Dichloroacetate Inhibits the Proliferation of a Human Anaplastic Thyroid Cancer Cell Line via a p53-independent Pathway
Yam Bahadur KC1, Sunil Poudel2, Eon Ju Jeon3, Ho Sang Shon3, Sung June Byun4 and Nam Ho Jeoung2*
1Department of Biomedical Science, Daegu Catholic University, Gyeongsan 38430, Korea
2Department of Pharmaceutical Science and Technology, Daegu Catholic University, Gyeongsan 38430, Korea
3Department of Internal Medicine, Daegu Catholic University School of Medicine, Daegu 42472, Korea
4Animal Biotechnology Division, National Institute of Animal Science (NIAS), RDA, Wanju-gun 55365, Korea Received November 3, 2018 /Revised November 11, 2018 /Accepted November 13, 2018
Occurrence of the Warburg effect in solid tumors causes resistance to cancer chemotherapy, and tar- geting energy metabolisms such as aerobic glycolysis is a potential strategy for alternative treatment.
Dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinase (PDK), shifts glucose metabo- lism from aerobic glycolysis to oxidative phosphorylation (OxPhos) in many cancers. In this study, we investigated the anticancer effect of DCA on a human anaplastic thyroid cancer (ATC) cell line, 8505C. We found that DCA selectively inhibits cell proliferation of the 8505C line but not of a normal thyroid line. In 8505C, the cell cycle was arrested at the G1/S phase with DCA treatment as a result of decreased antiapoptotic proteins such as HIF1α, PDK1, and Bcl-2 and increased proapoptotic pro- teins such as Bax and p21. DCA treatment enhanced the production of reactive oxygen species which consequently induced cell cycle arrest and apoptosis. Interestingly, DCA treatment not only reduced lactate production but also increased the expression of sodium-iodine symporter, indicating that it re- stores the OxPhos of glucose metabolism and the iodine metabolism of the ATC. Taken together, our findings suggest that PDK inhibitors such as DCA could be useful anticancer drugs for the treatment of ATC and may also be helpful in combination with chemotherapy and radiotherapy.
Key words : Anaplastic thyroid cancer, apoptosis, cell cycle arrest, dichloroacetate, Warburg effect
*Corresponding author
*Tel : +82-53-850-2563, Fax : +82-53-359-7431
*E-mail : [email protected]
This is an Open-Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Life Science 2018 Vol. 28. No. 12. 1469~1476 DOI : https://doi.org/10.5352/JLS.2018.28.12.1469
Introduction
Among all endocrine malignancies, thyroid cancer is the most common and accounts for 3.8% of new cancer cases in the United States [22]. The frequently found genetic alter- ations in anaplastic thyroid cancer (ATC) are mutations seen in the TP53 gene, a tumor suppressor gene, in more than 70% of ATC [14, 29]. There have been reported that p53-defi- cient primary cells increases the aerobic glycolysis, a Warburg effect, via activation of IKK-NFκB pathway [13]
and the BRAF(V600E) mutant cancer cells significantly in- crease the glucose uptake compared to that of wild type BRAF harboring cancer cells [7], suggesting that ATC may also alter the glucose metabolism from oxidative phosphor-
ylation (OxPhos) to the aerobic glycolysis. Therefore glucose metabolism shifting from OxPhos to aerobic glycolysis in- creases malignancy of thyroid cancer [6].
The mitochondrial pyruvate dehydrogenase complex (PDC) is one of major regulatory enzymes for glucose metab- olism and its activity should be tightly regulated. Pyruvate dehydrogenase kinases (PDKs) are responsible for the phos- phorylation of PDC [10]. So far, four PDK isoenzymes (PDK1, PDK2, PDK3, and PDK4) have been identified in mammalian tissues [4]. In thyroid cancer and other many cancers the expressions of the PDK1 as well as several other glycolytic enzymes are up-regulated by c-Myc and HIF-1α [14, 26]. Inhibition of PDKs stimulates the glucose OxPhos rather than aerobic glycolysis, which increases the mitochon- drial reactive oxygen species (ROS) which induces the apop- tosis of cancer cells [3, 16]. Dichloroacetate (DCA) is well-known as a PDKs inhibitor for long time [31, 32, 34].
DCA has engrossed attention as a potentially simple and economical means to target malignant tumors [3, 12, 31].
