저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Doctoral Thesis in the Department of
Biomedical Sciences
Lercanidipine enhances the cytotoxicity of
bortezomib in cancer cells by inducing
paraptosis-associated cell death
Ajou University Graduate School
Major in Cancer Biology
Lercanidipine enhances the cytotoxicity of
bortezomib in cancer cells by inducing
paraptosis-associated cell death
Kyeong Sook Choi, Advisor
I submit this thesis as the
Doctoral thesis in the Department of Biomedical Sciences
August, 2019
Graduate School of Ajou University
Major in Cancer Biology
The Doctoral thesis of A Reum Lee in the Department of
Biomedical Sciences is hereby approved.
Thesis Defense Committee Chair
Gyesoon Yoon Seal
Member Kyeong Sook Choi Seal
Member Hyeseong Cho Seal
Member You-Sun Kim Seal
Member Taeg Kyu Kwon Seal
Ajou University Graduate School
i - Abstract -
Lercanidipine enhances the cytotoxicity of bortezomib in cancer cells
via induction of paraptosis-associated cell death
Bortezomib (Btz), a proteasome inhibitor (PI), is effective in treating multiple myeloma and mantle cell lymphoma, but its anticancer effects are not satisfactory in solid tumors. In this study, we examined the possibility that the sensitivity to PIs can be enhanced by drug repositioning. We first show that lercanidipine (Ler), an antihypertensive agent, increases the cytotoxicity of various PIs including Btz, carfilzomib (Cfz), and ixazomib (Ixz) by inducing paraptosis-associated cell death, which is accompanied by severe dilation of mitochondria and the endoplasmic reticulum (ER) and in many solid tumor cell lines. We have found that mitochondrial Ca2+ overload induced by the combination of Btz and Ler is crucial for mitochondrial vauolation and subsequent mitochondrial membrane potential loss in MDA-MB 435S cells. In addition, Ler enhances Btz-mediated ER stress due to accumulation of misfolded proteins, contributing to ER vacuolization. Taken together, our results suggest that the combined regimen of PIs and Ler can effectively kill cancer cells through structural and functional disturbance in mitochondria and the ER.
Keywords: Bortezomib, Lercanidipine, ER stress, Mitochondrial Ca2+ overload, Paraptosis
ii
TABLE OF CONTENS
ABSTRACT --- i
TABLE OF CONTENTS --- ii
LIST OF FIGURES --- iv
LIST OF TABLES --- vii
I. INTRODUCTION
--- 1II. MATERIALS AND METHODS
--- 17A. Chemicals and antibodies --- 17
B. Cell culture --- 18
C. PI staining for cell viability assay --- 18
D. Determination of cell viability by MTT assay --- 19
E. Examination of morphologies of mitochondria and the ER employing the plasmids to specifically label the ER or mitochondria --- 19
F. Western blotting --- 19
G. Immunocytochemistry --- 20
H. Measurement of cytosolic and mitochondrial Ca2+ levels --- 20
I. Isobologram analysis --- 21
J. Statistical analysis --- 21
III. RESULTS
--- 231. Various dihydropyridine effectively enhance Btz-mediated cell death in breast cancer cells --- 23
iii
2. Lercanidipine effectively enhances PI-mediated cell death in a various cancer cells --- 27
3. Combined treatment with Ler and Btz induces apoptotic, necroptotic, and non-autophagic cell death --- 33
4. Combination of Ler and Btz induces paraptosis --- 40
5. Combined treatment with Ler and Btz enhances ubiquitinated protein accumulation and ER stress --- 47
6. Combination of Ler and Btz disrupts Ca2+ homeostasis via mitochondrial Ca2+ overload -- --- 50
7. Mitochondrial Ca2+ uptake critically contributes to Ler/Btz-induced paraptotic cell death - --- 56
IV. DISCUSSION
--- 59V. REFERENCES
--- 66iv
LIST OF FIGURES
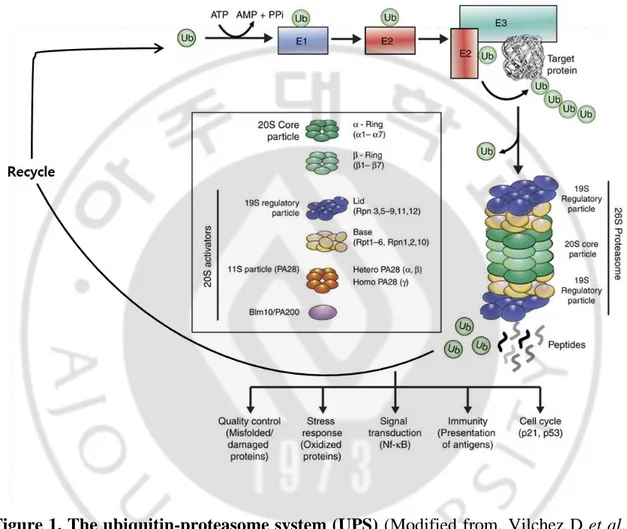
Fig. 1. The ubiquitin-proteasome system (UPS) --- 3
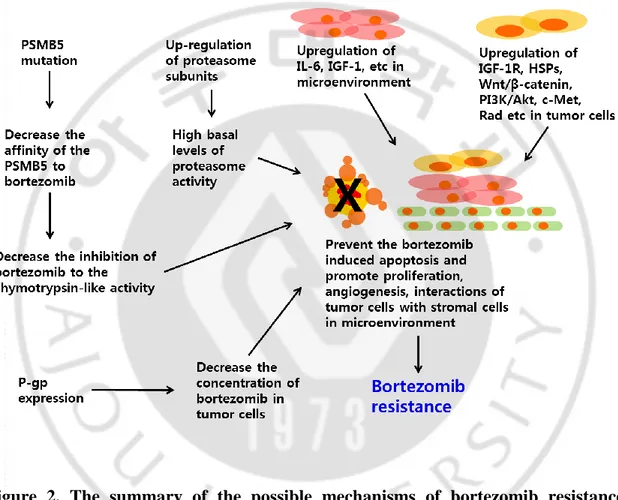
Fig. 2. The summary of the possible mechanisms of bortezomib resistance --- 7
