Fanconi 빈혈은 상염색체 열성으로 유전되는 선 천성 질환으로 골수부전과 함께 저신장과 비정상 적인 피부의 색소침착 및 발달기형을 특징으로 하며 경과 중 골수형성이상증후군이나 급성골수 성백혈병의 발병빈도가 높다1∼5). 치료로는 조혈 모세포이식만이 정상적인 조혈기능을 회복시킬 수 있는데5,6), 조직적합항원(human leukocyte anti-
gen, HLA)이 일치하는 혈연간 이식의 경우에는 70% 이상의 5년 생존율이 보고되고 있으나6,7), 조 혈모세포의 공급원으로서 비혈연간 제대혈을 이 용한 이식은 현재까지 치료 성적이 만족스럽지 못하다5,9). 한편 Fanconi 빈혈에서 조혈모세포이식 시 전처치로는 저용량의 cyclophosphamide와 전신 방사선조사(total body irradiation, TBI)가 일반적인 데, 최근에는 TBI 후 발생하는 여러 가지 부작용 및 이차 악성종양의 발생을 감소시키기 위하여 TBI 없이 fludarabine을 이용한 전처치가 시도되고
있다11∼13). 저자들은 Fanconi 빈혈로 진단된 6세
Pusan National University, Busan, Korea
Fanconi anemia is an autosomal recessive disorder characterized by progressive bone marrow failure, growth retardation, abnormal skin pigmentation, developmental anomalies and marked predisposition to myelodysplastic syndrome or acute myeloid leukemia. Allogeneic hematopoietic stem cell transplantation offers the only curative treatment for bone marrow complications of these patients. Recently umbilical cord blood transplantation has been tried as a successful alternative to bone marrow transplantation in patients with Fanconi anemia, but the results are somewhat disappointing yet. In this report, we describe a case of unrelated HLA 1 antigen mismatched cord blood transplantation using non-TBI conditioning regimen in a 6-year-old boy with Fanconi anemia who is alive with a perfect performance status and normal blood count currently 17 months after the transplantation.
(Clin Pediatr Hematol Oncol 2007;14:68∼72)
Key Words: Fanconi anemia, Unrelated cord blood transplantation
책임저자 임영탁, 부산시 서구 아미동 1가 10 부산대학교 의과대학 소아과학교실, 602-739 Tel: 051-240-7295, Fax: 051-248-6205 E-mail: [email protected]
임상소아혈액종양
제 14 권 제 1 호 2007 68
남아에서 전처치로 TBI 없이 HLA 1부위 불일치 비혈연간 제대혈이식을 시행하여 생착한 뒤 이식 후 17개월까지 무병 생존하고 있어 문헌고찰과 함께 보고하는 바이다.
증 례
환 아: 김○○, 6세, 남아
주 소: 창백과 전신에 쉽게 드는 멍
현병력: 환아는 영아기 이후부터 창백하게 보였 으며 유치원에 다니는 동안 사지에 쉽게 멍이 드 는 병력이 있었는데, 같은 나이의 친구들보다 키 가 작았고 체중이 적었으며 인근 병원에서 검사 한 혈액검사에서 범혈구감소증이 있어 본원으로 전원되었다.
가족력: 특이 소견이 없었다.
진찰소견: 신장은 108 cm로 3∼10백분위수, 체 중은 15.6 kg으로 3백분위수 미만, 두위는 47.5 cm 로 3백분위수 미만으로 저신장, 저체중 및 소두증 을 보였다. 환아의 체간부에는 거대 청색 반점 및 café-au-lait 반점이 있었지만, 다른 신체적인 기형 및 간비종대나 림프절비대는 없었다.
검사소견: 혈액검사에서 백혈구 1,030/μL (호중
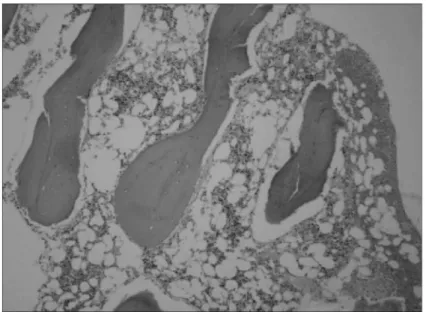
구 440/μL), 혈색소 4.9 g/dL, 교정망상적혈구 0.52%, 혈소판 18,000/μL로 범혈구감소증을 보였 다. 골수흡인에서 모세포나 이형성세포는 관찰할 수 없었으며, 골수생검에서 세포충실도는 20%였다 (Fig. 1). 말초혈액 세포유전학검사에서 mitomycin C에 의한 림프구의 염색체파열(chromosome breakage) 이 있어 중증 Fanconi 빈혈로 진단하였다(Fig. 2).