Na+/I- symporter (NIS) facilitates the active transport of iodine into the thyroid follicular cells as the crucial first step
for thyroid hormone synthesis [5, 20]. Since NIS expression is reduced in thyroid tumors [30], ATC is resistant to radio- iodine therapy [1, 33]. For the effectiveness of patient treat- ment, NIS can be a useful marker as better prognosis of thy- roid cancer treatment [35].
In this study, we examined whether DCA inhibits pro- liferation of a human anaplastic thyroid cancer, 8505C cell line, bearing p53 gene mutation [11]. Using promoter analy- sis and western blot analysis we observed that DCA induces the cell cycle arrest and apoptosis via p21 up-regulation, re- sulted from inhibition of Warburg effect. The results of this study indicate that PDKs are a good pharmacological candi- date for anaplastic thyroid cancer and PDK inhibitor might improve the efficacy of radiotherapy as well.
Materials and Methods
Cells and cell culture
N-Thy-ori, a normal thyroid cell line, and 8505C, an ana- plastic thyroid cell line, were purchased from ATCC and cultured in a humidified environment with 5% CO2 at 37℃
in RPMI-1640 containing 25 mmol/l HEPES (pH 7.4) supple- mented with 10% fetal bovine serum (FBS) (Hyclone) and 1% penicillin and streptomycin.
Chemicals, drugs, and antibodies
Propidium iodide (PI), dimethylsulfoxide (DMSO), [3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT), N-acetyl-L-cysteine (NAC), and sodium dichloroacetate (DCA) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
FBS (Hyclone, lot AYK173682), RPMI-1640 (lot 1643134), penicillin and streptomycin (lot 1546524) were purchased from Gibco-Life Technologies. Mouse polyclonal anti-human Bcl-2 (#2876), rabbit polyclonal anti-human Bax (#2772), cy- tochrome c (#4272), cyclin D1 (#2978), caspase-3 (#9661), HIF-1α (NB100-123), p-P38 (#9215), and p-Rb (39308) anti- bodies were purchased from Cell Signaling Technology (Beverly, MA, USA). p21 antibody (#556431) was purchased from BD Pharmingen. Antibodies specific to β-actin, GAPDH, and horseradish peroxidase-conjugated secondary antibodies (goat-anti-rabbit IgG, goat anti-mouse IgG) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Drug treatment
For treatments, each drug was added directly to complete cell culture media to achieve final concentrations of 25 mM
(DCA) and 10 mM (NAC). DCA stock solutions were diluted in dH2O and NAC stock solutions were diluted in 1 M so- dium bicarbonate (pH 7.0).
MTT assay
Cell viability was determined by MTT assay. Briefly, cells were seeded in 96-well plates at 6×103 cells/well and were treated with DCA (0, 5, 10, 15, 25, and 30 mM) for various time periods (24, 48, and 72 hr). After treatment, 25 μl MTT assay solution was added at a final concentration of 1 mg/ml to each well, and plates were incubated for 2-4 hr at 37℃. Thereafter, 100 μl of lysis buffer [50% DMF (N,N-di- methyl formamide), containing 20% SDS] was added to each well and incubated overnight at 37℃. The absorbance at 570 nm was measured using a microplate reader. Three in- dependent experiments were performed.
Cell cycle assay
Cell cycle analysis was carried out by measuring DNA content of cells using flow cytometry. Cells were incubated with DCA (25 mM) for 24 hr. After treatment, cells were collected, washed with PBS containing 2% FBS and fixed with cold absolute ethanol for overnight at 4℃. After wash- ing twice with ice cold PBS, the cells were incubated with 1 ml PI staining solution. Then, 50 μl of RNase A solution (100 mg/ml RNase A) was added and incubated for 30 min at room temperature in the dark. The DNA content of cells and cell-cycle distribution were analyzed by flow cytometry (Calibur, BD, USA.)
