Improved Therapeutic Effect against Leukemia by a Combination of the Histone Methyltransferase Inhibitor Chaetocin and the Histone Deacetylase Inhibitor Trichostatin A
SUV39H1 is a histone 3 lysine 9 (H3K9)-specific methyltransferase that is important for heterochromatin formation and the regulation of gene expression. Chaetocin specifically inhibits SUV39H1, resulted in H3K9 methylation reduction as well as reactivation of silenced genes in cancer cells. Histone deacetylase (HDAC) inhibitors inhibit deacetylases and accumulate high levels of acetylation lead to cell cycle arrest and apoptosis. In this study, we demonstrated that treatment with chaetocin enhanced apoptosis in human leukemia HL60, KG1, Kasumi, K562, and THP1 cells. In addition, chaetocin induced the expression of cyclin-dependent kinase inhibitor 2B (p15), E-cadherin (CDH1) and frizzled family receptor 9 (FZD9) through depletion of SUV39H1 and reduced H3K9 methylation in their promoters. Co-treatment with chaetocin and HDAC inhibitor trichostatin A (TSA) dramatically increased apoptosis and produced greater activation of genes. Furthermore, this combined treatment significantly increased loss of SUV39H1 and reduced histone H3K9 trimethylation responses accompanied by increased acetylation. Importantly, co- treatment with chaetocin and TSA produced potent antileukemic effects in leukemia cells derived from patients. These in vitro findings suggest that combination therapy with SUV39H1 and HDAC inhibitors may be of potential value in the treatment of leukemia.
Key Words: Histone Methyltransferase; Histone Deacetylase; Tumor Suppressor Genes;
Leukemia Huong Thi Thanh Tran,1,2 Hee Nam Kim,1
Il-Kwon Lee,1 Thanh-Nhan Nguyen-Pham,2 Jae-Sook Ahn,2 Yeo-Kyeoung Kim,2 Je-Jung Lee,2 Kyeong-Soo Park,3 Hoon Kook,4 and Hyeoung-Joon Kim1,2
1Genome Research Center for Hematopoietic Diseases, Chonnam National University Hwasun Hospital, Hwasun; 2Department of Hematology- Oncology, Chonnam National University Hwasun Hospital, Hwasun; 3Department of Preventive Medicine, College of Medicine, Seonam University, Namwon; 4Environmental Health Center for Childhood Leukemia and Cancer, Chonnam National University Hwasun Hospital, Hwasun, Korea Received: 11 September 2012
Accepted: 7 December 2012 Address for Correspondence:
Hyeoung-Joon Kim, MD
Department of Hematology-Oncology, Chonnam National University Hwasun Hospital, 322 Seoyangro, Hwasun 519-763, Korea
Tel: +82.61-379-7637, Fax: +82.61-379-7736 E-mail: [email protected]
This study was supported by a grant of the National Project for Personalized Genomic Medicine, Ministry for Health & Welfare, Republic of Korea (A111218-GM06) and a grant of the Environmental Health Center for Childhood Leukemia and Cancer, Chonnam National University Hwasun Hospital, Republic of Korean (HCRE/0035-6).
http://dx.doi.org/10.3346/jkms.2013.28.2.237 • J Korean Med Sci 2013; 28: 237-246
INTRODUCTION
Epigenetic deregulations that underlie the development of leu- kemia can be in one of two major categories: changes in the DNA methylation state and alterations in the histone modifica- tion pattern (1). The interaction of these two processes repre- sents an important mechanism of epigenetic tumor suppressor gene (TSG) inactivation in the pathogenesis of human cancer (2). In the clinical trials to date, demethylation of crucial genes, such as cell cycle regulation and pro-apoptotic genes by DNA methyltransferase (DNMT) inhibitors in myeloid neoplasm is thought to be mediated through the reversal of epigenetic silenc- ing (1, 3). However, since DNA methylation is thought to be a
secondary event, and histone methylation can be a trigger in the silent state, it is possible that histone methyltransferase (HMT) inhibitors may replace DNMT inhibitors in epigenetic therapies (4, 5).
Histone 3 lysine 9 (H3K9) methylation, which was catalyzed by the histone methylase SUV39H1 and followed by the recruit- ment of heterochromatin protein 1 (HP), is recognized to be an inactive mark associated with transcriptional repression and heterochromatic states. In addition, H3K9 is recognized as an inactive mark associated with transcriptional repression and heterochromatic states. Interaction of SUV39H1-HP1 with his- tone deacetylase (HDAC) is involved in this inhibition by reti- noblastoma (Rb) protein (6). Notably, SUV39H1 functions in
concert with to DNA methylation via MeCP2, MBD1, and DNMT binding (7). SUV39H1 double-null mice are characterized by genomic instability and further increased risk of lymphoma in response to oncogenic Ras (8). However, its mutation is rare in epithelial cancers. Meanwhile, SUV39H1 is upregulated and as- sociated with DNMT1 elevation in colorectal cancer (9). It was also found to be overexpressed in lung cancer cell lines, in which suppression of SUV39H1 by siRNA induced apoptosis in vitro (10). Suppression of SUV39H1 by siRNA also produced similar effects in acute myeloid leukemia (AML) cells (11, 12). In pa- tients with an acute phase of chronic myeloid leukemia (CML) and patient with AML, strong methylation of H3K9 and all iso- forms of HP1 are detected in granulocytes (13).
Epigenetic silencing of TSGs has been shown to occur in var- ious hematopoietic neoplasms associated with cell proliferation and differentiation (2). Such as loss of p15 expression is com- mon in AML and myeloid dysplastic syndrome (MDS) through several different mechanisms. Cancers characterized by the loss of E-cadherin (CDH1) undergo either the promoter hypermeth- ylation or methylation independent events, which may, for ex- ample, result from the loss of a transactivating protein. Frizzled family receptor 9 (FZD9), a TSG on chromosome 7, is most fre- quently found in aberrantly methylated genes and its aberrant methylation combined with cytogenetic abnormalities to pre- dict a poor clinical outcome in MDS (14). Thus, p15, CDH1, and FZD9 are TSGs that have been frequently linked to pathology in AML and MDS.
