Introduction Introduction
Cancer of the larynx constitutes a majority of the cancers affecting the laryngeal epithelium based on tumor location. It
directly spreads to structures adjacent to the cervical lymph node, and more distantly through the blood stream via metas- tasis to the regional lung [1]. The overall five-year survival rate was within 50–70% over the past decade [2]. Consequently,
Int J Oral Biol 45:169-178, 2020pISSN: 1226-7155 • eISSN: 2287-6618 https://doi.org/10.11620/IJOB.2020.45.4.169
L-ascorbic acid induces apoptosis in human laryngeal epidermoid Hep-2 cells by modulating the nuclear
factor kappa-light-chain-enhancer of activated B cells/
mitogen-activated protein kinase/Akt signaling pathway
Jung-Sun Park
1, Yoon-Jung Kim
1,2, Sam Young Park
1,2, Kyung-Yi Chung
3, Sang-Jin Oh
4, Won-Jae Kim
1*, and Ji-Yeon Jung
1,2*
1
Department of Oral Physiology, Dental Science Research Institute, School of Dentistry, Chonnam National University, Gwangju 61186, Republic of Korea
2
Hard Tissue Biointerface Research Center, School of Dentistry, Chonnam National University, Gwangju 61186, Republic of Korea
3
Department of Dental Hygiene, School of Health Science, Honam University, Gwangju 62399, Republic of Korea
4
School of Biological Sciences and Technology, College of Natural Sciences, Chonnam National University, Gwangju 61186, Republic of Korea
L-ascorbic acid (L-AA; vitamin C) induces apoptosis in cancer cells. This study aimed to elucidate the molecular mechanisms of L-AA-induced apoptosis in human laryngeal epidermoid carcinoma Hep-2 cells. L-AA suppressed the viability of Hep-2 cells and induced apoptosis, as shown by the cleavage and condensation of nuclear chromatin and increased number of Annexin V-positive cells. L-AA decreased Bcl-2 protein expression but upregulated Bax protein levels. In addition, cytochrome c release from the mitochondria into the cytosol and activation of caspase-9, -8, and -3 were enhanced by L-AA treatment. Furthermore, apoptosis-inducing factor (AIF) and endonuclease G (EndoG) were translocated into the nucleus during apoptosis of L-AA-treated Hep-2 cells. L-AA effectively inhibited the constitutive nuclear factor-κB (NF-κB) activation and attenuated the nuclear expression of the p65 subunit of NF-κB. Interestingly, L-AA treatment of Hep-2 cells markedly activated Akt and mitogen-activated protein kinase (MAPK; extracellular signal-regulated kinase 1/2, p38, and c-Jun N-terminal kinase [JNK]) and and LY294002 (Akt inhibitor), SB203580 (p38 inhibitor) or SP600125 (a JNK inhibitor) decreased the levels of Annexin V-positive cells. These results suggested that L-AA induces the apoptosis of Hep-2 cells via the nuclear translocation of AIF and EndoG by modulating the Bcl- 2 family and MAPK/Akt signaling pathways.
Keywords: Apoptosis, Ascorbic acid, Mitogen-activated protein kinase, NF-kappa B, Squamous cell
Received October 30, 2020; Revised November 10, 2020; Accepted November 11, 2020
*Correspondence to: Ji-Yeon Jung, E-mail: [email protected] https://orcid.org/0000-0002-4419-8077
*Correspondence to: Won-Jae Kim, E-mail: [email protected] https://orcid.org/0000-0001-6549-744X Copyright © The Korean Academy of Oral Biology
CC This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by- nc/4.0/), which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Original Article IJOB
numerous studies have been conducted to develop efficient strategies for the development of cancer chemopreventive and chemotherapeutic agents. Recently, apoptosis induced by chemotherapy has been shown to play a significant role in the elimination of cancer cells [3]. Thus, targeting the apop- totic pathway in epithelial cancer cells may effectively prevent laryngeal cancer. L-ascorbic acid (L-AA) exhibits strong anti- proliferative and antioxidant effects and is known to modulate various enzymes that are involved in cancer development and progression [4]. The effects of L-AA can also be attributed to its biological role as a cofactor for a number of enzymes and most hydroxylases related to collagen synthesis, and repre- sents a source of the signaling molecule and hydrogen perox- ide. Other studies have discussed the anti-carcinogenic effect of L-AA on cancer prevention [5]. The signals associated with the molecular mechanism of cancer prevention by L-AA have been attributed to nuclear factor- κB (NF-κB), phosphatidylino- sitol 3’-kinase (PI3K)/Akt, and mitogen-activated protein ki- nase (MAPK) signaling pathways [6-8]. These signal transduc- tion pathways are related to the regulation of cell proliferation and apoptosis, modulation of cell cycle, and activation of tumor migration [9,10]. The constitutive activation of NF- κB in sev- eral cancers, including pancreatic, lung, breast, gastric, mela- noma, and head and neck cancer, is an important contributor to cancer metastasis and progression [11,12]. A recent study suggested that the level of NF- κB activity may be important in laryngeal cancer cell survival [13]. The PI3K/Akt signaling pathway might be the most central pathway in the transmis- sion of anti-apoptotic signals in cell survival. Multiple pathways have been implicated in cell proliferation, survival, and meta- bolic processes. The MAPKs, including extracellular signal- regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38, are also known to play a basic role in survival, prolif- eration, transcription, and apoptosis. The PI3K/Akt and MAPKs signaling pathways are clearly involved in a variety of cellular functions, including cell growth, differentiation, development, and apoptosis [14]. However, the anticancer effects of L-AA in squamous carcinoma head and neck cancer cells are not clearly established.
