 실험 조건 하의 붕산 처리로 인한 활 성탄소 표면의 붕소 도입 반응에는 한계가 있는 것으로 사료된다.
그러나 XPS 스펙트럼과 표면화학 조성 분석 결과만으로 활성탄소 의 붕산 처리에 대한 효과를 판단하기에는 무리가 있다고 사료된다.
따라서 붕산 처리 전후의 활성탄소의 화학적 결합구조 변화를 상세하게 분석할 필요가 있다. 이를 위하여 활성탄소의 XPS O1s 코어 레벨 스캔 스펙트라(core level scan spectra)를 관능기 별로 분할하여 Figure 3에 나타내었고, Table 2에 각 관능기의 결합에너지 및 함량을 나타내었다.
결합구조의 변화를 확인하기 위해 피크분할 및 데이터 값은 pseudo- Voigt식 (1)을 이용하였으며, 식 (1)을 아래에 나타내었다[27].
(1)위 식 (1)에서 F(E)는 에너지 E에서의 intensity, H는 피크의 높이, E0는 피크 값, FWHM은 반폭 값, S는 symmetry와 Gaussian-Lorentzian mixing ratio에 관련된 shape function을 의미한다.
Figure 3에서처럼, 미처리 및 붕산 처리된 활성탄소의 O1s 피크의 결합에너지에 따른 표면 구조를 상기 pseudo-Voigt식으로부터 O=C (531.2 eV), O-C (532.4 eV), R-O-C=O (533.3 eV), O=C-OH (534.3 eV)로 나눌 수 있었다[28,29]. 상기 4개의 산소 관능기 중 O=C 관능기
(a) (b) (c) (d)
(e) (f) (g)
Figure 3. O1s deconvolution of (a) R-AC, (b) 10BA500, (c) 10BA700, (d) 10BA1000, (e) 5BA700, (f) 20BA700, and (g) 50BA700.
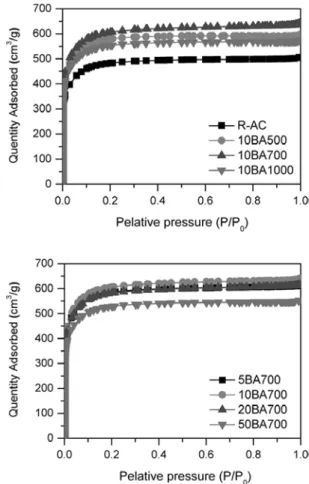
Figure 4. Nitrogen adsorption isotherms at 77 K of untreated and boric acid treated activated carbon.
는 활성탄소 표면에서 pseudo-faradaic 반응과 같은 전기화학적 특 성을 향상시키는 활성점으로 작용한다[30-32]. 활성탄소의 붕산 처 리는 조건이 바뀌어도 활성탄소 표면에 붕소를 도입시키는 반응에 는 한계가 있음을 위의 XPS 표면화학 조성 결과와 EDX mapping 결 과로부터 확인할 수 있었지만, 표면 산소 관능기의 변화에는 영향이 있는 것을 볼 수 있다. Figure 3과 Table 2를 보면, 붕산 처리 시 반응 온도가 증가할수록 O=C 관능기 비율이 증가하다가 반응온도가 1000
℃인 경우에는 O=C 결합이 오히려 감소하는 것을 확인할 수 있다. 또한 붕산 처리 시 붕산과 활성탄소의 비율이 증가할수록 O=C 관능기 비율 이 증가하다가 붕산과 활성탄소의 비율이 20 wt% 이상이 되면 O=C 결합이 감소하는 것을 확인할 수 있다. 이는 붕산 처리가 활성탄소 표면 의 산소 관능기 종류를 변화시킬 수 있으며, 이때 반응온도 및 붕산 과 활성탄소의 비율이 전기화학 활성점으로 작용하는 O=C 관능기의 비율 증가에 관여하는 것으로 여겨진다. 이와 같은 붕산 처리에 따른 O=C 관능기의 비율 증가 결과는 기존에 연구된 질산이나 과산화수소 등으로 산 처리 된 활성탄소의 표면 관능기 변화 연구결과와 흡사한 데[33], 붕산이 활성탄소 표면과 다른 종류의 산과 유사한 반응을 하기 때문으로 여겨진다. 본 조건의 실험에서는 10BA700가 붕산 처리로 활성탄소 표면의 O=C 관능기가 생성되도록 하는 최적 조건에서 얻어 진 샘플로 판단된다. 탄소재료 표면에 존재하는 이런 O=C 관능기는 재료의 전기적 특성 향상에 도움을 주는 것으로 알려져 있다[30]. 따 라서 이와 같은 붕산 처리된 활성탄소의 표면화학적 특성 변화는 활 성탄소의 전기화학적 특성을 변화시키는 주요한 요인이 될 것으로 사 료된다.