Western blot analysis
Cells were treated with DCA for various time periods, washed twice with PBS, and lysed for 30 min on ice using WIP cell-lysis buffer (150 mM NaCl, 50 mM Tris (pH 8.0), containing 1% Triton X-100, 1 mM Na2EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 mg/ml leupeptin). The insoluble cell debris was removed by centrifugation at the 10,000× g for 15 min at 4℃. The protein concentrations were determined using Bradford assay. Proteins were separated by 15%
SDS-PAGE and transferred to a PVDF membrane. After blocking with 5% (w/v) non-fat milk and washing with TBST buffer (Tris-buffered saline-containing 0.05% Tween-20), the membranes were incubated overnight at 4℃ with specific primary antibodies (1:1,000), followed by anti-rabbit IgG (1:2,000) or anti-mouse IgG (1:2,000) secondary antibodies for
1 h at room temperature and then washed again three times with TBST buffer. The membranes were incubated with en- hanced chemiluminescence substrate solution (Millipore) for 2 min according to the manufacturer’s instructions and vi- sualized (DNR, MicroChemi systems, Jerusalem).
p21 promoter assay
Cell were seeded in 6-well plates (2×103 cells/well) prior to co-transfection with 1 μg of p21 promoter reporter vector and 0.01 μg of the p-CMV-β-galactosidase plasmid vector as an internal control, using lipofectamine 2000 (Invitrogen).
Cells were transfected for 5 hr and waited for 12 hr before the drug treatment. After 24 hr of treatment, cells were lysed with passive lysis buffer (Promega). Cell lysates were then analyzed with the luciferase reporter assay system following the standard protocol using AB-2270 LuminescencerOcta (ATTO Corporation Japan). Luciferase activities were nor- malized to β-galactosidase activity of the co-transfected vector. All transfection experiments were repeated in- dependently at least three times.
Lactate measurement
8505C cells were seeded in 60-mm dishes (2×105 cells/
plate) 24 hr prior to the start of the experiment. The cells were washed with HBSS and incubated in RPMI-1640 me- dium without FBS, containing the drug (25 mM DCA), for 24 hr. After treatment, 1 ml medium was used for lactate measurements as described previously [9].
Statistical analysis
Results are expressed as mean ± SEM. Statistical sig- nificances were evaluated by Student’s t-test. p<0.05 was con- sidered significant.
Results
Dichloroacetate (DCA) inhibits the proliferation of anaplastic thyroid cancer cells
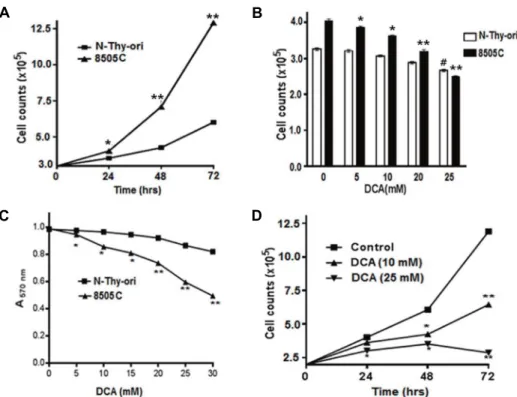
To examine the effect of DCA on different thyroid cell lines, we checked the growth pattern of 8505C and N-thy-ori cells where 8505C showed an aggressive behavior, as shown by its rapid growth compared to N-thy-ori (Fig. 1A). The cell proliferation rate of 8505C was decreased by DCA treat- ment in a dose-dependent manner while N-thy-ori was not (Fig. 1B). MTT assay showed the decreased viability of can- cer cells rapidly as compared to the N-thy-ori cells upon
DCA treatment (Fig. 1C). We observed that a low dose (10 mM) of DCA significantly inhibited the growth rate of 8505C cells within 72 hr of treatment whereas growth inhibitory effect of DCA was further increased at a higher dose (25 mM) (Fig. 1D). These findings suggest that DCA is a strong candidate as a therapeutic drug for human anaplastic thy- roid cancer.
DCA induces the cell cycle at G1 phase in ATC cells To elucidate the effect of DCA on the cell cycle in thyroid cancer cells, we performed flow cytometry analysis of DCA-treated 8505C cells using PI staining. DCA (25 mM) significantly enriched the cell populations in G1 and S phas- es, but reduced the number of cells in G2/M phase (Fig.
2A, Fig. 2B).
As shown in Fig. 2C, HIF-1α was down regulated by DCA treatment up to 24 hr. Likewise, PDK1 was also down regu- lated by DCA. Furthermore Bcl-2, an anti-apoptotic marker, was down regulated by DCA treatment after 12 hr. Bax, a pro-apoptotic protein, was up-regulated by DCA treatment at an early time point. These results suggested that DCA could alter glucose metabolism and cell survival via HIF-1a.
Similarly, DCA treatment increased the expression of p21 within 3 hr which persisted up to 24 hr (Fig. 2D). The ex- pression of cyclin D1 and phosphorylation of Rb (p-Rb) were markedly reduced by DCA treatment (Fig. 2D). Taken to- gether, these results indicated that DCA induces cell cycle arrest as well as apoptosis in human ATC cells.