In the present study, we explored the anti-tumor activity of L-AA in laryngeal Hep-2 cells and the mechanism underlying L-AA-induced growth inhibition and apoptosis.
Materials and Methods Materials and Methods
1. Materials
L-AA, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazo- lium bromide (MTT) and all other chemicals were of analyti- cal grade and were purchased from Sigma Chemical Co. (St.
Louis, MO, USA). The primary antibodies used were polyclonal goat anti-human Bcl-2, monoclonal mouse anti-human Bax, monoclonal mouse anti-human cytochrome c, polyclonal rabbit anti-human Bid, monoclonal mouse anti-human apoptosis- inducing factor (AIF), polyclonal goat anti-human endonuclease G (EndoG), and polyclonal goat anti-human actin (Santa Cruz, Santa Cruz, CA, USA). Monoclonal rabbit anti-human NF- κB P65 antibodies were acquired from Cell Signaling Technology (Danvers, MA, USA).
2. Cell culture and cell viability assay
The Hep-2 cell line, delivered from a human laryngeal epi- dermoid carcinoma of the larynx, was purchased from a Korean Cell Line Bank (KCLB No. 10023). Hep-2 cells were cultured in Dulbecco’s modified eagle media, supplemented with 10%
fetal bovine serum (FBS; Gibco, Waltham, MA, USA) and 1%
streptomycin-penicillin. The cells were grown at 37°C in a humidified atmosphere of 5% carbon dioxide in air. Cells were incubated with or without L-AA in 96-well plates (5 × 10
3cells/well) for 24 hours, and the cell viability was evaluated using MTT assay as described previously [15]. The relative cell viability was expressed as the ratio (%) of the absorbance in the experimental wells to that of the control wells (without test compound). The cytotoxic concentration for 50% cell death (CC50) was determined from the dose-response curve.
3. 4′,6-diamidino-2-phenylindole (DAPI) staining
Hep-2 cells (1 × 10
6/mL) were cultured in 60-mm diameter
dishes in RPMI containing 1% FBS with 10 mM L-AA for 24
hours, followed by fixation of the cells in 4% paraformaldehyde
for 15 minutes at room temperature before permeabilization
with 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA)
for 10 minutes. The cells were rinsed with phosphate-buffered
saline (PBS) and incubated for 15 minutes at room tempera-
ture with 0.01% DAPI stain (Sigma-Aldrich). Excess stain was
removed by washing three times in PBS. Stained nuclei were
visualized via fluorescence microscopy. The percentage of
apoptotic cells was determined by counting at least 200 cells within the treatment group.
4. Flow cytometry
An annexin V FLUOS staining kit (Sigma-Aldrich) was used to measure the level of annexin V binding according to the manufacturer’s instructions. Briefly, the cells were trypsinized in PBS, collected by centrifugation, and resuspended in 100 µL of binding buffer containing annexin V. The cells were then incubated at room temperature for 15 minutes in the dark, and analyzed by flow cytometry (Becton-Dickinson, Franklin Lakes, NJ, USA). Several controls were used to optimize the instru- ment settings and determine the gating for the Windows- based platform.
5. Caspase activity and inhibition of apoptosis with a caspase inhibitor
The activities of caspase-3, -8, and -9 in L-AA-treated cells were measured colorimetrically using a commercial assay kit (Calbiochem, San Diego, CA, USA) according to the manufac- turer’s instructions. To determine the inhibition of apoptosis, a general caspase inhibitor z-VAD-fmk (50 µM), was added to the culture medium 1 hour before L-AA treatment. The cyto- toxic effect of L-AA was measured using MTT in the presence or absence of caspase inhibitors.
6. Protein extraction and Western blot analysis
Cell lysates were prepared in NP-40 lysis buffer (30 mM Tris-Cl, pH 7.5, 1 mM ethylenediaminetetraacetic acid [EDTA], 150 mM NaCl, 1% NP-40, 1 mM phenylmethylsulfonyl fluo- ride [PMSF], and protease inhibitor mixture containing 1 µg/
mL aprotinin and leupeptin), and the total protein content was quantified using the BCA protein assay (Pierce, Rockford, IL, USA). Equal amounts of total protein (100 µg) were electro- phoretically separated via 12% sodium dodecyl sulphate- polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane (Amersham Pharmacia Biotech, Buckinghamshire, UK). The membrane was blocked with 5%
nonfat dry milk and incubated with primary antibodies for 1 hour at room temperature. The blots were then incubated with horseradish peroxidase-conjugated secondary antibodies (Sigma-Aldrich) for 2 hours. The blots were then washed and reacted with enhanced chemiluminescence (ECL) solutions
(Amersham Pharmacia Biotech) and exposed to ECL Hyper- film ((American BioSciences, Blauvelt, NY, USA)). The relative intensity of each band was determined using a computerized software program.
7. Extraction of nuclear protein
Hep-2 cells reaching 80% to 90% confluence were incu- bated in a medium containing 1% FBS in 0–10 mM of L-AA for 24 hours. The cells were resuspended in 500 µL of cold buffer A [50 mM Tris (pH 7.4), 150 mM NaCl, 0.2 mM EDTA, 3% (v/
v) glycerol, and 1.5 mM MgCl
2]. The cells were allowed to swell for 5 minutes on ice and then lysed with 500 µL of buffer B (buffer A containing 0.05% Nonidet P-40; Sigma-Aldrich). The cell lysates were gently layered onto an equal volume of buffer C [10 mM Tris (pH 7.4), 25% (v/v) glycerol, and 1.5 mM MgCl
2] and centrifuged for 5 minutes at 200 × g. The white nuclear pellet was resuspended in 75 µL of a cold high-salt lysis buf- fer [20 mM N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES) (pH 7.9), 400 mM NaCl, 1 mM EDTA, 1 mM di- thiothreitol, and 1 mM PMSF]. This suspension was agitated for 30 minutes at 4℃ and microcentrifuged for 15 minutes at 4℃. The resulting supernatant was stored in aliquots at –80℃.