3.2. 붕산 처리에 따른 페놀계 활성탄소 전극의 기공 특성 붕산 처리에 따른 활성탄소의 흡착등온선, 기공분포도 등 기공 특성 변화에 대하여 알아보고자 질소 흡⋅탈착을 통한 분석을 수행하였고 그 결과를 Figures 4, 5 및 Table 3에 나타내었다. Figure 4에서 보면, 미처리 및 붕산 처리된 활성탄소의 흡착등온선은 IUPAC에서 명시한 6가지 흡착등온선 중 Type I의 형태인 것을 확인할 수 있다[34]. 이런 Type I의 흡착등온선은 기공의 직경이 2 nm 이하인 미세기공이 발달한 다공성 물질이 나타내는 전형적인 특성이다[34]. 각각의 흡착등온선은 매우 유사한 형태를 보이는데, 이는 본 실험에서 사용한 여러 조건에
서 페놀계 활성탄소에 붕산 처리를 수행하여도 활성탄소의 그 기공 특성은 유지되고 변화가 없기 때문이라고 할 수 있다. 미처리 및 붕산 처리된 활성탄소의 기공특성을 상세히 알아보기 위하여 DFT으로 기 공분포도를 계산하여 Figure 5에 나타내었다. 미처리 및 붕산 처리된 활성탄소는 2.5 nm 이하의 직경을 갖는 기공이 주로 존재하는 것을
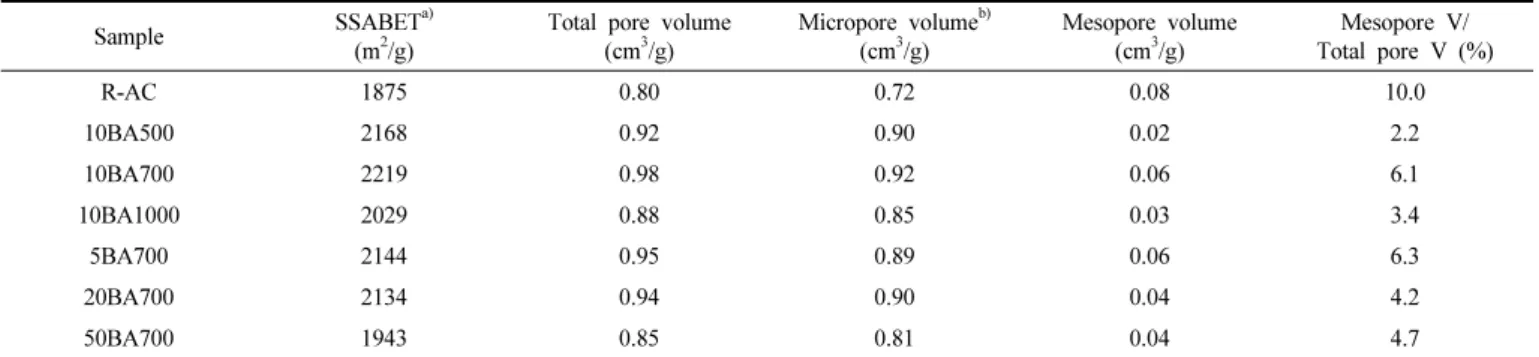
10BA700 2219 0.98 0.92 0.06 6.1
10BA1000 2029 0.88 0.85 0.03 3.4
5BA700 2144 0.95 0.89 0.06 6.3
20BA700 2134 0.94 0.90 0.04 4.2
50BA700 1943 0.85 0.81 0.04 4.7
a) BET Specific surface area
b) t-plot Micropore volume
Figure 5. DFT pore size distribution of untreated and boric acid treated activated carbon.