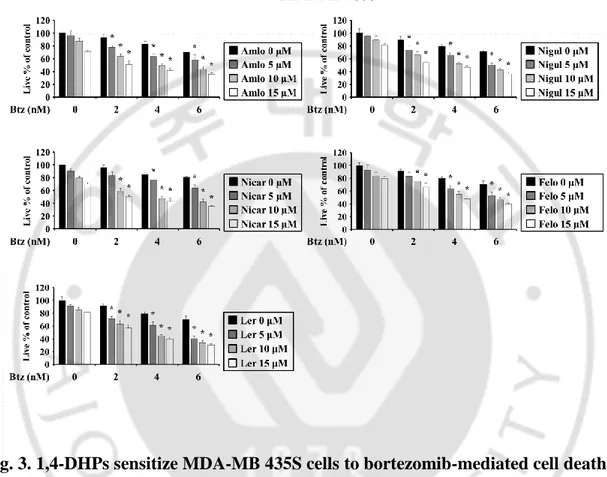
Fig. 3. 1,4-DHPs sensitizes MDA-MB 435S cells to bortezomib-mediated cell death --- 24
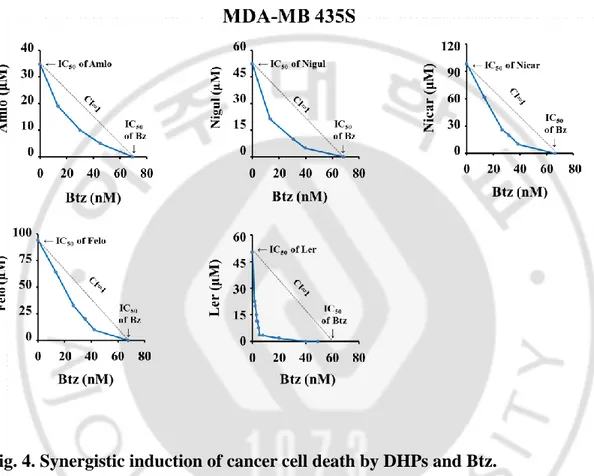
Fig. 4. Synergistic induction of cancer cell death by DHPs and Btz --- 25
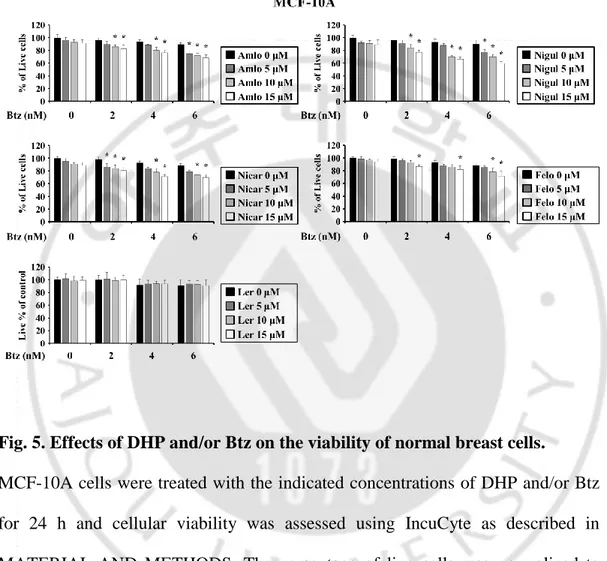
Fig. 5. Effects of DHP and/or Btz on the viability of normal breast cells --- 26
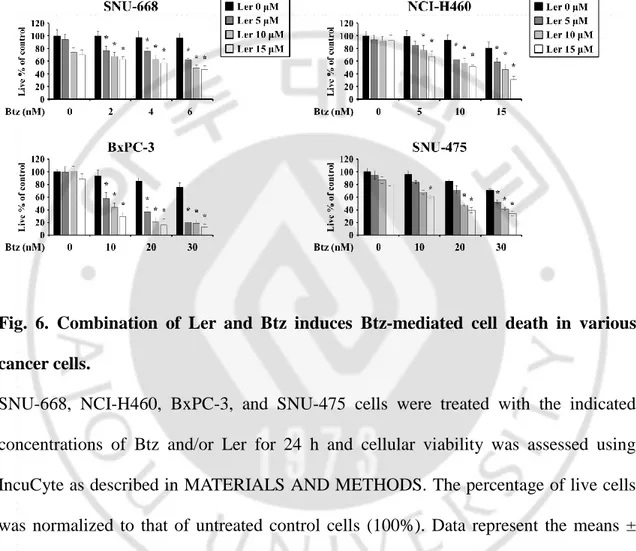
Fig. 6. Combination of Ler and Btz induces Btz-mediated cell death in various cancer cells --- --- 28
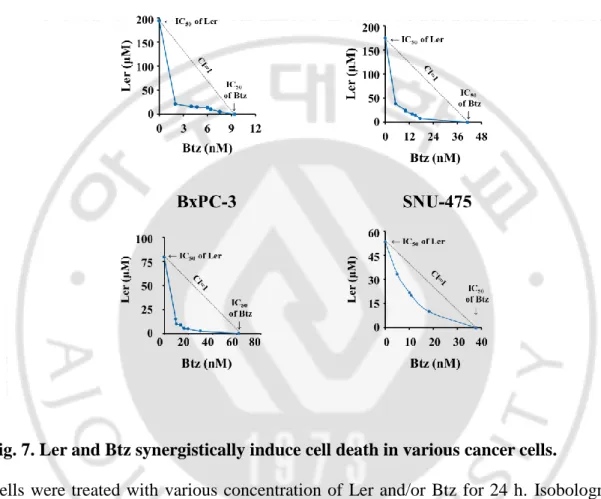
Fig. 7. Ler and Btz synergistically induce cell death in various cancer cells --- 29
Fig. 8. Ler sensitizes to Btz-mediated cell death in multiple myeloma cells --- 30
Fig. 9. Ler sensitizes to the proteasome inhibitor-mediated cell death in breast cancer cells --- 31
Fig. 10. Combined treatment with Ler and PIs did not increases cell death in normal cells - 32 Fig. 11. The morphological changes in MDA-MB 435S and MCF-10A cells treated with Amlo/Btz, Nicar/Btz, Nigul/Btz, or Felo/Btz --- 35
Fig. 12. Combined treatment with Ler and Btz induces cellular vacuolation in breast cancer cells, but not in normal breast cells --- 36
Fig. 13. Combined treatment with Ler and Btz induces cellular vacuolation in various cancer cells --- 37
Fig. 14. The morphological changes in breast cancer cells or normal breast cells treated with Ler/Cfz or Ler/Ixz --- 38
v
Fig. 15. Ler/Btz-induced vacuolization and cell death is not associated with apoptosis, necroptosis, or autophagic cell death --- 39 Fig. 16. Combined treatment with Ler/Btz induces the ER and mitochondrial dilation in
cancer cells --- 42 Fig. 17. Morphological changes in cells treated with Btz or Ler --- 43 Fig. 18. Mophologies and membrane potential of mitochondria in cells treated with Btz or Ler
alone --- 44 Fig. 19. Combined treatment with Ler and Btz induces mitochondrial vacuolization and MMP loss in cancer cells --- 45 Fig. 20. Pretreatment with CHX inhibits Ler/Btz-induced cytoplasmic vacuolation and cell
death in cancer cells --- 46 Fig. 21. Ler enhances Btz-mediated ubiquitinated protein and ER stress --- 48 Fig. 22. The expression levels of ER stress marker proteins in Ler/Btz-treated breast cancer
cells or breast cells --- 49 Fig. 23. Combined treatment with Ler and Btz markedly increases cytosolic Ca2+ levels --- 52 Fig. 24. Combined treatment with Ler and Btz significantly increases mitochondrial Ca2+ levels --- 53 Fig. 25. Pretreatment with MCU inhibitors significantly suppresses Ler/Btz-induced cell
death and dilation of mitochondria and the ER --- 54 Fig. 26. The effects of pretreatment with DIDS or 2-APB on Ler/Btz-induced cell death in MDA-MB 435S cells --- 55 Fig. 27. Mitochondrial Ca2+ overload is critical for Ler/Btz-induced paraptosis --- -- 57
vi
Fig. 28. Mitochondrial Ca2+ overload and the increase in cytosolic Ca2+ may localize downstream of ER stress in Ler/Btz-induced signals --- 58 Fig. 29. Hypothetical model of Ler/Btz-induced paraptosis --- 65
vii
LIST OF TABLS
Table. 1. Examples of the successful drug repositioning --- 9 Table. 2. Examples of the selected failed drug repositioning --- 10 Table. 3. Comparison of programmed cell death --- 14
1
I. INTRODUCTION
A. Ubiquitin Proteasome system (UPS)
In eukaryotes, the regulated hydrolysis of cellular proteins is mediated by a