Chaetocin, a specific inhibitor of SUV39H1, potently induces cellular oxidative stress, thus selectively killing cancer cells (15- 17). It has been reported to have potent anti-myeloma activity in vitro and in vivo (18). Inhibition of SUV39H1 results in reduced H3K9 methylation and enhanced expression of p15 and CDH1 in AML cell lines without promoter demethylation (11, 12). Mean- while, the histone deacetylase inhibitor trichostatin A can reac- tivate gene silencing and have efficacy against leukemia in pre- clinical (4). Thus combined treatment with an HMT inhibitor and an HDAC inhibitor might form the optimal basis for revers- ing epigenetic gene inactivation and resensitizing leukemia cells to anti-tumor treatments (12, 19). Combined epigenetic thera- py with the HMT inhibitor chaetocin and the HDAC inhibitor TSA has not yet been tested. In the present study, the effects of chaetocin alone and in combination with TSA, were evaluated in human leukemia cells.
MATERIALS AND METHODS Reagents
Chaetocin and TSA were obtained from Sigma Aldrich (Oakville, ON, Canada). Annexin V-FITC was obtained from BD Biosci- ences (San Diego, CA, USA). Monoclonal anti-trimethyl histone 3 lysine 9 was obtained from Abcam (Cambridge, UK). Anti-poly
(adenosine 5-diphosphate-ribose) polymerase (PARP) and anti- acetyl histone H3 lysine 9 antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Polyclonal anti-SU- V39H1 was purchased from Millipore (Temecula, CA, USA).
Anti-β-Actin, normal IgG, horseradish-peroxidase conjugated secondary antibodies were acquired from Santa Cruz Biotech- nology (Santa Cruz, CA, USA).
Cell lines and cell culture
All cell lines were obtained from American Type Culture Col- lection (Rockville, MD). HL60 (AML M3), KG1 (AML M6), K562 (chronic erythroleukemia in blast crisis), THP1 (AML M5), Ka- sumi (AML M2) carrying the t[8; 21] AML1-ETO fusion were used in this study. All cells were cultured in RPMI-1640 media supplemented with 1% penicillin/streptomycin and 10% heat inactivated fetal bovine serum (FBS) (all from Gibco-BRL, Grand Island, NY, USA) except Kasumi in media supplemented with 20% FBS. Cells were cultured in 5% CO2 at 37°C in humidified air. Medium was changed every 2 to 3 days, and cells were pas- saged at a density of 0.25 × 106/mL. Logarithmically growing cell cultures were used for all experiments described below.
Patient samples
AML samples were obtained with informed consent in accor- dance with the Declaration of Helsinki as part of a clinical pro- tocol approved by the Institutional Review Board of the Chon- nam National University Hwasun Hospital. Fresh bone marrow (BM) aspirate samples at diagnosis were collected and separated for mononuclear cells, as previous described (19). 1 × 106 cells were incubated in six-well plates in the presence or absent of 100 nM of chaetocin with or without 1 µM of TSA for 18 hr at 37°C in 5% CO2 humidified air. The drug concentrations were selected based on our results of cell lines.
Assessment of apoptosis of AML cells
For cytometric evaluation of apoptosis in AML cell lines, un- treated and drug treated cells were washed with cold PBS and resuspended in 1 × Binding buffer. Cells were stained by incu- bated with 5 µL of annexin V (Pharmingen, San Diego, CA, USA) and 10 µL of propidium iodide (PI) (Invitrogen) for 15 min in the dark at room temperature, and the percentage of apoptotic cells were determined by flow cytometry within 1 hr.
Western analysis
Western blotting analysis of total protein lysates was performed as previously described (19). Briefly, cell pellets were washed twice with 1 × PBS, resuspended in RIPA buffer supplemented with a protease inhibitors (1 mM phenylmethylsulfonyl fluo- ride, 1 µg/mL leupeptin, 1 µg/mL pepstatin-A, 1 µg/mL apro- tinin, 1 mM sodium orthovanadate, and 1 mM sodium fluoride), and incubated on ice. Cell lysates were centrifuged at 12,000
rpm for 15 min to remove the nuclear and cellular debris. Pro- tein concentration was determined with the BCA assay (Pierce Chemical, West Pico, Rockville, IL, USA).
One hundred micrograms of total cell lysate were separated by SDS-PAGE and blotted onto a membrane. The membranes were blocked in blocking solution, incubated with primary an- tibodies (e.g. PARP) over-night, and secondary antibodies for 1-2 hr. After that, the blots were developed by using a two-com- ponent ECL detection reagent (GE Healthcare, Buckingham- shire, UK) and exposed to scientific imaging film. Immunoblot analyses were performed at least twice, and representative blots were subjected to densitometric analysis. Densitometry was performed using Multi Gauge v3.2 software (Fuji film Corpora- tion, Tokyo, Japan).
Reverse transcriptase PCR and real-time PCR
Total RNA was harvested using TRIzol (Invitrogen) according to the manufacturer’s instructions and reverse transcribed using a SuperScript First-Strand Synthesis kit (Invitrogen). All samples were plated in triplicate, and then loaded onto a 72-well Rotor- Gene RG-3000 (Corbett Research, Sydney, Australia) with a 10 μL final reaction mixture containing 250 nM of each primer, 1 × SYBR Green (Takara, Tokyo, Japan), and cDNA. The primer se- quences are listed in Table 1. The reaction mixture was preheat- ed to 95°C for 10 sec, followed by 45 cycles of 95°C for 10 sec, 60°C for 20 sec, and 72°C for 20 sec. All reactions included nega- tive controls where reverse transcriptase was omitted.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assay were performed, according to the manufacturer’s protocol (Upstate Biotechnol- ogy, Temecula, CA, USA). The primary antibody (anti trimethyl H3K9) and rabbit IgG (negative control) were used. For quanti-
tative assessment of p15, CDH1, and FZD9 in the chromatin immunoprecipitates, real time PCR using SYBR Green (Takara, Tokyo, Japan) was performed. Amplified products were nor- malized to the non-specific glyceraldehyde 3-phosphate dehy- drogenase (GAPDH) promoter enrichment. Relative enrichment in the chromatin immunoprecipitates was normalized against p15, CDH1, and FZD9 in the input samples. Primer sequences are listed in Table 1.