The protein was quantified spectrophotometrically using the BCA assay (Pierce) with bovine serum albumin as the stan- dard.
8. Electrophoretic mobility shift assay (EMSA)
EMSA was performed using a gel shift assay (Prome-
ga, Madison, WI, USA). Briefly, the oligonucleotide with
the consensus sequence for NF- κB (5′-AGTTGAGGGGA
CTTTCCCAGG-3′) was end-labeled with [ g-32P]-adenosine
triphosphate (3 Ci/mmol; Amersham Pharmacia Biotech) using
T4 polynucleotide kinase. The labeled oligonucleotide was then
purified in a Microspin G-25 column (Sigma-Aldrich) and used
as an EMSA probe. The nuclear extract proteins (15 µg) were
pre-incubated with the binding buffer [10 mM Tris (pH 7.5), 50
mM NaCl, 0.5 mM EDTA, 1 mM MgCl
2, 0.5 mM dithiothreitol,
4% (v/v) glycerol, and 0.05 mg/mL poly(deoxyinosine-deox-
ycytidine)] for 5 minutes, and then incubated with the labeled
probe for 15 minutes at 37℃. Each sample underwent electro-
phoresis in a 5% non-denaturing polyacrylamide gel with a 0.5X
Tris-borate-EDTA buffer (pH 7.4) at 150 V for 4 hours. The gel
was dried and subjected to autoradiography. In the competition
study, a 50-fold excess of the unlabeled oligonucleotide was
incubated with the radiolabeled probe in the reaction mixture.
9. Statistical analyses
The data are expressed as means ± standard error of mean.
Statistical analyses were conducted using Student’s t-test and Turkey’s tests for the determination of post hoc differences between group means. Statistical significance was determined at p < 0.05.
Results Results
1. L-AA induced apoptotic cell death in human laryngeal Hep-2 cancer cells
The effect of L-AA on the viability of Hep-2 cells was evalu- ated via MTT assay with different concentrations of L-AA.
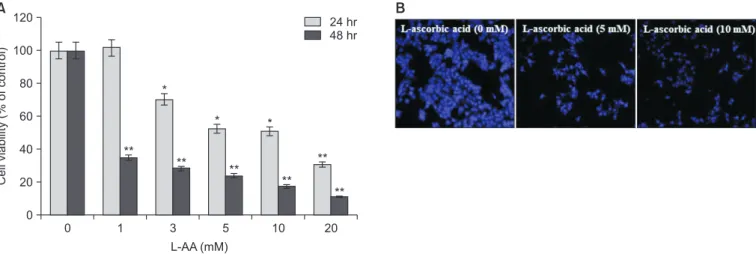
L-AA decreased the viability of Hep-2 cells in a time- and dose-dependent manner. As shown in Fig. 1A, the concentra- tion required for 50–60% inhibition over a period of 24 hours and 48 hours was 5 mM and 1 mM, respectively. The cells undergoing apoptosis showed profound structural changes, including nuclear disintegration and condensation. After a 24-hours treatment with 5 or 10 mM L-AA, the nuclei were stained with DAPI to assess the changes in nuclear morphol- ogy. The nuclei of the L-AA-treated cells contained clearly condensed chromatin, showing apoptotic changes (Fig. 1B). In addition, the L-AA-induced apoptosis of epithelial cells was
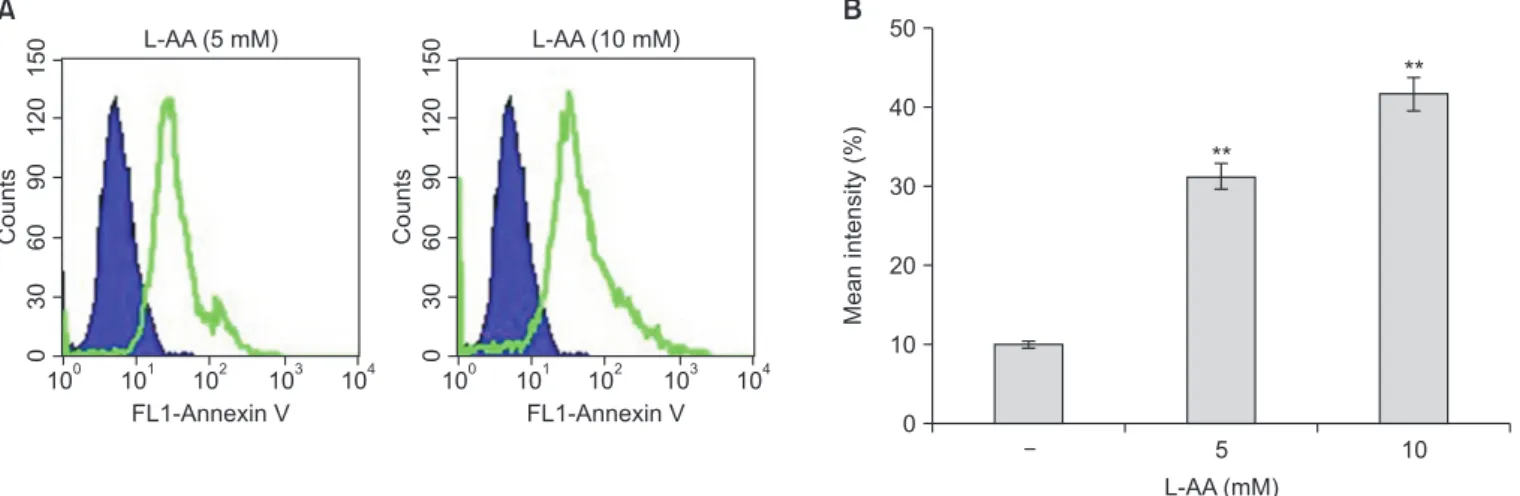
further analyzed for the presence of annexin V-conjugated cells via fluorescence analysis. As shown in Fig. 2, incubation with 5 to 10 mM L-AA induced a dose-dependent increment in the number of annexin V-positive apoptotic cells.