ubiquitous macromolecular enzymatic complex, the 26S proteasome (Murata S et al., 2009). The ubiquitin proteasome system (UPS) is the most important regulated intracellular protein degradation system, and is involved in processes such as apoptosis, cell survival, cell-cycle progression, DNA repair, and antigen presentation (Manasanch EE and Orlowski RZ, 2017). To ensure appropriate destruction of damaged or misfolded proteins, or of proteins that are no longer needed, the components of this system must act in a highly coordinated manner through different steps that include poly-ubiquitination, de-poly-ubiquitination, and degradation of the target proteins (Manasanch EE and Orlowski RZ, 2017). Degradation by the UPS is initiated by the conjugation of ubiquitin, a highly conserved 76 amino-acid residue polypeptide, to the substrate protein (Finley D, 2009). Proteins destined for proteasome-mediated destruction in eukaryotic cells are covalently tagged with ubiquitin (Ub), a small protein modifier (Kale AJ and Moore BS, 2012). The process of protein (poly) ubiquitination involves a cascade of 3 enzymatic steps; Ub-activating enzymes (E1), Ub-conjugating enzymes (E2), and Ub E3 ligases (Figure 1) (Roeten MSF et al., 2018). For protein degradation, the target protein must be linked with four or more ubiquitin units (Demarchi F and Brancolini C, 2005). Prior to actual degradation by the proteasome, deubiquitinases (DUBs) remove and recycle ubiquitin moieties from the tagged proteins (Komander D et al., 2009;
2
Streich FC Jr and Lima CD, 2014). The 26S proteasome consists of the 20S catalytic core domain and two 19S regulatory particles (Roeten MSF et al., 2018). The 19S cap of the proteasome removes the Ub moieties after which the substrate is degraded in the 20S into smaller peptides (Roeten MSF et al., 2018). The shorter peptides generated after proteasomal degradation can either be processed for antigen presentation on major histocompatibility complex (MHC) class 1 molecules, or being fully hydrolyzed into amino acids by aminopeptidases and then recycled for protein synthesis (Hitzerd SM et
al., 2014; Reits E et al., 2003; Rock KL et al., 2002). Given the high protein turnover
and the critical role of the UPS in the development, cell growth and survival of cancer cells (Adams J, 2004; Chen D et al., 2011), proteasome inhibition constitutes an attractive target for chemotherapeutic intervention (Dou QP and Li B, 1999; Almond JB and Cohen GM, 2002; Manasanch EE and Orlowski RZ, 2017).
3
Figure 1. The ubiquitin-proteasome system (UPS) (Modified from. Vilchez D et al.,
4 B. Proteasome inhibitor (PI)
Bortezomib (Btz) is a slowly reversible and peptide boronate inhibitor, which
binds with the catalytic site of the 26S proteasome, enabling inhibition of the β5/chymotrypsin-like and, to a lesser extent, the β2/trypsin-like and β1/post-glutamyl peptide hydrolyzing activities (Orlowski M and Wilk S, 2000; Crawford LJ et al., 2011). Btz represents the first-generation PI being approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) for treatment of multiple myeloma (MM) and mantle cell lymphoma (MCL) (Anderson KC, 2012). Nonetheless, limiting factors in Btz therapy included its non-availability of oral formulation, off-target activity and acquired resistance (Roeten MSF et al., 2018). The most prominent clinical adverse event included peripheral neuropathy, which is caused by off-target inhibition by BTZ of a neuronal survival protein, HtrA2/Omi (Arastu-Kapur S et al., 2011). These neurotoxic side effects could be diminished by alternative scheduling and route of administration (Roeten MSF et al., 2018). Btz resistance development was recognized as a relevant issue, as both a sub-population of patients had no response to Btz and a large proportion of patients relapsed on Btz treatment (Roeten MSF et al., 2018). To overcome these limiting factors, second-generation PIs were developed to improve the efficacy, reducing the toxicity, enhancing the oral availability and overcoming Btz resistance by targeting multiple β catalytic subunits and/or do this, other than Btz, in an irreversible manner (Allegra A et al., 2014; Moreau P et al., 2012).
