□ 원 저 □ Vol. 14, No. 2, November, 2006
1)
책임저자 : 황규근, 동아대학교 의과대학 소아과학교실
Tel : 051)240-2958, Fax : 051)242-2765
E-mail : [email protected]
Introduction
Status epilepticus (SE) is a medical emer- gency and more common in children than in
The Effect of Topiramate on Status Epilepticus- Induced Neurotoxicity in Immature Mouse Brain
Sang Soo Park, M.D., Hae Rahn Bae, M.D.*
and Kyu Geun Hwang, M.D.
Department of Pediatrics and Physiology
*, Dong-A University Medical School, Busan, Korea
= 국문 요약 =
미성숙 쥐의 뇌에서 간질중첩증에 의한 신경세포 손상에 미치는 Topiramate의 효과
동아대학교 의과대학 소아과학교실, 생리학교실
*
박상수·배혜란
*·황규근
목 적 : 미성숙 뇌는 경련에 대한 감수성, 경련의 특징 및 항간질약제의 반응성 등에서 성 숙 뇌와 다르다. 출생 초기의 간질 경련과 항간질약제의 사용이 이 후 뇌 성숙 과정에 어떠 한 영향을 주는지가 소아신경학 분야의 주요 관심사이다. 이에 본 연구자는 미성숙 쥐에서 간질중첩증을 유발시킨 후 신경세포의 손상 및 신경 흥분성 정도를 성숙 뇌와 비교하고 항 간질약제인 topiramate가 신경세포 손상 및 뇌 성숙 과정에 미치는 효과를 알아보고자 하였 다.
방 법 : 생후 14일된 미성숙 쥐에 kainate를 주입하여 간질중첩증을 유발시킨 뒤 실험군은 1주 혹은 1달 동안 topiramate를 투여한 뒤 뇌를 적출하여, 간질중첩증으로 인한 지속적인 신경흥분성은 western blot을 이용하여 glutamate 및 GABA 수용체의 발현 차이로 관찰하 였고, 신경세포 손상 유발 정도는 공촛점현미경을 이용하여 TUNEL 및 HE 염색으로 관찰 하였다.
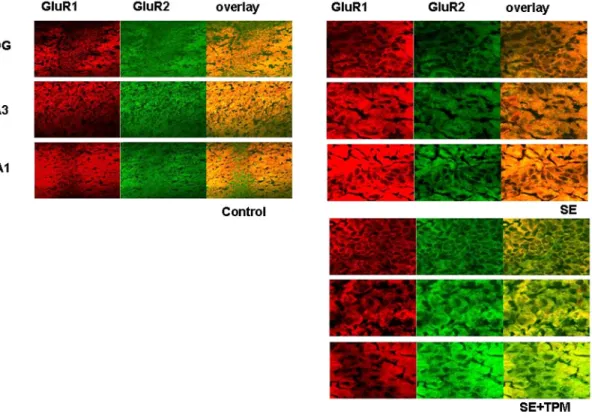
결 과 : 미성숙 쥐에서 간질중첩증 유발 1개월 후 해마의 유의한 세포손상과 구조적 변화 를 관찰하였다. 간질중첩증으로 인해 GluR2에 비해 GluR1의 발현이 현저히 증가하여 신경 세포 흥분독성이 유발될 수 있는 Ca
2+ 투과성 AMPA 수용체의 형성이 증가되었다. Topira- mate 치료군에서는 간질중첩증으로 인해 유발된 GluR1의 발현 증가가 억제되었다. GABA
B
수용체는 간질중첩증 1개월 후 유의한 변화가 없었으나, GABA
A
수용체의 발현은 현저히
증가되었으며 이는 topiramate 처리에 의해 억제되었다. Topiramate 치료군에서는 간질중첩 증으로 인해 유발된 해마 CA1 및 CA3 부위의 세포 손상과 구조적 변화가 감소됨으로써, topiramate는 미성숙 뇌에서 간질중첩증으로 인한 해마 손상을 보호하는 효과가 있었다.
결 론 : 이상의 결과를 종합하면 topiramate는 미성숙 쥐의 GluR1와 GABA
A
수용체의 발 현을 조절함으로써 신경보호작용을 가지는 것으로 보인다.
Key Words : Topiramate, Status epilepticus, Neurotoxicity, AMPA glutamate receptor,
GABA receptor