2. L-AA induced the release of cytochrome c from mitochondria into the cytosol and upregulated AIF and EndoG levels
Apoptosis and cell death in most cancer cells are activated by p53 during the initial stages of apoptosis. The Bcl-2 fam- ily also plays a major role in apoptosis, controlling the release of mitochondrial cytochrome c into the cytosol [16,17]. The expression of p53 and the Bcl-2 family was analyzed in L-AA- induced apoptosis of Hep-2 cells. As shown in Fig. 3A, after L-AA exposure, the expression of the p53 and Bax proteins was significantly higher, while the expression of the Bcl-2 pro- tein was significantly lower than that of the control group.
The release of cytochrome c, AIF, and EndoG from mito- chondria is essential for programmed cell death. To elucidate the mitochondrial role in Hep-2 cells undergoing L-AA-in- duced apoptosis, the release of cytochrome c, AIF, and EndoG from mitochondria into the cytosol was determined by West- ern blot. The levels of the mitochondrial and cytosolic fractions isolated from Hep-2 cells exposed to different doses of 5 or 10 mM L-AA for 24 hours were measured. Compared to the control, the contents of cytochrome c, AIF, and EndoG were significantly increased after exposure to L-AA (Fig. 3B).
0 120
100
80
60
40
Cellviability(%ofcontrol) 20
L-AA (mM) 0
1 3 5 10 20
24 hr 48 hr
**
**
*
*
**
*
**
**
**
A B
Fig. 1. L-ascorbic acid (L-AA)-induced apoptotic cell death in Hep-2 cells. (A) Cells were incubated with different concentrations of L-AA for 24 hours or 48 hours, and the cell viability was determined via 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. (B) 4′,6-diamidino-2-phenylindole (DAPI) staining of cells treated with 5 and 10 mM L-AA for 24 hours (×40 magnification). These data are represented as the mean ± standard deviation based on triplicate measurements.
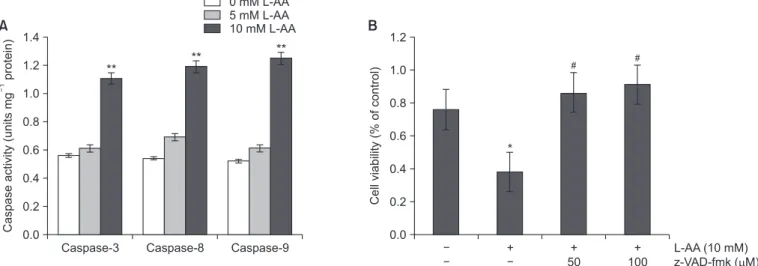
3. L-AA-induced apoptosis was caspase dependent in Hep-2 cells
Caspases play a crucial role during the apoptosis of various cancer cells. The activities of caspase-8, -9 and -3 were ex- amined in L-AA-treated Hep-2 cells. After exposure to differ- ent concentrations of L-AA for 24 hours, the activities of the three caspases were significantly increased (Fig. 4A). To con- firm the role of caspases in L-AA-induced cell death, the cells were pretreated with the general caspase inhibitor z-VAD-fmk before exposure to L-AA and cell viability was subsequently measured. Treatment with 50 or 100 µM z-VAD-fmk inhibited the cell death induced by L-AA in Hep-2 cells (Fig. 4B). These results indicated that L-AA-induced apoptosis in Hep-2 cells was mediated via caspase.
4. L-AA-induced apoptosis is mediated via NF-κB, MAPK, and Akt signal pathways
The PI3K/Akt and MAPKs signaling pathways play a crucial role in apoptosis of many cells [14]. The PI3K/Akt and MAPKs signaling pathways were investigated to determine the cel- lular apoptotic pathways associated with L-AA-induced Hep- 2 cell death. Fig. 5A showed that the phosphorylation of JNK, p38 MAPK and Akt was observed from 30 minutes to 36 hours after treatment with 10 mM L-AA, and peaked at 24 hours af- ter treatment, whereas that of ERK1/2 decreased at 36 hours.