DCA inhibits 8505C cell proliferation by ROS- mediated cell cycle arrest
DCA was found to switch the glucose metabolism from aerobic glycolysis to OxPhos in cancer cells, which induces the ROS production [12, 17]. Next, we examined the possi- bility that the effect of DCA on cell cycle arrest is dependent on ROS production. One hour pretreatment with N-acetyl cysteine (NAC), a ROS scavenger [2], increased the HIF-1a expression reduced by DCA treatment (Fig. 3A). The ex- pression of PDK1 and Bcl-2 was enhanced by NAC treat- ment compared to DCA alone treated cells (Fig. 3A). In addi- tion, cell cycle markers, p21 and cyclin D1, were restored by NAC treatment (Fig. 3B).
We also examined whether NAC could release the cell cycle arrest caused by DCA in 8505C cells. As expected, the cell cycle arrest by DCA was inhibited by NAC pretreatment (Fig. 3C, Fig. 3D). Phosphorylation of PDHE1α (p-PDHE1α)
A B
C D
Fig. 1. Growth of thyroid cancer cells and effect of DCA on cell viability. (A) Growth of normal and anaplastic thyroid cancer cells. Cells were grown in 35 mm culture dishes and counted at various time points. *p˂0.05, **p˂0.01 normal thyroid cells vs. 8505C cells (n=3). (B) Cells were treated with indicated concentrations of DCA and counted after 24 hr. *p˂0.05, **p˂0.01 untreated- vs. treated-8505C cells; #p˂0.05, #p˂0.05 untreated- vs. treated- N-Thy-ori cells (n=3). (C) MTT assay was performed in 96-well plates. *p˂0.05, **p˂0.01 treated N-Thy-ori cells vs. treated 8505C cells (n=3). (D) Concentration and time dependent effects of DCA on 8505C cells. Cells survive was measured by MTT analysis. *p˂0.05, **p˂0.01 untreated- vs. treated-8505C cells (n=3).
A B
C D
Fig. 2. Effect of DCA on cell cycle profile and expression of various genes. (A) Representative flow cytometry images (n=3) after PI staining. (B) Quantitative analysis. *p˂0.05 untreated- vs. treated-8505C cells. (C & D) Effect of DCA treatment on expression of genes. The cells were treated with 25 mM DCA for the indicated time points. The expression of proteins was analyzed by Western blotting.
A B
C D
E F
Fig. 3. NAC pretreatment reverses the effect of DCA. (A and B) Cells were pre-treated with NAC before one hour of DCA treatment.
Protein analysis was done by Western blot for cell signaling (A) and the cell cycle (B). Effect of NAC pre-treatment on cell cycle analysis: representative flow cytometric image (C) and quantitative analysis (D). *p˂0.05 untreated- vs. treated-8505C cells, #p˂0.05 DCA treated- vs. NAC+DCA treated-8505C cells. Effect of NAC pretreatment on DCA-treated cells on MAPK panel (E) and caspases cascade (F). GAPDH was used as a loading control.
was reduced by DCA treatment, which was not rescued by NAC pretreatment (Fig. 3E). BRAF (V600E) mutation-in- duced MAP kinases play a pivotal role in cell proliferation of cancers such as thyroid and breast cancers [5].
Phosphorylation of ERK (p-ERK1/2) was reduced by DCA treatment but was fully recovered by NAC pretreatment (Fig. 3E). However, phosphorylation of p38, an induction marker of cell cycle arrest, was dramatically increased by treatment of DCA. NAC pretreatment partially prevented the DCA-induced p38 activation (Fig. 3E). Phosphorylation of JNK was not altered by DCA treatment, indicating that
the JNK pathway is not involved in DCA-induced cell cycle arrest, at least in 8505C cells (Fig. 3E).
Next, we examined whether DCA-induced ROS could in- duce the mitochondrial apoptotic pathway in 8505C cells.
DCA treatment increased the cytosolic cytochrome C and cleavage of caspase-3, a pivotal marker of apoptosis (Fig.