Isolation of histones
Histones were isolated by a modification of a previously de- scribed method (20). After the designated treatments, cells were harvested and incubated on ice cold histone isolation buffer (PBS containing 0.5% Triton X-100 (v/v), 2 mM phenylmethyl- sulfonyl fluoride, and 0.02% NaN3) on ice for 10 min with gentle stirring. Nuclei were pelleted by centrifuge at 6,500 × g for 10 min at 4°C and washed briefly with the histone lysis buffer. The nuclei were resuspended in 0.2 N HCl and the tubes were incubated overnight at 4°C. The acid-treated nuclei were centrifuged for 10 min at 6,500 × g. The supernatants were removed to a clean microcentrifuge tube, and 1 mL acetone was added. Histones were precipitated from the acid extracts overnight at -20°C. The extracted histones were centrifuged briefly at 10,000 rpm, air dried, and resuspended in 50 μL water. Protein concentrations were quantified as described above. For Western blot, 1 to 3 μg of purified histones were used per condition.
Statistical analysis
All statistical analyses were performed with the program SPSS 13.0 for Windows. Significant differences between values ob- tained in a population of leukemic cells treated with different experimental conditions were determined using the Mann- Whitney U-test. P values < 0.05 were assigned significance.
Ethics statement
This study was approved by the institutional review board of the Chonnam National University Hwasun Hospital in Hwasun, Korea (IRB No. CNUHHIRB 2009-22). At the time of samples collection, all cases and control subjects provided informed consent to participate in this study.
RESULTS
Chaetocin treatment induces apoptosis and increases tumor suppressor gene expression in myeloid cell lines In the earlier studies, chaetocin had a cytotoxic effect on Dro- sophila cell (15), and on myeloma (18). Chaetocin and SUV39H1 shRNA substantially increased cell cycle arrest in human leuke- mia AML-193, KG1, and U937 cells (11, 12), as well as microglial cells (21). In this study, we first assessed the biologic effect of chaetocin on different representative cell lines-HL60, KG1, Table 1. Primers used for quantitative real-time PCR and ChIP assays
Name Sequence (5´-3´) Product size
(bps) Real-time PCR
p15 F p15 R CDH1 F CDH1 R FZD9 F FZD9 R β2 microglobin F β2 microglobin R
TGATTAGCACTTGGGTGACG CCTCCTCCACTTTGTCCTCA AGGAATCCAAAGCCTCAGGT TTGGGTTGGGTCGTTGTACT CTTCTTCCTGTGCTCGCTCT CCGAAGTTGAACTGCTCCAT ACCCCCACTGAAAAAGATGA ATCTTCAAACCTCCATGATG
129 129 129 114 ChIP assay
p15 F p15 R CDH1 F CDH1 R FZD9 F FZD9 R GAPDH F GAPDH R
GCAGGCTTCCCCGCCCTCGTGACGC ATTACCCTCCCGTCGTCCTTCTGC AGAGGGTCACCGCGTCTATG CTCACAGGTGCTTTGCAGTT TAAAATGAGGCGACCCAGTC GCCTCCCACTGGGCTTTA TACTAGCGGTTTTACGGGCG TCGAACAGGAGGAGCAGAGAGCGA
367 201 186 166
Kasumi, K562, and THP1-on apoptosis by Annexin V staining.
Treatment of these cells with increasing doses of chaetocin (0-
500 nM) for 24 hr induced greater apoptosis (Fig. 1A). Next, these results were confirmed by Western blotting after treatment with
Fig. 1. Chaetocin induces apoptosis in the leukemia cell lines. (A) After treated 24 hr, apoptotic cells were determined by flow cytometry. (B) Confirmatory Western blotting. (C) After treated 48 and 72 hr. Beta actin is protein loading control. C-48: 48 hr-untreated control, C-72: 72 hr-untreated control. *P < 0.05.
Chaetocin (nM) A
% apoptosis
HL60
* * *
0 100 200 500 100
50
0
KG1
* * *
0 100 200 500 100
50
0
Kasumi
*
* *
0 100 200 500 100
50
0
K562
* * *
0 100 200 500 100
50
0
THP1
*
* *
0 100 200 500 100
50
0
B HL60
PARP Cleaved PARP β-actin Chaetocin (nM), 24 hr
0 100 200 500 0 100 200 500 0 100 200 500 0 100 200 500 0 100 200 500
KG1 Kasumi K562 THP1
C HL60
PARP Cleaved PARP β-actin
Chaetocin (100 nM), hr C-48 48 C-72 72 C-48 48 C-72 72 C-48 48 C-72 72 C-48 48 C-72 72 C-48 48 C-72 72
KG1 Kasumi K562 THP1
Chaetocin (nM)
Chaetocin (nM)
HL60
K562
KG1
THP1
Kasumi
Relative expressionRelative expression
p15
CDH1 0 50 100 200
0 50 100 200 30
20 10 0
8 6 4 2 0
*
*
*
* 40
30 20 10 0
6 4 2 0
*
*
*
* 60
40 20 0
*
†
CDH1
FZD9 0 50 100 200
0 50 100 200 8
6 4 2 0
2
1
0
*
*
* 60
40 20 0
6 4 2 0
†
*
†
* 30
20 10 0
*
*
FZD9
0 50 100 200 5
4 3 2 1 0
*
30 20 10 0
*
*
9 6 3 0
*
*
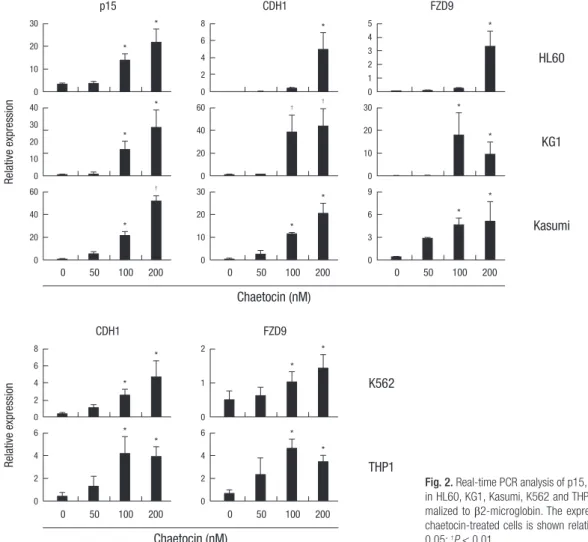
Fig. 2. Real-time PCR analysis of p15, CDH1 and FZD9 mRNA expression in HL60, KG1, Kasumi, K562 and THP1 cells. Gene expression was nor- malized to β2-microglobin. The expression of p15, CDH1 and FZD9 in chaetocin-treated cells is shown relative to that in untreated cells. *P <
0.05; †P < 0.01.