Many studies proposed that the regulation of NF- κB activity is important in cancer cell survival [18]. The known target genes of NF- κB include a number of anti-apoptotic genes, such as the Bcl-2 family. L-AA-induced apoptosis of Hep-2 cells in- hibited constitutive NF- κB activation. EMSA results showed that L-AA treatment dose-dependently decreased the DNA-
50
40
30
20
10
Meanintensity(%)
L-AA (mM) 0
10
B
5 Counts 1501200306090
100 101 102 103 104 L-AA (5 mM)
FL1-Annexin V
Counts 1501200306090
100 101 102 103 104 L-AA (10 mM)
FL1-Annexin V
**
**
A
Fig. 2. Induction of apoptosis via L-ascorbic acid (L-AA) in Hep-2 cells. (A) Histograms representing flow cytometry results of cells treated with 5 and 10 mM L-AA for 24 hours. The vertical axes (counts) represent the number of cells and the horizontal axes (annexin-V fluorescein) indicate cells labeled with annexin V.
(B) The percentage of annexin V-positive cells is expressed as the mean ± standard deviation from triplicate measurements.
**p < 0.01, compared with the untreated control.
Cytc EndoG AIF
-actin p53
Bax Bcl-2
-actin
Mitochondria fraction Cytosolic fraction
10 5 3
1 1 3 5 10
L AA (mM)- L-AA (mM)
10 5
3 1
L-AA (mM)
A B
Fig. 3. Effect of L-ascorbic acid (L-AA) on the expression of various mitochondrial apoptotic factors in Hep-2 cells. (A) The relative protein expression of apoptotic factors including p53, Bax, and Bcl-2 was analyzed via Western blot. (B) After isolating the cytosolic and mitochondrial fractions of cells treated with different concentrations of L-AA for 24 hours, the levels of cytochrome c (Cyt c), apoptosis-inducing factor (AIF), and endonuclease G (EndoG) were ana- lyzed by Western blot. The data represent the mean ± standard deviation from triplicate measurements.
Caspase-3 1.4
1.2 1.0 0.8 0.6 0.4 0.2
Caspaseactivity(unitsmgprotein)
0.0
Caspase-8 Caspase-9 0 mM L-AA 5 mM L-AA 10 mM L-AA
** ** **
A
1.21.0
0.8
0.6
0.4
Cellviability(%ofcontrol) 0.2
0.0
L-AA (10 mM) z-VAD-fmk ( M)
+ +
50
+ 100
*
# #
B
Fig. 4. Caspase activation in L-ascorbic acid (L-AA)-treated Hep-2 cells. (A) The caspase-3, -8, and -9 activity was determined by treating the cells with 5 and 10 mM L-AA for 24 hours and the cell lysates were analyzed via a colorimetric assay. (B) Apoptotic inhibitory effects by z-VAD-fmk, a general caspase inhibitor, were analyzed in 10 mM L-AA-treated cells using an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. The data represent the mean ± standard deviation from triplicate measurements.
*p < 0.05, **p < 0.01, compared with the untreated control; #p < 0.05, compared with the L-AA-treated cells group.
Fig. 5. Role of mitogen-activated protein kinase and Akt signaling in L-ascorbic acid (L-AA)-induced apoptosis of Hep-2 cells. (A) The cells were incubated with 10 mM L-AA for 1–36 hours, and cell lysates were assayed by Western blot. (B) The nuclear extracts of cells treated with 10 mM L-AA for 24 hours were analyzed by electrophoretic mobility shift assay to determine the activated nuclear factor-κB (NF-κB) using a radiolabeled oligonucleotide probe. Protein (50 µg) from these nuclear lysates was subjected to Western blot using the NF-κB (p65 subunit) antibody. (C) After pretreatment with 10 µM BAY11-7082 (Bay), 20 µM LY294002 (LY), 10 µM SB203580 (SB) and 10 µM SP600125 (SP) for 24 hours before 10 mM L-AA treatment, the number of apoptotic cells was de- termined by flow cytometry via annexin V staining. (D) The percentage of annexin V-positive cells indicated the ratio of apoptotic cells, based on flow cytom- etry results. The data represent the mean ± standard deviation from triplicate measurements.
Counts 1501200306090
100 101 102 103 104 Bay (10 M)
Annexin V
Counts 1501200306090
100 101 102 103 104 LY (20 M)
Annexin V
Counts 1501200306090
100 101 102 103 104 SB & SP (10 M)
Annexin V
C
5040 30 20 Meanintensity(%) 10
0
L-AA (10 mM) Bay (10 M) LY (20 M) SB & SP (10 M)
#
D
+ +
+ +
# #
NF- B
Free probe L-AA (mM) p65 L-AA (mM) p-ERK1/2
p-p38 p-JNK p-Akt (ser473) Akt
3 5 10
3 5 10
A B
0 1 3 6 12 24 36
Time (hr)
1
binding activity of NF- κB in the Hep-2 cells. In addition, the protein level of the p65 subunit of NF- κB in the nuclear lysates of Hep-2 cells was also decreased by L-AA-treatment (Fig.
5B).
The role of NF- κB, Akt, and MAPK signaling pathways in L- AA-induced apoptosis was analyzed by pretreating the cells with 10 µM BAY11-7082 (NF-κB inhibitor), 20 µM LY294002 (PI3K/Akt inhibitor), 10 µM SB203580 (p38 inhibitor) and SP600125 (JNK inhibitor) for 1 hour before L-AA addition.
After the inhibitor treatment, flow cytometry analysis of the apoptosis of L-AA-induced Hep-2 cells showed a decrease in the level of annexin-V-positive cells compared with the L-AA- treated group (Fig. 5C and 5D). These results strongly indicated that NF- κB, Akt, and MAPK signal pathways were involved in the L-AA-induced apoptosis of Hep-2 cells.