3F). NAC pretreatment inhibited the cytochrome C release and activation of caspase-3 induced by DCA (Fig. 3F).
p21 promoter activity, lactate production, and NIS expression
A B C
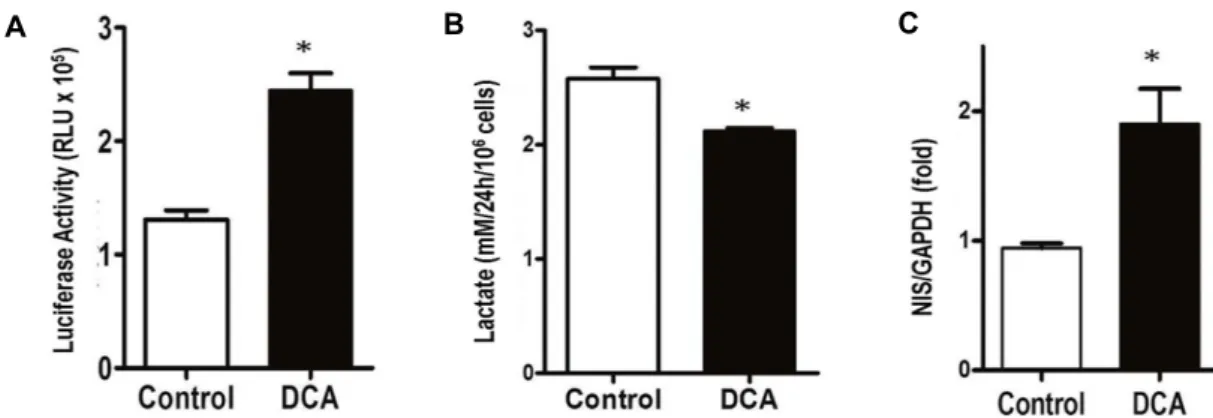
Fig. 4. DCA effect on p21 promoter activity, lactate production, and NIS expression. (A) The p21 promoter activity was measured after 24 hr of DCA treatment. β-gal assay was used as a control. *p˂0.01 untreated- vs. treated-8505C cells (n=3). (B) Lactate production in cultured medium was measured after DCA treatment for 24 hr. *p˂0.05 untreated- vs. treated-8505C cells (n=3). (C) NIS expression was measured by real time PCR after treatment with DCA for 24 hr. GAPDH was used as a control. *p˂0.01 untreated- vs. treated-8505C cells (n=3).
It has been reported that activation of p53 induces cell cycle arrest and apoptosis via activation of p21 and p27 [24].
However, most ATC cells have a mutant p53 which is not involved in the cell cycle arrest [23]. We examined whether DCA could directly increase p21 expression using transient transfection. Upon treatment with DCA after transient trans- fection of a p21-luciferase vector into 8505C cells, the pro- moter activity of p21 was increased by two folds (Fig. 4A), suggesting that DCA treatment increased the expression of p21 via a p53-independent manner.
Subsequently, DCA treatment significantly reduced the lactate production in human 8505C cells (Fig. 4B). In addi- tion, DCA treatment also increased the expression of NIS mRNA by two folds (Fig. 4C). These results suggested that DCA altered glucose and iodine metabolism of ATC cells towards that of normal thyroid cells.
Discussion
Since the radio-iodine uptake is limited in ATC due to reduced NIS levels, radiotherapy is an uneffective treatment of ATC [20, 33]. Therefore it is required to develop of an effective method to treat ATC. Activation of glycolytic genes by HIF-1a is considered critical for metabolic adaptation in cancer cells to hypoxia through increased Warburg effect.
PDK inhibition therefore might be an ideal target to treat the malignant tumor by restoration of glucose metabolic pathways [12, 26]. ATC treated with DCA showed a decrease in HIF-1a expression, leading suppression of the PDK1 ex- pression (Fig. 2C), and also reduced the phosphorylation of PDH-E1α (Fig. 3E), indicating increased PDC activity. Active
PDC is one of the major ROS sources in mitochondria [21].
Mitochondrial ROS induces the intrinsic apoptosis via the Bax-cyt C-caspase 3 signaling pathway [25]. Similar to these findings DCA treatment increased Bax and cyt C as well as cleaved caspase 3 levels (Fig. 3). Moreover, pre-treatment with NAC, a ROS scavenger, inhibited Bax expression and caspase 3 cleavage induced by DCA treatment, indicating the effect of DCA occurs in an ROS-dependent manner.