5
Carfilzomib (Cfz) is the second PI approved by the FDA in 2012 as a 3rd line treatment in MM and by the EMA in 2015 (Kortuem KM and Stewart AK, 2013; Roeten MSF et al., 2018). Cfz is structurally, chemically and mechanistically different from Btz, it does so in an irreversible manner, with greater selectivity and lesser off-target activity, contributing to an improved clinical safety profile as compared to Btz (Siegel D et al., 2013). Moreover, the covalent binding of CFZ ensures prolonged proteasome inhibition (Ruschak AM et al., 2011) and showed cytotoxic activity against Btz-resistant cells (Demo SD et al., 2007; Kuhn DJ et al., 2007).
Ixazomib (Ixa) is an orally bioavailable boronic ester prodrug, which reversibly binds to the β5- and β1-subunits (Dick LR and Fleming PE, 2010). The drug was the first oral PI to enter clinical trials and has been approved for MM treatment since September 2016. Ixa is clinically active in heavily pre-treated and refractory/relapsed MM, in vitro Ixa had the ability to overcome Btz resistance in MM cells (Chauhan D et al., 2011). However, in parallel with the development and clinical use of these novel proteasome inhibitor therapies, there is the emergence of novel mechanisms of resistance to various cancer, including multiple myeloma, non-Hodgkin’s lymphoma, and solid tumor (Figure 2, Lü S and Wang J, 2013; Wallington-Beddoe CT et al., 2018). Thus the clinical efficacy of PI as a single agent is limited in hematological malignancies and solid cancer (Cusack JC, 2003; Adams J, 2004). For this reason, many research groups are conducting a lot of study on combination therapy to overcome the resistance of the PI, including irinotecan, gemcitabine, and loperamide (Cusack JC Jr et al., 2001; Bold
6
RJ et al., 2001; Kim IY et al., 2019). In particular, combined treatment of non-anticancer drugs in clinical use may increase the efficacy of PIs reducing side effects
7
Figure 2. The summary of the possible mechanisms of bortezomib resistance
8 C. Drug repositioning
Drug repositioning (also called by drug repurposing) involves the investigation of
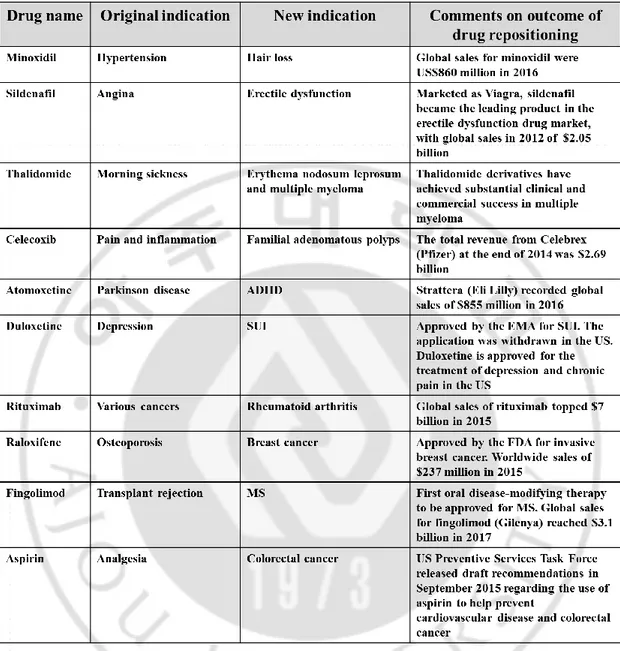
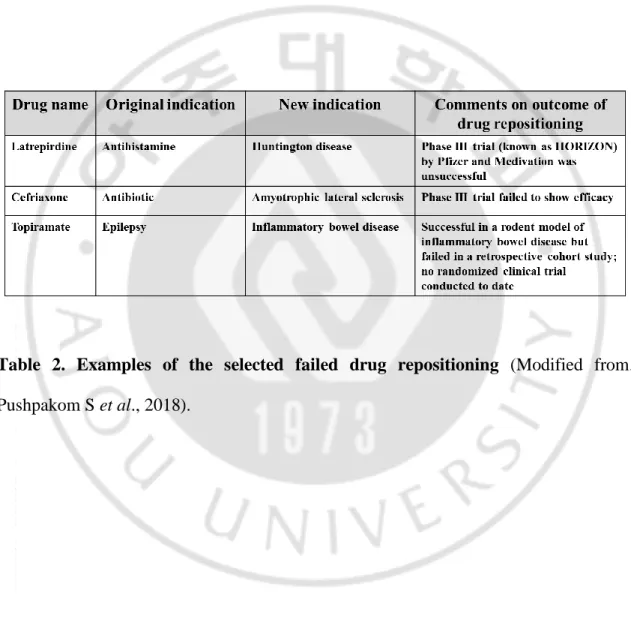
novel therapeutic indications for approved, discontinued, and archived drugs, as well as drugs currently undergoing clinical trials (Talevi A, 2018). Drug discovery is a time-consuming, taxing, costly, and high-risk process, whereas drug repositioning is an effective strategy to find new indications for existing drugs and is highly efficient, low-cost and riskless (Xue H et al., 2018). Recently, improvements in drug delivery speed, effectiveness, and safety by making changes to drug formulations or some chemical structures are also included in the category of drug repositioning. Therefore, drug repositioning is considered as a new drug development strategy (Padhy BM and Gupta YK, 2011; O'Connor KA and Roth BL, 2005). Table 1 shows the examples of some successful drug repositioning with the adopted repurposing approach, most so far derived from pharmacological understanding of the clinical effect of a drug when prescribed for its original indication (Pushpakom S et al., 2018). Nevertheless, drug repurposing is not always a success. Table 2 shows the selected drug cancicate for drug repositioning failed, mainly at the trials of the Phase III. As with the development of completely new drugs, some failures in late-stage development are clearly expected, which should not be attributed to toxicity because the candicate’s safety profile has previously been characterized. However, there are also failures in the drug repurposing field, including patent rebalancing, regulatory considerations, and organizational hurdles, which are associated with barriers specific to drug repositioning (Pushpakom S
9
ADHD, attention deficit hyperactivity disorder; EMA, European Medicines Agency; FDA, US Food and Drug Administration; MS, multiple sclerosis; SUI, stress urinary incontinence.
Table 1. Examples of the successful drug repositioning (Modified from. Pushpakom
10
Table 2. Examples of the selected failed drug repositioning (Modified from.