Fig. 1. The bone marrow biopsy at initial diagnosis shows hy- pocellular marrow with fo- cal hematopoietic cells (app- roximately 20% cellularity, H&E stain, ×40).
Fig. 2. The cytogenetic analysis of peripheral blood lymphocytes shows markedly increased chromo- some breakages and gaps (arrows) with mito- mycin C.
치료 및 경과: HLA가 일치하는 혈연 및 비혈연 공여자를 찾을 수 없어 HLA-A, -B 및 -DRB1 중 DRB1 1부위 불일치 비혈연간 제대혈이식을 시행 하였다. 전처치로 fludarabine (30 mg/m2/일, 6일), cyclophosphamide (10 mg/kg/일, 4일), thymoglobu- lin (3 mg/kg/일, 4일)을 투여하였다. 이식편대숙주 병의 예방을 위해 이식 전 2일부터 tacrolimus (0.03 mg/kg/일)와 이식 후 1일, 3일, 6일 및 11일 에 mini MTX (methotrexate, 5 mg/m2/일)를 투여했 다. 제대혈의 해동 후 이식된 총유핵세포는 1.9×
107/kg, CD34 양성 세포는 1.5×105/kg이었다. 백 혈구 생착 및 혈소판 생착은 각각 이식 후 24일과 42일에 이루어졌고, 이식 후 28일째 골수세포에서 VNTR (variable number of tandem repeats)을 시행 하여 공여자와 완전키메리즘이 확인되었으며, 이 식 후 24일 및 133일에 각각 골수검사에서 정상 골수소견을 보였다(Fig. 3). 환아는 이식 후 출혈 성 방광염 및 1등급 급성이식편대숙주병(피부 및 위장관)과 일과성혈소판감소증 등의 이식 합병증 이 있었으나 이식 후 17개월째 혈액검사에서 백 혈구 8,910/μL (호중구 40%), 혈색소 15.2 g/dL, 교정망상적혈구 0.99%, 혈소판 167,000/μL로 특별 한 이상 없이 건강한 상태로 추적 관찰 중에 있다.
고 찰
Fanconi 빈혈은 상염색체 열성으로 유전되는 질 환으로 골수부전에 의한 범혈구감소증과 여러 가 지 기형을 동반할 수 있으며, 경과 중 골수형성이 상증후군, 급성골수성백혈병으로 진행할 수 있으 며 상피종양의 발생빈도가 높은 질환이다1∼5). 환 자의 75%는 뚜렷한 선천성 기형을 동반하는데, café-au-lait 반점 형태의 피부착색 혹은 색소결핍, 저신장, 엄지와 요골의 골격이상, 소두증, 눈 및 귀의 이상, 신기형, 선천성 심질환 및 성선기능감 소증을 동반할 수 있지만, 약 25%에서는 이런 기 형을 동반하지 않을 수도 있다1,4,5).
Fanconi 빈혈 환아들은 범혈구감소증이 나타나 지만 대부분은 출생 당시나 초기 영아기에는 보 이지 않고 평균 5∼10세가 되면 의미 있게 나타 난다. 이후에 골수형성이상증후군 혹은 급성골수 성백혈병으로 진행할 수 있는데, 급성골수성백혈 병의 평균 발병연령은 14세로 보고되고 있고 정 상인보다 800배 높게 발병하고 있다5).
Fanconi 빈혈에서 범혈구감소증 이외의 검사소 견으로는 태아 혈색소의 증가와 적혈구의 대구증 Fig. 3. The bone marrow biopsy
at post-transplantation day 133 shows approximately 70% cellular marrow with hematopoietic cells (H&E stain, ×40).