확인하였고 그 분포 정도가 크게 다르지 않은 것으로 보여진다. 이는 Figure 4의 흡착등온선 결과와 유사한 결과이다.
Table 3에 미처리 및 붕산 처리된 활성탄소의 세공구조 및 기공 특 성을 나타내었다. 미처리 및 붕산 처리된 활성탄소는 흡착등온선 및 DFT 기공분포도의 결과에서도 보였듯이 대체적으로 미세기공을 가 지고 있는 것으로 나타났다. 붕산 처리된 활성탄소의 비표면적, 총 기 공 부피 및 미세공 부피는 반응온도가 증가함에 따라 증가하다가 감 소하는 경향을 보여주고 있다. 활성탄소와 붕산의 비율의 증가 역시 붕산 처리된 활성탄소의 기공특성에 반응온도와 같은 경향으로 영향을 미치는 것으로 판단된다. 이와 같은 활성탄소의 기공 특성의 변화는
붕산과 활성탄소의 반응과 열처리로 인한 표면 식각 효과로 인한 것 으로 여겨지며 붕산 처리 시 반응온도와 활성탄소와 붕산의 비율이 너무 높은 경우에 표면 식각 반응이 과도하게 일어나 그 비표면적이 감소하는 현상이 보이는 것으로 사료된다. Table 3에 따르면, 10BA700가 비표면적 2219 m2/g, 총 기공 부피 0.98 cm3/g, 미세공 부피 0.06 cm3/g 으로 이 실험에서 가장 큰 값을 가지는 것을 확인할 수 있다. 이는 10BA700 조건이 상기 3.1.절에서 설명한 O=C 관능기에 관한 조건뿐 만 아니라 기공 특성에서도 최적 조건이라고 보여진다. 이와 같은 비 표면적의 증가는 전기이중층 커패시터의 용량 향상에 긍정적인 영향 을 미칠 것으로 사료된다.
3.3. 붕산 처리에 따른 페놀계 활성탄소 전극의 전기화학적 특성 붕산 처리에 의한 페놀계 활성탄소 전극들에 대해서 0~1 V (vs.
Ag/AgCl)의 전극전위 범위에서 5와 50 mV/s의 전위주사속도로 cyclic voltammogram을 측정하였고 이를 Figures 6과 7에 나타내었다.
일반적으로 이론적인 축전지(capacitor)에서는 전위를 인가함에 따 라 초기에 전류가 급격히 증가하다가 일정한 전위 이상에서는 전류의 변화가 거의 일어나지 않기 때문에 박스 형태의 cyclic voltammogram 이 나타난다. 하지만 Figures 6과 7을 보면, 전위를 인가시켰을 때 초기 에는 전류가 급격히 증가하다가 점점 증가폭이 감소하는 것을 알 수 있다. 또한 전위를 감소시켰을 때도 초기 전류는 급격한 변화량을 보 이다가 변화폭이 점점 감소하고 있다. 이러한 형태의 cyclic voltammogram 이 나타나는 원인은 활성탄소 전극의 전기이중층에서 나타나는 이온 흡ㆍ탈착 메커니즘으로 설명할 수 있다[35]. 활성탄소 전극에 전위를 인가하게 되면 전극과 접촉하고 있는 전해질의 이온들이 전극 표면에 형성된 전기이중층에 흡착하게 된다. 이때 인가되는 전위주사속도가 이온들이 전기이중층에 흡착하는데 필요한 시간보다 빠를 경우에는 이온이 충분히 흡착되지 않아 계속해서 전류가 증가하는 경향이 나타 나게 되는 것이다. 이러한 결과는 활성탄소 전극에서 나타나는 일반 적인 cyclic voltammogram 경향이므로, 붕소 처리된 활성탄소 전극 표면에서 이온들이 원활하게 흡ㆍ탈착되고 있음을 알 수 있다. 또한, 5 mV/s의 전압주입속도에서 측정된 CV 그래프(Figure 6)에서는 0.3∼
0.4 V 영역에서 약한 레독스(redox) 피크가 존재하는 것을 확인할 수 있었다. 이런 레독스 피크는 산소 관능기, 특히 퀴논형 관능기(O=C) 의 레독스 반응에 의하여 나타나는 것으로 알려져 있다[36]. 따라서 위의 3.1.절에서 설명하였듯이, 붕산 처리로 인하여 바뀐 활성탄소 표 면 산소 관능기의 비율이 활성탄소의 전기화학적 특성 및 정전용량의 변화에 영향을 미치는 것으로 사료된다.