11
D. Calcium channel blockers and lercanidipine (Ler)
Calcium channel blockers (CCBs), calcium channel antagonist, are a number of drugs that interfere with the movement of calcium (Ca2+) through calcium channels (Olson and Kent 40. Calcium Channel Antagonists Ed. 2011 Edition. McGraw-Hill Medical ISBN978-0071668330). CCBs are widely used for the management of hypertension and angina (Bucci C et al., 2008). CCBs comprise three distinct subgroups: benzothiazepines (e.g. diltiazem), dihydropyridines (DHPs) (e.g. amlodipine, lercanidipine), and phenylalkylamines (e.g. verapamil) (Toal et al., 2012). The DHPs work primarily as vasodilators and have evolved from the first generation, short-acting compounds (eg, nifedipine and felodipine), which produced unwanted reflex tachycardia (Borghi C, 2005). Modified formulations were introduced to extend duration of action and limit adverse effects; however, amlodipine was the first DHP with an inherently long plasma half-life (Borghi C, 2005).
The scientific literature indicates that the effects of CCBs on apoptosis are complex and often contradictory (Mason RP, 1999). Although CCBs have been shown to inhibit apoptosis in certain non-transformed cell lines (Li LH et al., 1997; Escargueil-Blanc I et
al., 1997), there are numerous reports that CCBs promote apoptosis in cancer cell lines
(Vilpo J et al., 2000; Mason RP, 1999). Moreover, some CCBs (nifedipine, nicardipine, barnidipine, felodipine, lercidipine, verapamil, and diltiazem) have strong inhibitor effects on P-gp activity (Zhou SF et al., 2007). For this reason, when combined with chemotherapeutic agents, CCBs can help to promote intracellular drug accumulation
12
and overcome multidrug resistance (Broxterman HJ et al., 1996; Harmsze AM et al., 2010).
Lercanidipine is a lipophilic, third-generation dihydropyridine calcium channel blocker with a long receptor half-life, which has an established role in antihypertensive therapy (Borghi C, 2005; Grassi G et al., 2017). In addition, lercanidipine prevents renal damage caused by angiotensin II, and proves inflammatory, antioxidant, and anti-atherogenic properties through an increasing bioavailability of endothelial nitric oxide (Grassi G et al., 2017). Lercanidipine has shown a lower negative inotropic effect than other dihydropyridines including lacidipine, amlodipine, nitrendipine, nifedipine, and felodipine (Angelico P et al., 1999).
Lercanidipine is well tolerated with DHP-associated adverse effects occurring early in treatment. The incidence of pedal edema and subsequent withdrawals has been found to be lower with lercanidipine than with amlodipine or nifedipine gastrointestinal transport system (Borghi C, 2005). Preclinical and preliminary clinical findings suggest lercanidipine may have beneficial effects on atherosclerosis and left ventricular hypertrophy (Borghi C, 2005). The efficacy and tolerability profiles of lercanidipine make it a suitable choice for treating hypertension in a wide range of affected patients (Borghi C, 2005). However, there has been no report on the anti-cancer effect of lercanidipine.
13 E. Paraptosis
Paraptosis, (from para=next to or related to, and apoptosis) or paraptosis-like cell
death (PLCD) is a type of nonapoptotic programmed cell death, which reveals vacuolization of ER components and swelling of mitochondria (Sperandio S et al., 2000; Shubin AV et al., 2016). This cell death mode special feature is absence of caspase activation or nuclear changes, including pyknoisis and DNA fragmentation (Sperandio S
et al., 2000; Wyllie AH and Golstein P, 2001). Previously, IGF1R and overexpression of
TAJ/TROY receptor were shown to be associated with the induction of paraptosis (Pehar M et al., 2010; Wang Y et al., 2004). Recent studies have shown that various natural products, including curcumin, celastrol, 15d-PGJ2, ophiobolin A, and paclitaxel, demonstrate anti-cancer effects by inducing the paraptosis-assciated cell death (Yoon MJ et al., 2010; Wang Y et al., 2004; Yoon MJ et al., 2014; Kar R et al., 2009; Bury M
et al., 2013; Chen TS et al., 2008; Wang C and Chen T, 2012). The manifestations of
paraptosis could not be prevented by pre-treatment with caspase inhibitors, or by the overexpression of anatiapoptotic Bcl-2-like proteins (Sperandio S et al., 2000; Sperandio S et al., 2004) and seemingly resulted from a signaling cascade involving specific members of the mitogen-activated protein kinase family. Also paraptosis is characterized by the requirement for new gene transcription and translation, and inhibited by AIP-1/Alix (Tabel 3; Sperandio S et al., 2004). It is not yet clear whether paraptosis represents a route of cell death that distinguishes it from other cells, and the mechanism has not been clearly elucidated.
14
Table 3. Comparison of programmed cell death (modified from Kroemer G et al.,
15 F. Purpose of the study
Proteasome inhibition has been successfully targets for cancer therapy in a wide
spectrum of hematological and solid tumors (Rajkumar SV et al., 2005). However, despite the success of proteasome inhibitors (PIs) in the treatment of hematological malignancies including multiple myeloma, the clinical efficacy of these PIs as mono-therapies is limited in solid tumors (Johnson DE, 2015). Therefore, identifying sensitive agents that can enhnace the sensitivity of PIs can be an effective treatment strategy for solid tumors. In the present study, we investigated the sensitizers of PIs through drug repositioning.
In this study, we first demonstrate that lercanidipine (Ler), a third-generation 1,4-dihydropyridines (DHPs) calcium channel blocker (Burnier M et al., 2009), can synergistically enhance PIs’ cytotoxicity in various types of cancer cells. Ler contributes to peripheral vasodilation by preventing Ca2+ ions from passing through the L-type calcium channel of the cell membrane (Bang LM et al., 2003), and is highly effective in patients with high cardiovascular risk and atherosclerosis (Pruijm MT et al., 2008). Importantly, some compounds of the 1,4-DHP class have been shown to have potential to kill cancer cells by inhibiting P-glycoprotein pump and reverses multidrug resistance, an important factor in the failure of conventional chemotherapy in many cancer types (Shekari F et al., 2015; Viale M et al., 2011; Voigt B et al., 2007). In particular, MM patients are known to be vulnerable to cardiovascular complications, including high blood pressure, due to individual risk factors (age and predisposing comorbidities) and myeloma-associated effects (Mathur P et al., 2017). Therefore, combination therapy
16
with Btz and Ler may improve the efficacy and safety of PI-based therapies for various cancer treatments. Interestingly, we found that the combination of Btz and Ler (Btz/Ler) kills cancer cells by inducing paraptosis, which is accompanied by vauolation derived from mitochondria and the endoplasmic reticulum (ER) (Sperandio S et al., 2000; Sperandio S et al., 2004). Here we show that Ler and Btz synergistically kill various cancer cells by inducing the enhancement of ER stess and intracellular Ca2+ imbalance. In this process, Ca2+ accumulation in mitochondria leads to mitochondrial swelling, loss of mitochondrial membrane potential (MMP), and subsequently ER dilation, leading to paraptosis-associated cell death.