(macrocytosis)이 흔히 나타나는데, 결정적인 선별 검사는 말초혈 림프구나 피부의 섬유모세포들이 소량의 diepoxybutane (DEB)이나 mitomycin C (MMC)에 의해 염색체파열(chromosome breakage) 이 오거나 세포주기 중 G2의 현저한 증가를 보이 는 소견으로 확인이 가능하다3∼5).
치료는 조혈모세포이식만이 골수부전에 대한 완치와 골수형성이상증후군, 백혈병을 예방하기 위한 유일한 치료인데, HLA 일치 혈연간 동종조 혈모세포이식의 경우 5년 생존율은 70% 이상으 로 보고되고 있다6,7). 그러나 불행히도 많은 수의 Fanconi 빈혈 환자는 HLA가 완전 일치하는 혈연 공여자를 찾을 수 없으므로 비혈연간 조혈모세포 이식을 고려하게 되는데 예후는 HLA 일치 혈연 간 조혈모세포이식보다 훨씬 좋지 못하다. Fan- coni 빈혈의 HLA 일치 비혈연간 조혈모세포이식 에 대한 연구는 European Group for Blood and Marrow Transplantation (EBMT)에서 69례의 환자 를 대상으로 거의 전례에서 전처치로 TBI를 하였 으며 3년 생존율을 33%로 보고하였다8). 특히 전 처치와 관련된 독성, 이식거부 및 이식편대숙주병 등이 이식실패의 중요한 원인인데, 환자의 나이가 많을수록(>10세), 동반되는 기형이 많을수록, 이 전에 androgen 치료를 받은 과거력이 있는 경우, 공여자가 여성인 경우 등이 예후가 더 나쁜 것으 로 보고되었다5,8).
비혈연간 조혈모세포이식의 다른 공급원으로서 제대혈이식은 아직 제한된 수의 환자만 보고되고 있는데, 최근 생착과 생존율이 비혈연간 골수이식 과 큰 차이가 없는 것으로 보고되고 있다5). Gluc- kman 등9)은 HLA 1부위 불일치 29명, 2부위 혹은 3부위 불일치 33명을 포함한 모두 72명의 Fanconi 빈혈 환자에서 비혈연간 제대혈이식을 시행하여 생착률 54%, 2등급 이상의 급성이식편대숙주병 32%, 만성이식편대숙주병 11% 및 평균 23개월 생존율 36%를 보고하였다. 이들 중 많은 환자에 서 전처치로 TBI가 포함되었으며 HLA 불일치 항 원 수가 많을수록, 이식된 유핵세포 수가 적을수 록, fludarabine이 전처치에 포함되지 않을수록 예
후가 좋지 않았다.
한편 Fanconi 빈혈의 조혈모세포이식 시 예후는 전처치로 사용되는 항암제나 방사선 치료의 양에 따라 다르다. 이는 Fanconi 빈혈 환자의 세포가 DNA 교차결합(cross-linking) 약제와 방사선에 대 한 감수성이 높기 때문인데, 통상적으로 재생불량 성빈혈 환자에게 사용되는 cyclophosphamide 200 mg/kg와 TBI에는 높은 독성과 이차 악성종양의 발생빈도를 보인다7,10). 이식 전처치의 독성을 줄 이기 위해 저용량 cyclophosphamide 20 mg/kg과 4∼6 Gy의 TAI (thoraco-abdominal irradiation) 혹은 cyclophosphamide 40 mg/kg, ATG (antithymocyte globulin)와 4.5 Gy의 TBI와 같은 저강도의 전처치 가 일반적으로 이용되었지만, 이 전처치조차도 생 착실패 및 2∼4등급 급성이식편대숙주병의 발병 률이 각각 30% 및 50%로 보고되었다7,8). 이를 극 복하기 위해 최근에는 TBI 없이 fludarabine이 포 함된 전처치가 시도되고 있는데, 이는 강력한 T- 세포 면역억제를 통하여 이식거부를 줄이면서, 약 제와 관련된 독성은 훨씬 적은 것으로 보고되기
때문이다11∼13). Motwani 등14)은 HLA 1부위 및 2
부위 불일치 2명을 포함한 4명의 Fanconi 빈혈 환 아에게 비혈연간 제대혈이식을 시행하였는데 전 처치로 TBI 없이 fludarabine (25 mg/m2/일, 5일), cyclophosphamide (7.5 mg/kg/일, 4일), ATG (horse, 12.5 mg/kg/일, 4일)을 투여하여, 4명 모두 2∼3주 이내에 생착되었으며 평균 37개월째 심한 이식관 련 합병증 없이 생존하고 있다고 보고하였다. 저 자들도 본 증례의 환아에게 HLA 일치 혈연 및 비 혈연 공여자를 찾을 수 없어 HLA 1부위 불일치 제대혈이식을 시행하였는데 전처치로 방사선조사 없이 fludarabine (30 mg/m2/일, 6일), cyclophospha- mide (10 mg/kg/일, 4일), thymoglobulin (3 mg/kg/
일, 4일)을 투여하였으며, 이식된 총유핵세포 수 가 1.9×107/kg로 비교적 충분하지는 않았으나 생 착이 잘 되었고 심한 이식관련 합병증 없이 17개 월째 생존하고 있다.