Discussion Discussion
Several years of investigation into the effect of L-AA on vital cellular functions and the role of key physiological antioxidants revealed that many signaling intermediates play an anticancer role. However, other studies have demonstrated that L-AA may occasionally act as a pro-oxidant inducing the destruction of tumors. Kang et al. [19] demonstrated that L-AA induced apoptosis of cancer cells by inhibiting the iron uptake. Apop- tosis is important in controlling the cell number and prolifera- tion as part of normal development. It is also essential for the elimination of cancer cells [20]. The present study showed that the decreased cell viability following L-AA treatment of Hep- 2 cells was the result of apoptosis induction. Morphological analyses of the L-AA-induced Hep-2 cells revealed apoptotic characteristics, such as the appearance of apoptotic bodies and chromatin condensation, when compared to control cells via DAPI staining. Moreover, flow cytometry with annexin-V staining confirmed that L-AA dose-dependently increased the cellular the ratio of apoptotic death.
The apoptotic process is mainly intrinsic, whereas the ex- trinsic pathway is mediated by cysteine proteases known as caspases. The extrinsic pathway is triggered by the death re- ceptor, which induces the activation of the initiator caspase-8 and -10, followed by the activation of caspase-3. Active caspase-8 also cleaves cleaves BID to truncated BID (tBID), which induces the release of cytochrome c by the mitochon- drial membrane in the intrinsic pathway.
The intrinsic pathway of apoptosis is triggered by cellular stress. The mitochondrial stress is induced by factors such as
DNA damage. One of the stress signal regulators of the mito- chondria-mediated apoptosis is the Bcl-2 family of proteins.
The Bcl-2 family includes pro-apoptotic molecules such as Bax, Bak, and Bad, which are directly involved in pore formation at the mitochondrial membrane. Bax promotes mitochondrial integrity, dissipates mitochondrial membrane potential, and facilitates the release of mitochondrial proteins such as cyto- chrome c, AIF, and EndoG. Anti-apoptotic molecules such as Bcl-2 and Bcl-x
Lact as repressors of apoptosis by preventing mitochondrial pore formation and binding with pro-apoptotic protein. In the present study, L-AA treatment not only reduced the expression of Bcl-2, but also increased the expression of Bax. Furthermore, L-AA treatment dose-dependently in- creased p53 protein in Hep-2 cells. Many studies have shown that p53 regulates the transcription genes that play an impor- tant role in cell cycle and apoptosis.
Apoptosis is induced by p53 via several pathways, includ- ing the Bcl-2 family. Other reports showed that L-AA induced apoptosis via caspase activation in various cells [21,22]. The present results showed that L-AA exposure dose-dependently activated the expression of caspase-8, -9, and -3. Interest- ingly, the general caspase inhibitor z-VAD-fmk completely inhibited the L-AA-induced cell death, indicating that L-AA activated the classical caspase-dependent signaling pathway.
Thus, we assumed that L-AA regulated the expression of Bcl- 2 family of proteins and increased the release of cytochrome c, AIF, and EndoG, leading to the activation of caspase-9, -8, and -3 and thereby induced apoptosis in Hep-2 cells.
The MAP kinases, including ERK, p38, and JNK, are activated in response to a variety of stimuli and mediate signals impor- tant for the generation of biological responses [23]. In general, ERK plays a vital role in cell growth and division, and mediates survival. However, Park et al. [24] reported the roles of ERK and p38 in L-AA-induced apoptosis of acute myeloid leukemia cells. In addition, JNK and p38 MAPKs are activated by diverse stimuli such as oxidative stress and UV [25,26]. However, the functions of these MAPK signals depend on the type of cell system. Many studies have shown that the MAPK signaling pathway plays an important role in the pharmacological action of chemotherapeutic drugs [27,28].
The Akt cellular pathway is particularly relevant to prolifera-
tion and cell survival in various cancer cells. Activation of this
pathway via phosphorylation and functional inactivation of sev-
eral pro-apoptotic targets, including the Bcl-2 family member
BAD and the protease caspase-9, in vitro prevented apoptosis
of malignant cells [29]. In addition, Akt exerts its anti-apoptotic
effects by maintaining mitochondrial interactions and prevent- ing both Bax conformational change and its mitochondrial translocation, which appears to be crucial to Akt-mediated inhibition of cytochrome c release and apoptosis [30]. In the present study, L-AA increased the levels of phosphorylated Akt in Hep-2 cells, demonstrating that the downregulation of cytochrome c release may be mediated via Akt signaling.
These results suggest that the Akt pathway may be involved in L-AA-induced apoptosis.
NF- κB exists in a latent form in the cytoplasm of unstimulat- ed cells. The NF- κB family in mammals includes p50, p65, c- Rel, p52, and RelB, which constitute the p50/p65 dimer. The transcription factor NF- κB plays a pivotal role in the carcino- genesis of various cancers, serving as one of the key elements in the apoptotic pathway, via regulation of its target gene products including Bcl-2 and Bcl-xl. Thus, it is reasonable to identify NF- κB as a target of L-AA for induction of apoptosis in Hep-2 cells. Our data showed that L-AA treatment sig- nificantly downregulated the expression of p65 and inhibited the nuclear translocation of NF- κB, suggesting that NF-κB mediates the induction of apoptosis in Hep-2 cells by L-AA.