chaetocin for 24 hr. Exposure to chaetocin dose dependently induced caspase-dependent cleavage of PARP to a greater ex- tent in myeloid cells (Fig. 1B). Moreover, chaetocin induced apoptosis in a time dependent manner (Fig. 1C).
Re-expression of epigenetically silenced TSGs as a result of SUV39H1 inhibition has been reported (11). We ourselves pre- viously described increased p15 and CDH1 mRNA expression in KG1 and Kasumi cells. We next determined the effects of var- ious concentrations of chaetocin treatment on expression of the p15, CDH1, and FZD9 genes in the myeloid cell lines. The results showed that treatment with 100-200 nM chaetocin resulted in strong re-expression of epigenetically silenced/weakly expressed p15, CDH1 and FZD9 genes in HL60, KG1, and Kasumi cells, as well as re-expression of CDH1 and FZD9 in K562 and THP1 cells (Fig. 2) (P < 0.05). They also revealed p15 deletions in K562 and THP1 cells, as indicated by the lack of a p15 signal in those cell lines, according to the characteristic of these cell lines.
Chaetocin dose-dependently reduces histone
methyltransferase protein levels and subsequently lowers histone H3K9 methylation in tumor suppressor gene promoters
In Drosophila SL-2, chaetocin has been demonstrated to deplete the activity of SUV39H1 (15). Consistent with this report, treat- ment of HL60, KG1, Kasumi, K562 and THP1 myeloid cells with chaetocin dose-dependently reduced SUV39H1 protein levels (Fig. 3A), which can lead to the inhibition of H3K9 methylation.
Recently, chaetocin was shown to reduce SUV39H1 and H3K9 trimethylation in the promoter regions of the p21 (21), p15, and
CDH1 (11) genes. In our study, ChIP assays were performed us- ing anti-trimethyl-H3K9 to analyze the effect of chaetocin on the p15, CDH1 and FZD9 promoters in these cell lines. The re- sult showed that the levels of trimethylation of H3K9 in the p15, CDH1 and FZD9 promoter regions decreased relative to the untreated control cells in HL60, KG1 and Kasumi cells (Fig. 3B).
Also, this association with the CDH1 and FZD9 promoters was down- in regulated K562, and THP1 cells compared to untreat- ed control cells (Fig. 3B).
Co-treatment with chaetocin and TSA dramatically induces apoptosis and enhances tumor suppressor gene re-expression
The IC50 values of the HDAC inhibitor TSA for apoptosis in dif- ferent cell lines were determined by flow cytometry (data not shown). The concentration 1 µM of TSA caused cell death in these cells. To determine whether TSA enhances the effects of chae- tocin in leukemia cell lines, the effects of the combination of the two compounds on apoptosis were evaluated. As shown in Fig.
4A, apoptosis was higher in myeloid cell lines treated with both chaetocin and TSA than in cells treated with chaetocin alone.
These responses were accompanied by synergistically increased PARP cleavage as verified by Western blotting (Fig. 4B), indicat- ing that the antileukemic activity of the combined treatment was higher than that of the individual compounds. Additionally the combination of compounds caused more apoptosis in HL60, Kasumi, and K562 cells than in KG1 or THP1 cells (Fig. 4A).
In HL60, K562, and particularly in Kasumi cells, the combi- nation of chaetocin and TSA produced a markedly stronger re-
Fig. 3. Chaetocin reduces SUV39H1 protein levels and H3K9 methylation in p15, CDH1 and FZD9 promoters. (A) Western blotting was showed with beta actin as a loading control. (B) The effect of chaetocin (100 nM) on H3K9 trimethylation in the promoters in HL60, KG1, Kasumi, K562 and THP1 cells was analyzed by ChIP assays using anti-tri- methyl H3K9 (H3K9trime). Histograms show antibody/input ratios for PCR products, quantified by real-time PCR. *P < 0.05.
B A HL60
SUV39H1
β-actin Chaetocin (nM), 24 hr
0 100 200 500
1 0.66 0.27 0.12 1 0.58 0.28 0.18 1 0.6 0.32 0.47 1 0.85 0.56 0.47 1 0.71 0.28 0.07
0 100 200 500 0 100 200 500 0 100 200 500 0 100 200 500
KG1 Kasumi K562 THP1
H3K9trime Ab/input ratio H3K9trime Ab/input ratio
Control Chaetocin Control Chaetocin
Control Chaetocin
Control Chaetocin
Control Chaetocin
p15
* 8
6 4 2 0
CDH1
* 5
4 32 1 0
FZD9
* 4
3 2 1 0
CDH1
* 6
4 2 0
FZD9
* 3
2 1 0
* 0.6
0.4 0.2 0
* 2
1.5 1 0.5 0
* 15
10 5 0
* 8
6 4 2 0
* 0.8
0.4 0
* 20
15 10 5 0
* 25
20 1510 5 0
* 25
20 1510 5 0
HL60 K562
KG1 THP1
Kasumi
expression of these genes than that achieved through treatment with chaetocin alone (Fig. 4C). In addition, KG1 cells showed a significant increase in p15 expression. In contrast, combined treatment somehow did not increase activation of CDH1 and
FZD9 genes in KG1 or THP1 cells, the results exposed to higher than untreated controls (Fig. 2 and 4C). Taken together, these findings can support greater activation of silencing genes in leu- kemia cells treated with a combination of chaetocin and TSA
Fig. 4. Chaetocin and TSA co-treatmentlly induce apoptosis and gene expression after 24 hr treatment in myeloid leukemia cell lines. (A) Apoptosis was determined by flow cy- tometry. (B) Confirmatory Western blotting with beta actin as a loading control. (C) Real-time PCR of p15, CDH1 and FZD9 in chaetocin and TSA-treated cells is shown relative to that in chaetocin-treated cells. *P < 0.05; †P < 0.01; Ch100: Chaetocin concentration at 100 nM.