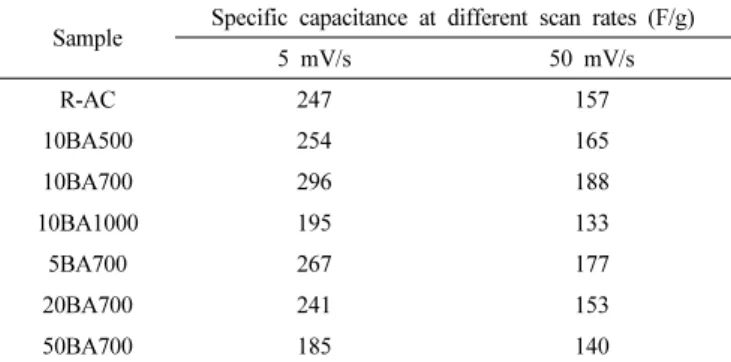
한편 붕산 처리된 활성탄소 전극의 비 정전용량 변화를 알아보기
Figure 6. Cyclic voltammograms of untreated and boric acid treated activated carbon obtained at 5 mV/s.
Figure 7. Cyclic voltammograms of untreated and boric acid treated activated carbon obtained at 50 mV/s.
Table 4. Specific Capacitance for Untreated and Boric Acid Treated Activated Carbon Based Electrodes at Different Scan Rate
Sample Specific capacitance at different scan rates (F/g)
5 mV/s 50 mV/s
R-AC 247 157
10BA500 254 165
10BA700 296 188
10BA1000 195 133
5BA700 267 177
20BA700 241 153
50BA700 185 140
위하여 활성탄소 전극의 전위주사속도 별 비 정전용량을 Table 4에 나타내었다. 비 정전용량은 cyclic voltammogram 결과로부터 식 (2)를 통해 계산되었다[37].
∆ (2)
여기서, C는 정전용량, w는 활물질의 무게, i는 시간의 경과(dt)에 따른 전류, ΔV는 인가한 전압의 변화량을 의미한다.
붕산 처리된 활성탄소의 비 정전용량은 반응 온도가 증가함에 따라
증가하다가 반응온도가 1000 ℃인 경우에는 감소하는 것으로 나타났다.