The activation of ERK1/2, JNK, and p38 MAPKs and of Akt and NF- κB by L-AA in various cancer cells precedes the cleav- ages of caspase-9 and -3, indicating that the MAPK, Akt and NF- κB signaling pathways are upstream of the caspase cas- cades, as reported in the human placenta, leukemia, anemia, and carcinoma cell lines [8,31,32]. L-AA-induced activation of MAPK, Akt, and NF- κB was sustained for 24 hours, suggest- ing that the signaling may have played a role in L-AA-induced apoptosis [6,24,33]. In our experiments investigating the func- tional role of these cascades, we found that inhibition of this pathway using p38- and JNK-specific inhibitors SB203580 and SP600125, Akt-specific inhibitor LY, and NF- κB specific- inhibitor BAY11-7082 suppressed the L-AA dependent in- duction of apoptosis. Thus, it appears that important signals regulating the L-AA-dependent responses are mediated by MAPK, Akt, and NF- κB. All these events appear to contribute
to the induction of apoptosis, and the upstream regulatory mechanisms of such pro-apoptotic signals are analyzed in the present study. Our data clearly establish that L-AA treatment induces phosphorylation of MAPK and Akt and downregulation of NF- κB in Hep-2 cells.
In conclusion, L-AA induced apoptosis via intrinsic and ex- trinsic pathways by targeting the Bcl-2 family, resulting in the mitochondrial release of cytochrome c, followed by the nuclear translocation of AIF and EndoG in Hep-2 cells. The target signal of L-AA-induced apoptosis may be mediated via MAPK, PI3K, and NF- κB signaling pathways. Our finding on the mechanism of L-AA-induced apoptosis in Hep-2 cells may contribute to the process of developing molecular signal- targeted therapy for laryngeal epithelial cancer Hep-2 cells.
In this study, we explored the anti-tumor activity of L-AA in squamous carcinoma head and neck cancer cells and dem- onstrated that L-AA efficiently inhibited growth and induced apoptosis of laryngeal Hep-2 cells. These anti-tumor activities of L-AA were mediated via activation of the NF- κB/Akt/MAPK pathway. Thus, we propose L-AA as a promising chemothera- peutic agent for the treatment of human squamous carcinoma head and neck cancer via regulation of the NF- κB/Akt/MAPK signaling pathway.
Acknowledgements Acknowledgements
This work was supported by the National Research Founda- tion of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2019R1A5A2027521, 2019R1A2C1087744 and 2020R1A2C1003310) and by a grant from the Chonnam Na- tional University Hospital Research Institute of Clinical Medi- cine (BCRI19028).
Conflicts of Interest Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
References References
1. Forastiere AA, Goepfert H, Maor M, Pajak TF, Weber R, Mor- rison W, Glisson B, Trotti A, Ridge JA, Chao C, Peters G, Lee DJ, Leaf A, Ensley J, Cooper J. Concurrent chemotherapy and radiotherapy for organ preservation in advanced laryngeal cancer. N Engl J Med 2003;349:2091-8. doi: 10.1056/NEJ- Moa031317.
2. Dechaphunkul T. Epidemiology, risk factors, and overall sur- vival rate of laryngeal cancer in Songklanagarind Hospital. J Med Assoc Thai 2011;94:355-60.
3. Mashima T, Naito M, Kataoka S, Tsuruo T. [Cancer chemo- therapy and apoptosis]. Nihon Rinsho 1996;54:1935-42.
Japanese.
4. Schwartz LH, Urban T, Hercberg S. [Antioxidant minerals and vitamins. Role in cancer prevention]. Presse Med 1994;23:
1826-30. French.
5. Charalabopoulos K, Karkabounas S, Charalabopoulos AK, Papalimneou V, Ioachim E, Giannakopoulos X. Inhibition of benzo(a)pyrene-induced carcinogenesis by vitamin C alone and by vitamin C/vitamin E and selenium/glutathione. Biol Trace Elem Res 2003;93:201-12. doi: 10.1385/BTER:93:1-3:201.
6. Bowie AG, O’Neill LA. Vitamin C inhibits NF-kappa B acti- vation by TNF via the activation of p38 mitogen-activated protein kinase. J Immunol 2000;165:7180-8. doi: 10.4049/
jimmunol.165.12.7180.
7. Cárcamo JM, Pedraza A, Bórquez-Ojeda O, Golde DW. Vita- min C suppresses TNF alpha-induced NF kappa B activation by inhibiting I kappa B alpha phosphorylation. Biochemistry 2002;41:12995-3002. doi: 10.1021/bi0263210.
8. Su YT, Chang HL, Shyue SK, Hsu SL. Emodin induces apop- tosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway.
Biochem Pharmacol 2005;70:229-41. doi: 10.1016/j.bcp.
2005.04.026.
9. Dackour R, Carter T, Steinberg BM. Phosphatidylinositol 3-ki- nase regulates early differentiation in human laryngeal kerati- nocytes. In Vitro Cell Dev Biol Anim 2005;41:111-7. doi: 10.
1290/0501003.1.
10. Garavello W, Nicolini G, Aguzzi A, Maggioni D, Leone BE, Viganò P, Gaini RM, Tredici G. Selective reduction of extracel- lular signal-regulated protein kinase (ERK) phosphorylation in squamous cell carcinoma of the larynx. Oncol Rep 2006;16:
479-84. doi: 10.3892/or.16.3.479.
11. Chandler NM, Canete JJ, Callery MP. Increased expression of NF-kappa B subunits in human pancreatic cancer cells. J Surg Res 2004;118:9-14. doi: 10.1016/S0022-4804(03)00354-8.
12. Yan M, Xu Q, Zhang P, Zhou XJ, Zhang ZY, Chen WT. Corre- lation of NF-kappaB signal pathway with tumor metastasis of human head and neck squamous cell carcinoma. BMC Cancer 2010;10:437. doi: 10.1186/1471-2407-10-437.