C
Relative expression Relative expression
Ch100 Ch100 + TSA Ch100 Ch100 + TSA
Ch100 Ch100 + TSA
Ch100 Ch100 + TSA
Ch100 Ch100 + TSA
p15
80 *
40 0
CDH1
40 * 20
0
FZD9
20 * 10
0
CDH1
20 * 10
0
FZD9
5 * 2.5
0 40 *
20 0
60 40 20 0
40 20 0
* 8
4 0
* 6
4 2 0 500 †
250
0
500 †
250 0
500 †
250 0
HL60 K562
KG1 THP1
Kasumi
A
% apoptosis
HL60
*
Control
Control
Control
Control
Control
TSA Ch100 TSA TSA TSA TSA
Ch100
Ch100
Ch100
Ch100 Ch100 + TSA
Ch100 + TSA
Ch100 + TSA
Ch100 + TSA
Ch100 + TSA 100
80 60 40 20 0
KG1
* 100
80 60 40 20 0
Kasumi
100 * 80 60 40 20 0
K562
* 100
80 60 40 20 0
THP1
* 100
80 60 40 20 0
B HL60
PARP
β-actin TSA (1 µM), 24 hr Chaetocin (100 nM), 24 hr
KG1 Kasumi K562 THP1
- + - +
- - + +
- + - +
- - + +
- + - +
- - + +
- + - +
- - + +
- + - +
- - + +
Cleaved PARP
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1 0.7
0.79
1.75
0.56
0.81
0.69
0.66
0.86
0.58
0.83
0.6
1.29
0.73
0.9
0.94 0.56
0.72
2.37
0.91
0.95
3.93
0.7
1.02
1.6
1.06
0.87
1.14
0.92
0.95
0.95 0.52
0.46
2.45
0.30
0.42
4.01
0.21
0.24
1.81
0.67
0.4
1.39
0.53
0.71
1.19
A
B HL60
HL60 SUV39H1
H3K9 trime
H3K9 ac β-actin
Ponceau stain TSA (1 µM), 24 hr
TSA (1 µM), 24 hr Chaetocin (100 nM), 24 hr
Chaetocin (100 nM), 24 hr
KG1
KG1
Kasumi
Kasumi
K562
K562
THP1
THP1
- + - +
- - + +
- + - +
- - + +
- + - +
- - + +
- + - +
- - + +
- + - +
- - + +
- + - +
- - + +
- + - +
- - + +
- + - +
- - + +
- + - +
- - + +
- + - +
- - + +
Fig. 5. Co-treatment with chaetocin and TSA alter the expression of methyltransferase related molecules. Myeloid cell lines were harvested after 24 hr incubation with indicated drug. Western blotting analyses were performed and representative blots were subjected to densitometric analysis. Beta actin was used as a loading control for cell lysates. Ponceau- stained histones were used as a loading control for acid-extracted histones. (A) Combined treatment with chaetocin and TSA increases SUV39H1 protein depletion. (B) The com- bined treatment reduces histone H3K9 trimethylation, and increases histone H3K9 acetylation. H3K9 trime: histone H3K9 trimethylation; H3K9 ac: histone H3K9 acetylation.
than in cells treated with chaetocin alone.
Co-treatment with chaetocin and TSA significantly increases loss of histone methyltransferase protein, resulting in reduced histone H3K9 methylation and increased histone acetylation
A link between the growth inhibitory and apoptotic antileuke- mic activities that are associated with epigenetic inhibition and the reactivation of genes has been identified (19). As shown in Fig. 5A, although TSA reduced histone methyltransferase levels in some myeloid cells, co-treatment with chaetocin and TSA pro-
duced a stronger reduction in SUV39H1 protein levels than treat- ment with either agent alone. Treatment of HL60, KG1, Kasumi, K562, and THP1 cells with a combination of chaetocin and TSA more strongly reduced histone H3K9 trimethylation, and more strongly induced increased histone H3K9 acetylation, than treat- ment with chaetocin alone (Fig. 5B).
Co-treatment with chaetocin and TSA enhances antileukemic activity in cells derived from patients with AML
The effects of chaetocin and TSA were assessed in leukemia cells
Primary AML Primary AML
PARP PARP
Chaetocin (nM), 24 hr Chaetocin (100 nM), 24 hr
TSA (1 µM), 24 hr
Cleaved PARP Cleaved PARP
β-actin β-actin
SUV39H1 SUV39H1
0 100 200 500
- + - +
- - + +
1 0.88 0.48 0.02 1 0.63 0.83 0.38
A
% apoptosis % apoptosis
100 80 60 40 20 0
100
80
60
40
20
Control TSA Ch100 Ch100 + TSA
B C
Pa1 Pa2 Pa3 Pa4 Pa5 Pa6 Pa7 TSA Ch100 nM Ch100 nM + TSA
*
*
*
Fig. 6. Co-treatment with chaetocin and TSA produces stronger antileukemic effects in cells from patients with AML. (A) Western blotting analyses were performed and repre- sentative blots were subjected to densitometric analysis. (B) Apoptosis of cells from bone marrow seven patients with AML. (C) Box plots showing the percentage of apoptotic cells in the AML cell populations of the seven analyzed patients after treatment with the indicated doses. *P < 0.01. Pa: patient; Ch100 nM: Chaetocin concentration at 100 nM.