또한, 활성탄소 대비 붕산의 비율이 증가함에 따라서 활성탄소의 비 정전용량이 증가하다가 붕산의 비율이 20 wt% 이상인 경우부터 비 정전용량이 감소하게 된다. 이는 활성탄소의 비표면적의 변화와 표면 화학적 특성 변화에 의한 결과로 보여진다. 이 실험에서 붕산 처리로 인하여 활성탄소의 비표면적 및 총 기공 부피는 미처리 활성탄소에 비하여 증가하였다. 활물질인 활성탄소의 비표면적이 증가하면 EDLC의 비 정전용량이 증가하는 것은 매우 잘 알려진 사실이다[38]. 그러나 10BA1000, 20BA700, 50BA700의 경우에는 R-AC보다 비표면적과 총 기공 부피가 증가하였음에도 불구하고 비 정전용량이 R-AC보다 낮은 것으로 나타났다. 이는 이 실험에서는 활성탄소의 붕산 처리에 따른 표면 관능기 제어와 기공 제어 중 표면 관능기 제어의 효과가 더 전기 화학적 특성에 영향을 미친다고 할 수 있다. 붕산 처리로 인한 O=C 관능기 비율의 증가는 전기화학 활성점으로 작용하여 활성탄소의 전 기화학적 특성을 증가시켰기 때문인 것으로 보여진다. 그러나 붕산 처리로 인하여 활성탄소 표면에 도입된 붕소는 전기화학적 특성에 큰 영향을 미치지 않는 것으로 보이며, 붕산 처리로 인한 활성탄소의 표 면 산소 관능기 비율 변화와 식각에 의한 비표면적의 변화가 활성탄 소의 비 정전용량 증가 원인으로 사료된다. 이 실험에서는 10BA700이 5 mV/s의 전압주입속도에서 296 F/g, 50 mV/s의 전압주입속도에서 188 F/g으로 가장 큰 비 정전용량을 갖는데, R-AC의 비 정전용량에 비하여 약 20% 증가하였다. 이와 같은 비 정전용량의 증가는 위에서 설명하였듯이, 10BA700 조건이 본 실험에서 가장 비표면적이 크고
4. 결 론
본 연구에서는 전기이중층 커패시터의 전극 활물질로 사용되는 페 놀계 활성탄소의 비 정전용량을 증가시키기 위하여 붕산을 이용하여 표면처리를 하였다. 활성탄소의 붕산 처리 시, 처리온도와 활성탄소와 붕산의 비율 변화가 활성탄소의 표면화학 특성 및 물리적 특성에 큰 영향을 미치는 것을 확인할 수 있었다. 붕산 처리 표면개질 방법은 활 성탄소 표면의 다양한 산소 관능기 중에서 전기화학적 특성 향상에 도움이 되는 퀴논형 관능기(O=C)의 비율을 효과적으로 증가시킬 수 있었다. 또한, 붕산 처리된 활성탄소는 본래의 기공 특성인 미세공을 주로 갖는 세공특성을 유지하면서 비표면적과 총 기공 부피 및 미세공 부피가 증가되었다. 본 실험에서는 반응온도 700 ℃, 활성탄소 대비 붕산 농도 10 wt%일 때가 전기화학 특성 향상을 위한 붕산 처리의 최적 조건으로 보여지고, 이 조건 하에서 처리된 활성탄소는 미처리 활성탄소에 비해 비 정전용량이 약 20% 증가하였다. 이는 붕산 처리 시 활성탄소 표면의 O=C 관능기 비율 증가와 비표면적 및 기공부피 증가에 의한 것이라고 할 수 있다. 이러한 결과로부터 활성탄소의 붕 산 처리는 활성탄소의 비 정전용량을 효과적으로 증가시킬 수 있는 것으로 기대된다.
감 사
본 연구는 지식경제부 및 한국산업기술평가관리원의 산업원천기술 개발사업의 일환으로 수행하였음. [KI002177-2013-05, 유비쿼터스 전 원용 Pouch/Radial형 리튬이온커패시터]
참 고 문 헌
1. L. Wei and G. Yushin, Carbon, 49, 4830 (2011).
2. P. Simon and Y. Gogotsi, Nat. Mater., 7, 845 (2008).
3. S. Yoon, J. Lee, T. Hyeon, and S. M. Oh, J. Electrochem. Soc., 147, 2507 (2000).
4. Q. Li, F. Liu, L. Zhang, B. J. Nelson, S. Zhang, C. Ma, X. Tao, J. Cheng, and X. Zhang, J. Power Sources, 207, 199 (2012).
5. J. K. Sun, E. H. Um, and C. T. Lee, Appl. Chem. Eng., 21, 11.
(2010).
6. C. T. Lee, J. H. Kim, and B. W. Cho, Prospectives Ind. Chem., 2, 16 (1999).
7. A. S. Arico, P. Bruce, J. M. Tarascon, and W. Van-Schalkwijk, Nature Mater., 4, 366 (2005).
8. J. P. Zheng, P. J. Cygan, and T. R. Jow, J. Electrochem. Soc., 142, 2699 (1995).
9. A. Yamada and J. B. Goodenough, J. Electrochem. Soc., 145, 737 (1998).
10. H. P. Stadniychuk, M. A. Anderson, and T. W. Chapman, J.
Electrochem. Soc., 143, 1629 (1996).