13. Yoshida K, Sasaki R, Nishimura H, Okamoto Y, Suzuki Y, Kaw- abe T, Saito M, Otsuki N, Hayashi Y, Soejima T, Nibu K, Sug- imura K. Nuclear factor-kappaB expression as a novel marker of radioresistance in early-stage laryngeal cancer. Head Neck 2010;32:646-55. doi: 10.1002/hed.21239.
14. Trisciuoglio D, Iervolino A, Zupi G, Del Bufalo D. Involvement of PI3K and MAPK signaling in bcl-2-induced vascular endo- thelial growth factor expression in melanoma cells. Mol Biol Cell 2005;16:4153-62. doi: 10.1091/mbc.e04-12-1087.
15. Hsu S, Singh B, Schuster G. Induction of apoptosis in oral cancer cells: agents and mechanisms for potential therapy and prevention. Oral Oncol 2004;40:461-73. doi: 10.1016/j.
oraloncology.2003.09.012.
16. Ola MS, Nawaz M, Ahsan H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem 2011;351:41-58. doi: 10.1007/s11010-010-0709-x.
17. Gottlieb E, Vander Heiden MG, Thompson CB. Bcl-x(L) pre- vents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol Cell Biol 2000;20:5680-9. doi: 10.1128/mcb.20.15.5680-5689.2000.
18. Beg AA, Baltimore D. An essential role for NF-kappaB in pre- venting TNF-alpha-induced cell death. Science 1996;274:
782-4. doi: 10.1126/science.274.5288.782.
19. Kang JS, Cho D, Kim YI, Hahm E, Kim YS, Jin SN, Kim HN, Kim D, Hur D, Park H, Hwang YI, Lee WJ. Sodium ascor- bate (vitamin C) induces apoptosis in melanoma cells via the down-regulation of transferrin receptor dependent iron up- take. J Cell Physiol 2005;204:192-7. doi: 10.1002/jcp.20286.
20. Vaux DL, Korsmeyer SJ. Cell death in development. Cell 1999;
96:245-54. doi: 10.1016/s0092-8674(00)80564-4.
21. Reed JC, Jurgensmeier JM, Matsuyama S. Bcl-2 family pro- teins and mitochondria. Biochim Biophys Acta 1998;1366:
127-37. doi: 10.1016/s0005-2728(98)00108-x.
22. Martinou JC, Green DR. Breaking the mitochondrial barrier.
Nat Rev Mol Cell Biol 2001;2:63-7. doi: 10.1038/35048069.
23. Plotnikov A, Zehorai E, Procaccia S, Seger R. The MAPK cas- cades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim Biophys Acta 2011;1813:
1619-33. doi: 10.1016/j.bbamcr.2010.12.012.
24. Park S, Park CH, Hahm ER, Kim K, Kimler BF, Lee SJ, Park HK, Lee SH, Kim WS, Jung CW, Park K, Riordan HD, Lee JH. Activation of Raf1 and the ERK pathway in response to l-ascorbic acid in acute myeloid leukemia cells. Cell Signal 2005;17:111-9. doi: 10.1016/j.cellsig.2004.06.006.
25. McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling path- ways. Antioxid Redox Signal 2006;8:1775-89. doi: 10.1089/
ars.2006.8.1775.
26. Kim EK, Choi EJ. Pathological roles of MAPK signaling path- ways in human diseases. Biochim Biophys Acta 2010;1802:
396-405. doi: 10.1016/j.bbadis.2009.12.009.
27. Fan M, Chambers TC. Role of mitogen-activated protein ki- nases in the response of tumor cells to chemotherapy. Drug Resist Updat 2001;4:253-67. doi: 10.1054/drup.2001.0214.
28. Boldt S, Weidle UH, Kolch W. The role of MAPK pathways in the action of chemotherapeutic drugs. Carcinogenesis 2002;
23:1831-8. doi: 10.1093/carcin/23.11.1831.
29. Lin J, Guan Z, Wang C, Feng L, Zheng Y, Caicedo E, Bearth E, Peng JR, Gaffney P, Ondrey FG. Inhibitor of differentiation 1 contributes to head and neck squamous cell carcinoma sur- vival via the NF-kappaB/survivin and phosphoinositide 3-ki- nase/Akt signaling pathways. Clin Cancer Res 2010;16:77- 87. doi: 10.1158/1078-0432.CCR-08-2362.
30. Yamaguchi H, Wang HG. The protein kinase PKB/Akt regu- lates cell survival and apoptosis by inhibiting Bax conforma- tional change. Oncogene 2001;20:7779-86. doi: 10.1038/sj.
onc.1204984.
31. Pearl-Yafe M, Halperin D, Scheuerman O, Fabian I. The p38
pathway partially mediates caspase-3 activation induced by reactive oxygen species in Fanconi anemia C cells. Biochem Pharmacol 2004;67:539-46. doi: 10.1016/j.bcp.2003.09.024.
32. Cindrova-Davies T, Spasic-Boskovic O, Jauniaux E, Char- nock-Jones DS, Burton GJ. Nuclear factor-kappa B, p38, and stress-activated protein kinase mitogen-activated protein kinase signaling pathways regulate proinflammatory cytokines and apoptosis in human placental explants in response to oxidative stress: effects of antioxidant vitamins. Am J Pathol 2007;170:1511-20. doi: 10.2353/ajpath.2007.061035.
33. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Op- posing effects of ERK and JNK-p38 MAP kinases on apop- tosis. Science 1995;270:1326-31. doi: 10.1126/science.270.
5240.1326.