Earlier studies have shown that p21 is one of the major cyclin dependent kinase inhibitors (CKIs), which directly in- hibits the activity of CDKs, thereby leading to cell cycle ar- rest in the G1 phase [8, 37]. Even though p53 is mutated in 8505C cells, we observed that DCA treatment increased p21expression and suppressed cyclin D1 and p-Rb ex- pression (Fig. 2D), which induced cell cycle arrest in the G1
phase. In addition, DCA treatment directly increased the p21 promoter activity (Fig. 4A). These results indicate that DCA could induce cell cycle arrest in a p53-independent manner in 8505C cells, which agrees to previous reports [18, 28].
DCA treatment increased the phosphorylation of p38 and inhibited the phosphorylation of ERK, while the phosphor- ylation of JNK was not affected (Fig. 3E), indicating that MAPK signaling pathway might be regulated by oxidative stress in DCA treated 8505C cells.
Lactate is the by-product of rapid glycolysis, which occurs due to the Warburg effect [19, 36]. Lactate production is as- sociated with the proliferation and aggressiveness of cancer cells and poor prognosis [27]. In this study, DCA treatment significantly reduced the lactate production in 8505C cells (Fig. 4B). In addition, we found that DCA treatment in- creased the NIS expression in 8505C cells, suggesting that
restoration of OxPhos glucose metabolism could also re- stored the iodine metabolism in ATC. Conclusively, PDK can be used as a good target for development of anti-ATC drug by restoration of glucose metabolism as well as radio-iodine therapy for anaplastic thyroid cancers.
Acknowledgement
This research was partially supported by the Basic Science Research Program through the National Research Founda- tion of Korea, which is funded by the Ministry of Education (NRF-2014R1A1A2056130) (N.H.J.).
References
1. Are, C. and Shaha, A. R. 2006. Anaplastic thyroid carcinoma:
biology, pathogenesis, prognostic factors, and treatment approaches. Ann. Surg. Oncol. 13, 453-464.
2. Atkuri, K. R., Mantovani, J. J., Herzenberg, L. A. and Her- zenberg, L. A. 2007. N-Acetylcysteine-a safe antidote for cys- teine/glutathione deficiency. Curr. Opin. Pharmacol. 7, 355- 359.
3. Bonnet, S., Archer, S. L., Allalunis-Turner, J., Haromy, A., Beaulieu, C., Thompson, R., Lee, C. T., Lopaschuk, G. D., Puttagunta, L., Bonnet, S., Harry, G., Hashimoto, K., Porter, C. J., Andrade, M. A., Thebaud, B. and Michelakis, E. D.
2007. A mitochondria-K+ channel axis is suppressed in can- cer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11, 37-51.
4. Bowker-Kinley, M. M., Davis, W. I., Wu, P., Harris, R. A.
and Popov, K. M. 1998. Evidence for existence of tissue-spe- cific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329 (Pt 1), 191-196.
5. Carrasco, N. 1993. Iodide transport in the thyroid gland.
Biochim. Biophys. Acta. 1154, 65-82.
6. Coelho, R. G., Fortunato, R. S. and Carvalho, D. P. 2018.
Metabolic reprogramming in thyroid carcinoma. Front.
Oncol. 8, 82
7. Falck Miniotis, M., Arunan, V., Eykyn, T. R., Marais, R., Workman, P., Leach, M. O. and Beloueche-Babari, M. 2013.
MEK1/2 inhibition decreases lactate in BRAF-driven human cancer cells. Cancer Res. 73, 4039-4049.
8. Gartel, A. L. and Tyner, A. L. 2002. The role of the cyclin-de- pendent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther.
1, 639-649.
9. Gutmann, I. and Wahlefeld, A. W. 1974. Lactate determi- nation with lactate dehydrogenase and NAD, pp. 1416-1468.
In: Methods of Enzymatic Analysis, Bergmeyer HU (2ed.), Academic Press, Inc.: New York, USA.
10. Harris, R. A., Bowker-Kinley, M. M., Huang, B. and Wu, P. 2002. Regulation of the activity of the pyruvate de- hydrogenase complex. Adv. Enzyme Regul. 42, 249-259.
11. Ito, T., Seyama, T., Hayashi, Y., Dohi, K., Mizuno, T., Iwa-
moto, K., Tsuyama, N., Nakamura, N. and Akiyama, M.
1994. Establishment of 2 human thyroid-carcinoma cell-lines (8305c, 8505c) bearing p53 gene-mutations. Int. J. Oncol. 4, 583-586.
12. Jeoung, N. H. 2015. Pyruvate dehydrogenase kinases:
Therapeutic targets for diabetes and cancers. Diabetes Metab.