Table 2. Characteristics of AML patients in the study
ID Age/Sex Classification* Blast (%)† Cytogenetic FISH FLT3-ITD
mutation C/EBPα mutation
NPM1 mutation
c-kit mutation
P1 38/M AML M2 51 45,X,-Y,t(8;21)(q22;q22) AML1–ETO
rearrangement
(+) (-) (-) (-) (-)
P2 18/M Mixed leukemia
(T/myeloid) 95 46,XY MLL
rearrangement (-) (-) (-) (-) (-)
P3 70/M AML secondary 62 46,XY,inv(16)(p13;q22) CBFB
rearrangement (+) (-) (+) (-) (-)
P4 22/M AML M2 87 47,XY,+21/47,idem,
inv(16)(p13q22)/46,XY
CBFB rearrangement
(+) (-) ( - ) (-) (-)
P5 24/M AML M4 52 46,XY,inv(16)
(p13.1q22)/46,XY CBFB
rearrangement (+) (-) (+) (-) (-)
P6 57/M AML M2 72 46,XY MLL
rearrangement (-) (+) (+) (+) (-)
P7 21/M AML M2 90 46,XY,t(16;21)(p11;q22) ND (-) (-) (-) (-)
*Following the French-American-British system (if possible); †Percentage in bone marrow. FISH, fluorescence in situ hybridization; ND, not done.
isolated from fresh BM from seven patients with AML. The char- acteristics of the patients are summarized in Table 2. As shown in Fig. 6A, chaetocin killed primary AML cells through induc- tion of PARP cleavage and reduced SUV39H1 protein levels in a dose-dependent manner. The expression of SUV39H1 under the treatment of chaetocin at a concentration of 100 nM seem to be higher than no treatment, however, the densitometric analysis by Multi Gauge v3.2 software reveals that the expression of SU- V39H1 was decreased in a dose-dependent manner. In addition, combined treatment with chaetocin and TSA induced greater PARP cleavage and loss of SUV39H1 protein in AML cells than single agent alone. As shown in Fig. 6B, as measured by annex- in V staining, indicating the stronger antileukemic activity of this combined treatment compared to that of the individual agents;
although levels of apoptosis in normal leukocytes were no in- crease in the combination (data not shown). Note that all AML samples were sensitive to combined treatment, despite the het- erogeneity of their biologic features: patients 1, 4, 6, and 7 were newly diagnosed with FAB M2; patient 2 had a mixed leukemia (T/myeloid); patient 3 had secondary AML arising from a can- cer treated with chemoradiotherapy for 9 yr; and patient 5 had M4. Cells from the patients with M2 (P1, 4, 6, and 7), blast > 85%
(P2, 4, and 7) or AML1-ETO fusion (P1) were more effectively killed by the combined treatment. Interestingly, the effect of combine treatment was observed not only in primary AML but also effective in secondary AML (P3). Concerning patient mu- tation status, only patient 6 carried three out of four gene muta- tions screened (FLT3-ITD, C/EBPα and NPM1). In addition, com- bined analysis of the activity of chaetocin and TSA in the total cells from the seven patients showed the combined treatment to be significantly superior to the individual agents (Fig. 6C).
DISCUSSION
Chaetocin was significantly more potent in myeloid cells, as in- dicated by its dose- and time-dependent enhancement of apop- tosis in human leukemia cells. Chaetocin induces apoptosis in leukemia cell lines in vitro and primary AML cells ex vivo has been reported (12). Identification of an optimal combination therapy, in particular epigenetic th erapy is ongoing. To our knowledge, our study is the first to describe the combined effects of the HMT inhibitor chaetocin and the HDAC inhibitor TSA on apoptosis. Moreover, chaetocin was also effective in AML cells from patients, particularly when co-administered with TSA.
As reported previously, chaetocin upregulates the transcrip- tion of death-receptor- related genes, leading to death receptor- dependent apoptosis (12, 17). Pro-apoptotic activity of chaeto- cin may be mediated, at least in part, by inhibition of SUV39H1 HMT activity at the TSG promoter (11, 21). Overexpression of SUV39H1 mRNA was found in AML cell lines (data not shown).
Treatment of leukemia cell lines with the SUV39H1 inhibitor
chaetocin reduced SUV39H1 levels and lowered H3K9 methyl- ation in the p15, CDH1, and FZD9 promoters, thereby reactivat- ing their expression. Furthermore, we demonstrated that chae- tocin-mediated depletion of SUV39H1 was associated with in- creased apoptosis in AML cells from patients. The development of aberrant TSG silencing may stem from increased SUV39H1 binding and H3K9 trimethylation in their promoters as a result of interactions with DNA-binding proteins (5). A possible can- didate for targeting SUV39H1 to the p15, Evi1 interacts with SU- V39H1 and represses TGF-β-induced activation of the p15 pro- moter (22). CDH1 promoter contains AML1-binding sites, and thus AML1 may recruit SUV39H1 to repress CDH1 expression (23). Through methyl-H3K9 binding, MPP8 targets CDH1 gene promoter and modulates CDH1 gene expression (24). FZD9 is activated by Wnt-2 and functions in Wnt signaling (25). Although the mechanism by which recruitment of SUV39H1 inhibits FZD9 expression remains unknown, our findings demonstrate that chaetocin reduced SU39H1 protein, thereby reducing H3K9 methylation in the FZD9 promoter and resulting in FZD9 re-ex- pression in leukemia cell lines.
Given the multiple effects of HDAC inhibitors on malignant cells, their true therapeutic potential most likely lies in combi- nation with other anticancer drugs (26). TSA is an antileukemic agent that has been reported to be a potent inducer or enhanc- er of differentiation in AML (27). We therefore focused on the anticancer effect of the HMT inhibitor chaetocin in combination with the HDAC inhibitor TSA. Interestingly, high levels of apop- tosis (at least 50% apoptotic cells) was detected in TSA-treated HL60, Kasumi and K562 cells, but not similarly treated KG1 or THP1 cells. So, while TSA showed potential as a single agent, combined treatment with chaetocin and TSA produced signifi- cantly stronger effects in HL60, Kasumi, and K562 cells com- pared to KG1 and THP1 cells (Fig. 4A). Notably, the effect of these compounds on apoptosis was outstanding in Kasumi cells.