17
II. MATERIALS AND METHODS
A. Chemicals and antibodies
Lercanidipine, Amlodipine, Nicardipine, Niguldipine, Felodipine was purchased from Tocris (Tocris Bioscience, Bristol, UK). Bortezomib and Carfilzomib were from Selleckchem (Houston, TX, USA). Ixazomib (MLN9708) was purchased from Cayman chemical (Ann Arbor, MI, USA). 3-methyladenine (3-MA), bafilomycin A1, cycloheximide (CHX), 1,2-bis(o-aminophenoxy)ethane-N,N,N’N’-tetraacetic acid acetoxymethyl ester (BAPTA-AM), necrostatin-1, and ruthenium red were purchased from Sigma (St. Louis, MO). Ru360 and 2-Aminoethosxydiphenyl borate (2-APB) were from Calbiochem (EDM Millipore Corp., Billerica, MA, USA). MitoTracker-Red (MTR), propidium iodide (PI), tetramethylrhodamine methyl ester (TMRM), Fluo-3-AM, and Rhod-2-AM were purchased from Molecular Probes (Eugene, OR, USA). Caspases inhibitors benzyloxy-carbonyl-Val-Ala-Asp-(OMe) fluoromethyl ketone (z-VAD-fmk) was from R&D systems (Minneapolis, MN). The following antibodies were used: monoclonal β-actin (Abcam); p62 (BD biosciences pharmingen); anti-ubiquitin and anti-ATF4 (Santa Cruz Biotechnologies, Santa Cruz, CA); anti-PERK, anti-p-eIF2α, anti-eIF2α, anti-LC3B, and anti-CHOP (GADD153) (Cell Signaling, Beverly, MA); anti-Grp78 and anti-caspase-3 (Stressgen, BC, Canada); HRP-conjugated anti-rabbit IgG and HRP-conjugated anti-mouse IgG (Molecular Probes).
18 B. Cell culture
The MDA-MB 435S (breast cancer), BxPC-3 (pancreatic cancer), SNU-668 (stomach cancer), NCI-H460 (lung cancer), MCF-10A (breast epithelial), and Chang (liver epithelial) were purchased from American Type Culture Collection (ATCC, Manassas, VA). The SNU-475 was purchased from Korean Cell Line Bank (KCLB, Seoul, Korea). MDA-MB 435S cells were cultured in DMEM and BxPC-3, NCI-H460, SNU-668, SNU-475, and Chang cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and antibiotics (GIBCO-BRL, Grand Island, NY). MDF-10A cells were maintained in DMEM/F12 supplemented with pituitary extract, insulin, human epidermal growth factor, hydrocortisone, and choleratoxin (Calbiochem). Cells were incubated in 5% CO2 at 37℃.
C. PI staining for cell viability assay
Cells were cultured in 24-well plates and treated as indicated. The cells were then fixed with methanol/acetone (1:1) at -20℃ for 5 min, washed three times with PBS, and stained with propidium iodide (PI; final concentration, 1 μg/ml) at room temperature for 10 min. The plates were imaged on an IncuCyte device (Essen Bioscience, Ann Arbor, MI, USA) and analyzed using the IncuCyte ZOOM 2016B software. The processing definition of the IncuCyte program was set to recognize attached (live) cells by their red-stained nuclei. The percentage of live cells was normalized to that found in untreated control cultures (100%).
19
D. Determination of cell viability by an MTT assay
Cells were plated in 96-well plates at a concentration of 1X104 cells/ml. After treatments, MTT assay was performed according to the manufacturer’s protocol (Sigma). Absorption at 570 nm was normalized to that of untreated control (100%), and the results were expressed as viability % of control.
E. Examination of the morphologies of mitochondria and the ER employing the plasmids to specifically label the ER or mitochondria
To establish the stable cell lines expressing the fluorescence specifically in the ER, MDA-MB 435S cells were transfected with the pEYFP-ER or pEYFP-mitochondria vector (Clontech, Mountain View, CA, USA). Stable cell lines expressing pEYFP-ER (YFP-ER) or pEYFP-mitochondria (YFP-Mito) were selected with fresh medium containing 500 μg/ml G418 (Calbiochem). After treatments, YFP-ER cells were stained with 100 nM MitoTracker Red (MTR) or YFP-Mito cells were stained with 200 nM TMRM for 10 min and morphological changes of the ER and mitochondria were observed under a K1-Fluo confocal laser scanning microscope (Nanoscope Systems, Daejeon, Korea).
F. Western blotting
Cells were washed in PBS and lysed in boiling sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (6.25 mM Tris [pH 6.8], 1% SDS, 10% glycerol, and 5% β-mercaptoethanol). The lysates were boiled for 5 min, separated by
20
SDS-PAGE, and transferred to an Immobilion membrane (Millipore, Bredford, MA, USA). After blocking nonspecific binding sites for 1 h using 5% skim milk, membrane were incubated for 2 h with specific Antibodies. Membranes were then washed three times with TBST and incubated further for 1 h with horseradish peroxidase-conjugated anti-rabbit, -mouse antibody. Visualization of protein bands was accomplished using ECL (Advansta).
G. Immunocytochemistry
After treatments, cell were fixed with 50% MeOH plus 50% Acetone for 5 min at -20℃ and blocking in 5% BSA in PBS for 30 min. Fixed cells were incubated overnight at 4℃ with primary antibody [anti-ubiquitin (1:500, mouse, Santa Cruz Biotechnology) diluted in PBS and then washed three times in PBS and incubated for 1 h at room temperature with anti-rabbit Alexa Fluor 488 (1:200, Molecular Probes). Or anti-mouse Alexa 594 was used as a secondary antibody (Molecular Probes). Next, cells were washed with PBS. Slides were mounted with ProLong Gold antifade mounting reagent (Molecular Probes) and cell staining was visualized with a fluorescence microscope (Axiovert 200M, Carl Zeiss) or the K1-Fluo confocal laser-scanning microscope.