J. 39, 188-197.
13. Kawauchi, K., Araki, K., Tobiume, K. and Tanaka, N. 2008.
p53 regulates glucose metabolism through an IKK-NF- kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 10, 611-618.
14. Kim, J. W., Tchernyshyov, I., Semenza, G. L. and Dang, C.
V. 2006. HIF-1-mediated expression of pyruvate dehydroge- nase kinase: a metabolic switch required for cellular adapta- tion to hypoxia. Cell Metab. 3, 177-185.
15. Manzella, L., Stella, S., Pennisi, M. S., Tirrò, E., Massimino, M., Romano, C., Puma, A., Tavarelli, M. and Vigneri, P.
2017. New insights in thyroid cancer and p53 family proteins. Int. J. Mol. Sci. 18, 1325.
16. Lee, M. and Yoon, J. H. 2015. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse Warburg effect and its therapeutic implication. World J. Biol.
Chem. 6, 148-161.
17. Liou, G. Y. and Storz, P. 2010. Reactive oxygen species in cancer. Free Radic. Res. 44, 479-496.
18. Lodygin, D., Menssen, A. and Hermeking, H. 2002. Induction of the Cdk inhibitor p21 by LY83583 inhibits tumor cell pro- liferation in a p53-independent manner. J. Clin. Invest. 110, 1717-1727.
19. Lopez-Lazaro, M. 2008. The warburg effect: why and how do cancer cells activate glycolysis in the presence of oxygen?
Anticancer Agents Med. Chem. 8, 305-312.
20. Mian, C., Lacroix, L., Alzieu, L., Nocera, M., Talbot, M., Bidart, J. M., Schlumberger, M. and Caillou, B. 2001. Sodium iodide symporter and pendrin expression in human thyroid tissues. Thyroid 11, 825-830.
21. Muntean, D. M., Sturza, A., Danila, M. D., Borza, C., Duicu, O. M. and Mornos, C. 2016. The role of mitochondrial re- active oxygen species in cardiovascular injury and pro- tective strategies. Oxid. Med. Cell. Longev. 2016, 8254942.
22. Nguyen, Q. T., Lee, E. J., Huang, M. G., Park, Y. I., Khullar, A. and Plodkowski, R. A. 2015. Diagnosis and treatment of patients with thyroid cancer. Am. Health. Drug Benefits 8, 30-40.
23. Nikiforov, Y. E. 2004. Genetic alterations involved in the transition from well-differentiated to poorly differentiated and anaplastic thyroid carcinomas. Endocr. Pathol. 15, 319- 327.
24. Oren, M. 2003. Decision making by p53: life, death and cancer. Cell Death. Differ. 10, 431-442.
25. Pang, Y., Qin, G., Wu, L., Wang, X. and Chen, T. 2016. Arte- sunate induces ROS-dependent apoptosis via a Bax-medi- ated intrinsic pathway in Huh-7 and Hep3B cells. Exp. Cell.
Res. 347, 251-260.
26. Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L. and Denko, N. C. 2006. HIF-1 mediates adaptation to hypoxia
초록:Dichloroacetate의 p53 비의존적 경로를 통한 인간 역분화 갑상선 암세포주의 성장억제 효과
얌 바하더 케이씨1․수닐 포우델2․전언주3․손호상3․변승준4․정남호2*
(1대구가톨릭대학교 의생명과학과, 2대구가톨릭대학교 제약산업공학과, 3대구가톨릭대학교 의과대학 내과, 4농촌
진흥청 국립축산과학원 동물바이오공학과)
Warburg 효과의 발생은 고형암에서 화학적 항암제의 내성을 발생시킨다. 따라서 호기성 해당과정과 같은 에너 지 대사과정은 암 치료의 중요한 표적으로 알려져 있다. Pyruvate dehydrogenase kinase (PDK) 활성 억제물질로 알려진 dichloroacetate (DCA)는 많은 암세포에서 포도당의 호기성 해당과정을 산화적인산화 과정으로 전환시킬 수 있음이 보고되었다. 이 연구는 치료가 매우 어렵다고 알려진 인간 역분화 갑상선 암세포주인 8505C의 성장에 미치는 DCA효과를 조사하였다. DCA는 정상 갑상선 세포주의 성장에는 영향을 주지 않은 반면 8505C 세포주의 성장을 특이적으로 저해하였다. DCA의 처리에 의해 8505C 세포주는 HIF1α, PDK1, Bcl-2와 같은 항-세포자살 관 련 단백질들의 발현이 감소하고, Bax와 p21과 같은 세포자살 유도 단백질과 세포주기 억제 단백질의 증가로 인하 여 세포주기 정지와 세포자살 유도에 의해 성장이 억제되었다. 이런 세포의 변화는 DCA 처리에 의한 활성산소족 의 생산이 증가하고, 포도당 대사가 호기성 해당과정에서 산화적인산화 과정으로 전환되었기 때문이란 것을 확인 하였다. 흥미롭게도, DCA는 포도당 대사과정의 변화뿐만 아니라 sodium/iodine symporter (NIS)의 발현양도 증가시킨다는 것을 확인하였다. 이 연구의 결과로 PDK 활성 저해제는 치료하기 힘든 역분화 갑상선 암 치료제로 이용할 수 있고, 또한 역분화 갑상선 암에 대한 방사능 치료의 효능을 높일 수 있을 것으로 기대된다.