About 70% apoptosis was observed in cells treated with individ- ual agent, while apoptosis was almost maximal in cells treated with both agents. This may have been due to the fact that Kasu- mi cells carry an AML1-ETO fusion that predisposes myeloid precursors to transformation, and that turnover of the myeloid oncoprotein was induced by TSA (28). Although TSA dramati- cally degrades AML1-ETO fusion prior to inducing apoptosis in Kasumi cells (28), additional mechanisms underlying the in- duction of apoptosis by TSA and chaetocin in t(8;21) AML cell lines should be further investigated. Nevertheless, these finding also suggest that chaetocin and/or TSA may be a potent target- ed therapies for leukemia, especially t(8;21) AML, as they target multiple pathways, including those inducing apoptosis.
Re-expression of epigenetically inactivated genes can result in the suppression of tumor growth or a sensitization to antican- cer therapy. Compared to either agent alone, co-treatment with chaetocin and TSA produced greater responses. Therefore, chae-
tocin and TSA cooperated in inducing TSG expression, which may potentially have contributed to the superior antileukemic activity of the combined treatment in HL60, Kasumi and K562 cells compared to KG1 and THP1 cells. Simultaneously, the greater decline in trimethylated H3K9 levels and increase in hy- peracetylated H3K9 levels were found to relieve gene repres- sion. Notably, the induction of apoptosis by the combination of chaetocin and TSA was correlated with the increased expression of these genes, in HL60, Kasumi and K562 compared to KG1 and THP1 cells (Fig. 4C). The combination of TSA and chaetocin may induce transcriptional activation and activity of apoptotic proteins, thereby sensitizing cells to apoptosis (26). In addition, the AML1-ETO fusion acts to repress the transcription of key regulatory genes such as C/EBPα, which regulates the cell cycle control and myeloid differentiation (29). Inhibitors target this oncoprotein, resulting in greater derepression and cancer cell death. Kasumi cells co-treated with TSA and chaetocin exhibit- ed greater expression of p15, CDH1, and FZD9 than similarly treated HL60 and K562 cells. The latter is known to trigger cell death though multiple mechanisms in transformed versus nor- mal hematopoietic progenitor cells via ROS production selec- tively in cancer cells (12, 16-18). The p15 gene regulates Rb func- tion by modulating cyclin D-CDK4/6 complexes, thereby inhib- iting cell growth (30). The expression of CDH1 were decreased during tumor progression and metastasis in a variety of cancers (31, 32). The loss of FZD9 in acute lymphoblastic leukemia was associated with increased in cell proliferation and cell cycle en- try (33). Chaetocin may modulate several of these mechanisms to increase apoptosis in response to TSA treatment. Reduced p53-triggered transactivation of p21 allows cells to reenter the cell cycle. TSA activates p53, thereby inducing apoptosis in co- lon cancer cells (34). TSA upregulates p73 and induces Bax-de- pendent apoptosis in ovarian cancer cells (35).
Consistent with the published reports (12, 18, 36, 37), chae- tocin and TSA has little effect in non-cancer cells, suggesting cancer-cell-specific cytotoxicity. In conclusion, the present study demonstrated that a combination of the HMT inhibitor chaeto- cin and the HDAC inhibitor TSA may provide effective treatment and enhance TSG reactivation in epigenetic therapies in leuke- mia cells. The reversal of SUV39H1-mediated gene repression by chaetocin in leukemia still needs to be clarified. In addition, more selective HDAC inhibitors should be tested. Recently, simi- lar combined epigenetic therapy with an HMT EZH2 inhibitor and an HDAC inhibitor was reported to be effective in AML cells (19).
Taken together, the findings presented in this study provide the rationale to design and implement further studies to inves- tigate the in vivo antileukemic effects of combined chaetocin and TSA treatment.
ACKNOWLEDGMENTS
The authors have no conflicts of interest to disclose.
REFERENCES
1. Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer 2010; 10: 23-36.
2. Krug U, Ganser A, Koeffler HP. Tumor suppressor genes in normal and malignant hematopoiesis. Oncogene 2002; 21: 3475-95.
3. Tran HT, Kim HN, Lee IK, Kim YK, Ahn JS, Yang DH, Lee JJ, Kim HJ.
DNA methylation changes following 5-azacitidine treatment in patients with myelodysplastic syndrome. J Korean Med Sci 2011; 26: 207-13.
4. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4: 143-53.
5. Lachner M, Jenuwein T. The many faces of histone lysine methylation.
Curr Opin Cell Biol 2002; 14: 286-98.
6. Vaute O, Nicolas E, Vandel L, Trouche D. Functional and physical inter- action between the histone methyl transferase Suv39H1 and histone deacetylases. Nucleic Acids Res 2002; 30: 475-81.
7. Fujita N, Watanabe S, Ichimura T, Tsuruzoe S, Shinkai Y, Tachibana M, Chiba T, Nakao M. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation- based transcriptional repression. J Biol Chem 2003; 278: 24132-8.
8. Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberg- er B, Stein H, DÖrken B, Jenuwein T, Schmitt CA. Oncogene-induced se- nescence as an initial barrier in lymphoma development. Nature 2005;
436: 660-5.
9. Kang MY, Lee BB, Kim YH, Chang DK, Park SK, Chun HK, Song SY, Park J, Kim DH. Association of the SUV39H1 histone methyltransferase with the DNA methyltransferase 1 at mRNA expression level in primary colorectal cancer. Int J Cancer 2007; 121: 2192-7.
10. Watanabe H, Soejima K, Yasuda H, Kawada I, Nakachi I, Yoda S, Naoki K, Ishizaka A. Deregulation of histone lysine methyltransferases contrib- utes to oncogenic transformation of human bronchoepithelial cells. Can- cer Cell Int 2008; 8: 15.
11. Lakshmikuttyamma A, Scott SA, DeCoteau JF, Geyer CR. Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 in- hibition. Oncogene 2010; 29: 576-88.