H. Measurement of cytosolic and mitochondrial Ca2+ levels
To measure cytosolic [Ca2+]c levels, treated cells were incubated with 1 μM Fluo-3-AM
at 37 °C for 20 min, washed with HBSS (without Ca2+ or Mg2+), and analyzed immediately by flow cytometry using a FACSAriaTM
21
confocal laser scanning microscope. To measure mitochondrial mitochondrial Ca2+ levels ([Ca2+]mt), treated cells were incubated with 1 μM Rhod-2-AM at 4 °C for 30 min,
washed with HBSS (without Ca2+ or Mg2+), further incubated with HBSS at 37 °C for 20 min, and then analyzed by flow cytometry or visualized by the K1-Fluo confocal laser scanning microscope.
I. Isobologram analysis
To determine how the combinations of PIs and DHPs affected the cancer cell lines, dose-dependent effects were determined for each compound alone and with a fixed concentration of the other co-treated agent. The interactions of the PIs and DHPs were quantified by determining the combination index (CI), in accordance with the following classic isobologram equation [Adv. Enzyme Regul 1984]: CI = (D)1/(Dx)1 + (D)2/(Dx)2,
where (Dx)1 and (Dx)2 indicate the individual doses of PIs and DHPs, respectively,
required to produce an effect, and (D)1 and (D)2 are the doses of PIs and DHPs,
respectively, that produce the same effect when applied in combination. From this analysis, the combined effects of the two drugs can be summarized as follows: CI < 1 indicates synergism; CI = 1 indicates summation (additive and zero interaction); and CI > 1 indicates antagonism.
J. Statistical analysis
All data are presented as mean ± S.D. (standard deviation) from at least three separate experiments. To perform statistical analysis, GraphPad Prism (GraphPad Software Inc,
22
Sandiego, CA) was used. Normality of data was assessed by Kolmogorov–Smirnov testes and equal variance using Bartlett’s test. For a normal distribution, statistical differences were determined using an analysis of variance (ANOVA) followed by followed by Bonferroni multiple comparison tests. If the data were not normally distributed, Kruskal–Wallis test was performed followed by Dunn’s test.
23
III. REULTS
1. Various dihydropyridine effectively enhance Btz-mediated cell death in breast cancer cells
Multidrug resistance (MDR) is frequently appeared by the overexpression of P-glycoprotein (P-gp) pump in cancer cells (Zhou J et al., 2006). Therefore, we have explored the drug repositioning approach to identify candidate modulators of anticancer activities. Since some compounds of the 1,4-dihydropyridines (1,4-DHP), which are known to be anti-hypertensive agents with calcium channel blocker activities, have shown to have potential to kill cells by inhibiting P-gp pump (Viale M et al., 2011), we investigated whether 1,4-DHP could sensitize bortezomib (Btz) to breast cancer cells. We found that when MDA-MB 435S (breast cancer) cells were treted with amlodipine (Amlo), niguldipine (Nigul), nicardipine (Nicar), and felodipine (Felo), and lercanidipine (Ler) up to 15 μM, cytotoxicity was not observed. But combined treatment with subtoxic doses of Btz and DHPs dose-dependently enhanced cell death in these cell lines (Figure 3). Isobologram analysis revealed that Btz and any DHPs synergistically induced cell death in these cells (Figure 4). Next, we tested whether combination of DHP and Btz affects the viability of normal cells. The combined treatment with Ler and Btz (Ler/Btz) did not affect the viability of MCF-10A cells, whereas Amlo/Btz, Nigul/Btz, Nicar/Btz, or Felo/Btz slightly reduced it (Figure 5). These results suggest that Ler may more effectively and safely sensitize cancer cells to Btz-mediated cell death than other DHPs.
24
Fig. 3. 1,4-DHPs sensitize MDA-MB 435S cells to bortezomib-mediated cell death.
MDA-MB 435S cells were treated with the indicated concentrations of Btz and/or Amlo, Nigul, Nicar, Felo, or Ler for 24 h and cellular viability was assessed using IncuCyte. The percentage of live cells was normalized to that of untreated control cells (100%). Data represent the means ± S.D. (n=7). One way ANOVA and Bonferroni’s post hoc test. *p <0.001 vs. Btz-treated cells.
25
Fig. 4. Synergistic induction of cancer cell death by DHPs and Btz.
MDA-MB 435S cells were treated with various concentrations of various DHPs and/or Btz for 24 h. Isobologram analysis was performed as described in MATERIALS AND METHODS. Isoboles for the combination of CCB and Btz, which were iso-effective (IC50) for inhibition of cell viability, are shown.
26
Fig. 5. Effects of DHP and/or Btz on the viability of normal breast cells.
MCF-10A cells were treated with the indicated concentrations of DHP and/or Btz for 24 h and cellular viability was assessed using IncuCyte as described in MATERIAL AND METHODS. The percentage of live cells was normalized to that of untreated control cells (100%). Data represent the means ± S.D. (n=7). One-way ANOVA and Bonferroni’s phost hoc test. *p <0.001 vs. Btz-treated cells.
27
2. Lercanidipine effectively enhances PI-mediated cell death in various cancer cells
We investigated whether Ler can effectively sensitize other types of cancer cells to Btz-mediated cell death. Cytotoxicity of Ler up to 15 μM was not observed in SNU-668 (stomach cancer), NCI-H460 (lung cancer), BxPC-3 (pancreatic cancer), or SNU-475 (liver cancer) cells, but combined treatment with subtoxic doses of Btz and Ler dose-dependently enhanced cell death in these cancer cell lines (Figure 6). Isobologram analysis revealed that Btz and Ler synergistically induced cell death in these cells (Figure 7). Since Btz is widely used in the patients with multiple myeloma (MM), we investigated whether Ler could enhance the Btz-mediated cell death in the MM cells, RPMI 8226. We found that treatment with Ler dose-dependently increased Btz-mediated cell death in these MM cells (Figure 8A), showing synergistic effects (Figure 8B). We further tested whether Ler affects the viability of MDA-MB 435S cells treated with two other FDA-approved PIs, including calfilzomib (Cfz) and ixazomib (Ixz). We found that Ler effectively enhanced the cell death mediated by these proteasome inhibitors (PIs) too (Figure 9A), showing synergistic effects (Figure 9B). Interestingly, Ler did not increase the death of MCF-10A (normal breast) cells treated with Cfz or Ixa (Figure 10). Furthermore, Ler had no cytotoxic effects on Chang (normal liver) cells treated with Btz (Figure 10). These results suggest that Ler can safely and effectively sensitize to various cancer cells to PIs.