by actively downregulating mitochondrial oxygen con- sumption. Cell Metab. 3, 187-197.
27. Pertega-Gomes, N., Felisbino, S., Massie, C. E., Vizcaino, J.
R., Coelho, R., Sandi, C., Simoes-Sousa, S., Jurmeister, S., Ramos-Montoya, A., Asim, M., Tran, M., Oliveira, E., Lobo da Cunha, A., Maximo, V., Baltazar, F., Neal, D. E. and Fryer, L. G. 2015. A glycolytic phenotype is associated with prostate cancer progression and aggressiveness: a role for monocarboxylate transporters as metabolic targets for therapy. J. Pathol. 236, 517-530.
28. Qiu, X., Forman, H. J., Schonthal, A. H. and Cadenas, E.
1996. Induction of p21 mediated by reactive oxygen species formed during the metabolism of aziridinylbenzoquinones by HCT116 cells. J. Biol. Chem. 271, 31915-31921.
29. Quiros, R. M., Ding, H. G., Gattuso, P., Prinz, R. A. and Xu, X. 2005. Evidence that one subset of anaplastic thyroid carcinomas are derived from papillary carcinomas due to BRAF and p53 mutations. Cancer 103, 2261-2268.
30. Ringel, M. D., Anderson, J., Souza, S. L., Burch, H. B., Tam- bascia, M., Shriver, C. D. and Tuttle, R. M. 2001. Expression of the sodium iodide symporter and thyroglobulin genes are reduced in papillary thyroid cancer. Mod. Pathol. 14, 289- 296.
31. Sanchez, W. Y., McGee, S. L., Connor, T., Mottram, B., Wilkinson, A., Whitehead, J. P., Vuckovic, S. and Catley, L. 2013. Dichloroacetate inhibits aerobic glycolysis in multi-
ple myeloma cells and increases sensitivity to bortezomib.
Br. J. Cancer 108, 1624-1633.
32. Shahrzad, S., Lacombe, K., Adamcic, U., Minhas, K. and Coomber, B. L. 2010. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett. 297, 75- 83.
33. Spitzweg, C., Harrington, K. J., Pinke, L. A., Vile, R. G. and Morris, J. C. 2001. Clinical review 132: The sodium iodide symporter and its potential role in cancer therapy. J. Clin.
Endocrinol. Metab. 86, 3327-3335.
34. Stacpoole, P. W., Kurtz, T. L., Han, Z. and Langaee, T. 2008.
Role of dichloroacetate in the treatment of genetic mitochon- drial diseases. Adv. Drug Deliv. Rev. 60, 1478-1487.
35. Tavares, C., Coelho, M. J., Eloy, C., Melo, M., da Rocha, A.
G., Pestana, A., Batista, R., Ferreira, L. B., Rios, E., Selmi- Ruby, S., Cavadas, B., Pereira, L., Sobrinho-Simões, M. and Soares, P. 2018. NIS expression in thyroid tumors, relation with prognosis clinicopathological and molecular features.
Endocr. Connect. 7, 78-90.
36. Warburg, O. 1956. On the origin of cancer cells. Science 123, 309-314.
37. Zhang, Z., He, H., Chen, F., Huang, C. and Shi, X. 2002.
MAPKs mediate S phase arrest induced by vanadate through a p53-dependent pathway in mouse epidermal C141 cells. Chem. Res. Toxicol. 15, 950-956.