결론적으로 Fanconi 빈혈 환자의 비혈연간 제대 혈이식은 비록 생착이 늦고 이식되는 유핵세포수
고 HLA 1부위 불일치 비혈연간 제대혈이식을 시 행하였으며, 성공적인 생착 이후 17개월까지 무병 생존하고 있어 문헌고찰과 함께 보고하는 바이다.
참 고 문 헌
1. Cho SH, Kook H, Kim GM, Yoon WS, Cho TH, Hwang TJ. A clinical study of Fanconi's anemia.
Korean J Pediatr Hematol Oncol 1997;4:70-7 2. Cho TH, Ryu NE, Kim CJ, Lee JH, Hwang TJ. In-
fant with Fanconi anemia presenting with myelodys- plastic syndrome. Korean J Pediatr Hematol Oncol 1998;5:322-7
3. Ryang DW, Cho D, Hong WP, Kook H, Hwang TJ.
Chromosome breakage test for the diagnosis of Fanconi's anemia. Korean J Clin Pathol 1998;18:
101-6
4. Alter BP. Inherited bone marrow failure syndromes.
In: Nathan DG, Oskin SH, Ginsburg D, Look AT, editors. Hematology of infancy and childhood. 6th ed. Philadelphia. WB Saunders Co, 2003:280-365 5. Kook H. Fanconi anemia: current management. He-
matol 2005;10(Suppl 1);108-10
6. Gluckman E, Auerbach AD, Horowitz MM, Sobo- cinski KA, Ash RC, Bortin MM, et al. Bone marrow transplantation for Fanconi anemia. Blood 1995; 86:2856-62
7. Socie G, Devergie A, Girinski T, Piel G, Ribaud P,
R, Wagner J, et al. Results of unrelated cord blood transplants in Fanconi anemia. Blood 2004;104(Suppl 1):590
10. Deeg HJ, Socie G, Schoch G, Henry-Amar M, Witherspoon RP, Devergie A, et al. Malignancies after marrow transplantation for aplastic anemia and Fanconi anemia: A joint Seattle and Paris analysis of results in 700 patients. Blood 1996;87:386-92 11. de la Fuente J, Reiss S, McCloy M, Vulliamy T,
Roberts IAG, Rahemtulla A, et al. Non-TBI stem cell transplantation protocol for Fanconi anaemia using HLA-compatible sibling and unrelated donors.
Bone Marrow Transplant 2003;32:653-6
12. George B, Mathews V, Shaji RV, Srivastava V, Srivastava A, Chandy M. Fludarabine-based condi- tioning for allogeneic stem cell transplantation for multiply transfused patients with Fanconi's anemia.
Bone Marrow Transplant 2005;35:341-3
13. Bitan M, Or R, Shapira MY, Aker M, Resnick IB, Ackerstein A, et al. Fludarabine-based reduced inten- sity conditioning for stem cell transplantation of Fanconi anemia patients from fully matched related and unrelated donors. Biol Blood Marrow Transplant 2006;12:712-8
14) Motwani J, Lawson SE, Darbyshire PJ. Successful HSCT using nonradiotherapy-based conditioning re- gimens and alternative donors in patients with Fan- coni anemia-experience in a single UK centere. Bo- ne Marrow Transplant 2005;36:405-10