12. Chaib H, Nebbioso A, Prebet T, Castellano R, Garbit S, Restouin A, Vey N, Altucci L, Collette Y. Anti-leukemia activity of chaetocin via death re- ceptor-dependent apoptosis and dual modulation of the histone methyl- transferase SUV39H1. Leukemia 2012; 26: 662-74.
13. Lukásová E, Koristek Z, Falk M, Kozubek S, Grigoryev S, Kozubek M, Ondrej V, Kroupová I. Methylation of histones in myeloid leukemias as a potential marker of granulocyte abnormalities. J Leukoc Biol 2005; 77:
100-11.
14. Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O’Keefe C, Sekeres M, Saunthararajah Y, Maciejewski JP. Aberrant DNA methylation is a dom- inant mechanism in MDS progression to AML. Blood 2009; 113: 1315-25.
15. Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat Chem Biol 2005; 1: 143-5.
16. Tibodeau JD, Benson LM, Isham CR, Owen WG, Bible KC. The antican-
cer agent chaetocin is a competitive substrate and inhibitor of thioredox- in reductase. Antioxid Redox Signal 2009; 11: 1097-106.
17. Isham CR, Tibodeau JD, Bossou AR, Merchan JR, Bible KC. The anti- cancer effects of chaetocin are independent of programmed cell death and hypoxia, and are associated with inhibition of endothelial cell pro- liferation. Br J Cancer 2012; 106: 314-23.
18. Isham CR, Tibodeau JD, Jin W, Xu R, Timm MM, Bible KC. Chaetocin: a promising new antimyeloma agent with in vitro and in vivo activity me- diated via imposition of oxidative stress. Blood 2007; 109: 2579-88.
19. Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells.
Blood 2009; 114: 2733-43.
20. Fiskus W, Pranpat M, Balasis M, Herger B, Rao R, Chinnaiyan A, Atadja P, Bhalla K. Histone deacetylase inhibitors deplete enhancer of zeste 2 and associated polycomb repressive complex 2 proteins in human acute leukemia cells. Mol Cancer Ther 2006; 5: 3096-104.
21. Cherrier T, Suzanne S, Redel L, Calao M, Marban C, Samah B, Muker- jee R, Schwartz C, Gras G, Sawaya BE, et al. P21(WAF1) gene promoter is epigenetically silenced by CTIP2 and SUV39H1. Oncogene 2009; 28:
3380-9.
22. Spensberger D, Delwel R. A novel interaction between the proto-onco- gene Evi1 and histone methyltransferases, SUV39H1 and G9a. FEBS Lett 2008; 582: 2761-7.
23. Chakraborty S, Sinha KK, Senyuk V, Nucifora G. SUV39H1 interacts with AML1 and abrogates AML1 transactivity: AML1 is methylated in vivo.
Oncogene 2003; 22: 5229-37.
24. Kokura K, Sun L, Bedford MT, Fang J. Methyl-H3K9-binding protein MPP8 mediates E-cadherin gene silencing and promotes tumour cell motility and invasion. EMBO J 2010; 29: 3673-87.
25. Karasawa T, Yokokura H, Kitajewski J, Lombroso PJ. Frizzled-9 is acti- vated by Wnt-2 and functions in Wnt/beta -catenin signaling. J Biol Chem 2002; 277: 37479-86.
26. Bots M, Johnstone RW. Rational combinations using HDAC inhibitors.
Clin Cancer Res 2009; 15: 3970-7.
27. Kosugi H, Towatari M, Hatano S, Kitamura K, Kiyoi H, Kinoshita T, Tan- imoto M, Murate T, Kawashima K, Saito H, et al. Histone deacetylase in-
hibitors are the potent inducer/enhancer of differentiation in acute my- eloid leukemia: a new approach to anti-leukemia therapy. Leukemia 1999; 13: 1316-24.
28. Yang G, Thompson MA, Brandt SJ, Hiebert SW. Histone deacetylase in- hibitors induce the degradation of the t(8;21) fusion oncoprotein. Onco- gene 2007; 26: 91-101.
29. Pabst T, Mueller BU, Harakawa N, Schoch C, Haferlach T, Behre G, Hid- demann W, Zhang DE, Tenen DG. AML1-ETO downregulates the gran- ulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia.
Nat Med 2001; 7: 444-51.
30. Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA.
Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996; 85: 27-37.
31. Karayiannakis AJ, Syrigos KN, Chatzigianni E, Papanikolaou S, Alexiou D, Kalahanis N, Rosenberg T, Bastounis E. Aberrant E-cadherin expres- sion associated with loss of differentiation and advanced stage in hu- man pancreatic cancer. Anticancer Res 1998; 18: 4177-80.
32. Sulzer MA, Leers MP, van Noord JA, Bollen EC, Theunissen PH. Reduced E-cadherin expression is associated with increased lymph node metasta- sis and unfavorable prognosis in non-small cell lung cancer. Am J Respir Crit Care Med 1998; 157: 1319-23.
33. Khan NI, Bradstock KF, Bendall LJ. Activation of Wnt/beta-catenin path- way mediates growth and survival in B-cell progenitor acute lympho- blastic leukaemia. Br J Haematol 2007; 138: 338-48.
34. Habold C, Poehlmann A, Bajbouj K, Hartig R, Korkmaz KS, Roessner A, Schneider-Stock R. Trichostatin A causes p53 to switch oxidative-dam- aged colorectal cancer cells from cell cycle arrest into apoptosis. J Cell Mol Med 2008; 12: 607-21.
35. Muscolini M, Cianfrocca R, Sajeva A, Mozzetti S, Ferrandina G, Costan- zo A, Tuosto L. Trichostatin A up-regulates p73 and induces Bax-depen- dent apoptosis in cisplatin-resistant ovarian cancer cells. Mol Cancer Ther 2008; 7: 1410-9.
36. Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 2006; 6:
38-51.
37. Zhang Y, Yu G, Wang D, Hu Y, Lei W. ERK1/2 activation plays impor- tant roles in the opposite effects of Trichostatin A in non-cancer and can- cer cells. Toxicon 2011; 57: 932-7.