14. J. H. Huh, M. K. Seo, H. Y. Kim, I. J. Kim, and S. J. Park, Polymer (Korea), 36, 756 (2012).
15. Y. Yamada, T. Sasaki, N. Tatsuda, D. Weingarth, K. Yan, and R.
Kötz, Electrochim. Acta, 81, 138 (2012).
16. J. H. Lee, G. Y. Heo, and S. J. Park, Polymer (Korea), 36, 756 (2012).
17. N. A. Fathy and I. Y. El-Sherif, Carbon Lett., 12, 1 (2011).
18. J. W. Lim, E. Jeong, M. J. Jung, S. I. Lee, and Y. S. Lee, J. Ind.
Eng. Chem., 18, 116 (2012).
19. D. Tashima, H. Yoshitama, T. Sakoda, A. Okazaki, and T. Kawaji, Electrochim. Acta, 77, 198 (2012).
20. D. Tashima, A. Sakamoto, M. Taniguchi, T. Sakoda, and M.
Otsubo, Surf. Coat. Technol., 202, 5560 (2008).
21. M. Endo, T. Maeda, T. Takeda, Y.J. Kim, K. Koshiba, H. Hara, and M. S. Dresselhaus, J. Electrochem. Soc., 148, A910 (2001).
22. M. J. Jung, E. Jeong, S. Cho, S. Y. Yeo, and Y. S. Lee, J. Colloid Interface Sci., 381, 152 (2012).
23. M. J. Jung, E. Jeong, S. I. Lee, and Y. S. Lee, J. Ind. Eng. Chem., 18, 642 (2012).
24. Y. S. Lee, Y. H. Kim, J. S. Hong, J. K. Suh, and G. J. Cho, Catal.
Today, 120, 420 (2007).
25. J. Kim, H. Hong, K. Y. Oh, and C. Lee, J. Korea Vacuum Soc., 11, 159 (2002).
26. P. Redlich, J. Loeffler, P. M. Ajayan, J. Bill, F. Aldinger, and M.
Rühle, Chem. Phys. Lett., 260, 465 (1996).
27. T. W. Little and F. S. Ohuchi, Surf. Sci., 445, 235 (2000).
28. C. Zhang, Y. Duan, B. Xing, L. Zhan, W. Qiao, and L. Ling, Mining Science and Technology (China), 19, 259 (2009).
29. M. J. Jung, E. Jeong, J. W. Lim, S. I. Lee, and Y. S. Lee, Colloid Surf. A-Physicochem. Eng. Asp., 389, 274 (2011).
30. V. Khomenko, E. Raymundo-Piñero, and F. Béguin, J. Power Sources, 195, 4234 (2010).
31. J. Lang, X. Yan, W. Liu, R. Wang, and Q. Xue, J. Power Sources, 204, 220 (2012).
32. T. E. Rufford, D. Hulicova-Jurcakova, Z. Zhu, and G. Q. Lu, Electrochem. Commun., 10, 1594 (2008).
33. C. Moreno-Castilla, M. A. Ferro-Garcia, J. P. Joly, I. Bautista-Toledo, F. Carrasco-Marin, and J. Rivera-Utrilla, Langmuir, 11, 4386 (1995).
34. S. J. Gregg and K. S. W. Sing, Adsorption Surface Area and Porosity, second ed., Academy Press, London (1982).
35. K. L. Yang, S. Yiacoumi, and C. tsouris, J. Electroanal. Chem., 540, 159 (2003).
36. R. B. Mathur, V. Gupta, O. P. Bahl, A. Tressaud, and S. Flandrois, Synth. Met., 114, 197 (2000).
37. M. Ramani, B. S. Haran, R. E. White, and B. N. Popov, J.
Electrochem. Soc., 148, A374 (2001).
38. W. Shen, Z. Li, and Y. Liu, Recent Patents on Chemical Engineering, 1, 27 (2008).