28
Fig. 6. Combination of Ler and Btz induces Btz-mediated cell death in various cancer cells.
SNU-668, NCI-H460, BxPC-3, and SNU-475 cells were treated with the indicated concentrations of Btz and/or Ler for 24 h and cellular viability was assessed using IncuCyte as described in MATERIALS AND METHODS. The percentage of live cells was normalized to that of untreated control cells (100%). Data represent the means ± S.D. (n=7). One-way ANOVA and Bonferroni’s post hoc test. *p<0.001 vs. Btz-treated cells.
29
Fig. 7. Ler and Btz synergistically induce cell death in various cancer cells.
Cells were treated with various concentration of Ler and/or Btz for 24 h. Isobologram analysis was performed as described in MATERIALS AND METHODS. Isoboles for the concentration of Btz and Ler that proved iso-effective (IC50) for inhibiting cell
30
Fig. 8. Ler sensitizes to Btz-mediated cell death in multiple myeloma cells.
(A) RPMI 8226 cells were treated with the indicated concentrations of Btz and/or Ler for 24 h and cellular viability was assessed using MTT assay as described in MATERIALS AND METHODS. The percentage of live cells was normalized to that of untreated control cells (100%). Data represent the means ± S.D. (n=7). One-way ANOVA and Bonferroni’s post hoc test. *p<0.001 vs. Btz treated cells. (B) Isobologram was performed as described in MATERIALS AND METHODS. Isoboles for the concentration of Btz and Ler that proved iso-effective (IC50) for inhibiting cell
31
Fig. 9. Ler sensitizes to the proteasome inhibitor-mediated cell death in breast cancer cells.
(A) MDA-MB 435S cells were treated with the indicated concentrations of PIs and/or Ler for 24 h and cellular viability was assessed using IncuCyte as described in MATERIALS AND METHODS. The percentage of live cells was normalized to that of untreated control cells (100%). Data represent the means ± S.D. (n=7). One-way ANOVA and Bonferroni’s post hoc test. *p<0.001 vs. Btz-treated cells. (B) Isobologram was performed as described in MATERIALS AND METHODS. Isoboles for the concentration of PIs and Ler that proved iso-effective (IC50) for inhibiting cell
32
Fig. 10. Combined treatment with Ler and PIs did not increase cell death in normal cells.
MCF-10A and Chang cells were treated with the indicated concentrations of PIs and/or Ler for 24 h and cellular viability was assessed using IncuCyte as described in MATERIALS AND METHODS. The percentage of live cells was normalized to that of untreated control cells (100%). Data represent the means ± S.D. (n=7).
33
3. Combined treatment with Ler and Btz induces apoptotic, non-necroptotic, and non-autophagic cell death
To understand how Ler overcomes the resistance of the cancer cells to PIs, we observed cell morphology after treatment with DHP and/or Btz. We found that treatment with 4 nM Btz or 10 μM Amlo, Nicar, Nigul, Felo, and Ler for 16 h did not induce morphological change in MDA-MB 435S cells. Combinations of DHP and Btz induced the extensive vacuolation in MDA-MB 435S cells (Figure 11 and Figure 12). Btz-Ler did not induce any vacuolation or cell death in MCF-10A cells. Btz/Amlo, Btz/Nicar, Btz/Nigul, or Btz/Felo induced cytoplasmic vacuolation in MCF-10A cells, but at a much lesser extent than that in MDA-MB 435S cells. In contrast, treatment with Ler/Btz, but not in single treatment, induced a dramatic cellular vacuolization in SNU-668, NCI-H460, BxPC-3, and SNU-475 cells (Figure 13). Moreover, not only Ler/Cfz or Ler/Ixa but also any tested DHP/Btz induced an extensive vacuolation and subsequent cell death in MDA-MB 435S cells, but not in MCF-10A cells (Figure 14). These results suggest that the combined treatment with DHP and PI may commonly induce vacuolation-mediated cell death in these cancer cells, while conserving normal breast cells. Next, we tested whether Ler/Btz induces apoptosis. Ler/Btz-induced cell death was not accompanied by the apoptotic morphologies, including cell blebbing, formation of apoptotic bodies (Figure 15A) and pretreatment with the pan-caspase inhibitor, z-VAD-fmk, did not affect Ler/Btz-induced cellular vacuolation and subsequent cell
34
death in MDA-MB 435S cells (Figure 15A and 15B). In addition, while doxorubicin treatment induced the cleavage of p32 procaspase-3 into p20 (intermediate form) and p17 subunit of caspase-3, Ler/Btz treatment did not affect the proteolytic processing of procaspase-3 in MDA-MB 435S cells (Figure 15C). Furrthermore, pretreatment with necroptosis inhibitor, necrostatin-1 (Nec-1), or autophagy inhibitors, 3-methyladenine (3-MA) and bafilomycin A1 (BafA1), did not inhibit Ler/Btz-induced cellular vacuolation (Figure 15A) and cell death (Figure 15B). Moreover, Ler did not notably affect Btz-meditaed upregulation of LC3 and p62, suggesting that the sensitizing effect of Ler on Btz-mediated cell death is not associated with autophagy (Figure 15D). Taken together, these results suggest that apoptosis, necroptosis, or autophagic cell death is not critically involved in the anticancer effects of Ler/Btz.
35
Fig. 11. The morphological changes in MDA-MB 435S and MCF-10A cells treated with Amlo/Btz, Nicar/Btz, Nigul/Btz, or Felo/Btz.
Morphologies of MDA-MB 435S or MCF-10A cells treated with Amlo, Nicar, Nigul, Felo and/or Btz for 24 h were observed by phase-contrast microscopy. Bars, 20 μm.
36
Fig. 12. Combined treatment with Ler and Btz induces cellular vacuolation in breast cancer cells, but not in normal breast cells.
Cells were treated with 10 μM Ler and/or 4 nM Btz for indicated time points and cellular morphologies were observed by phase-contrast microscopy. Bars, 20 μm.
37
Fig. 13. Combined treatment with Ler and Btz induces cellular vacuolation in various cancer cells.
Cellular morphologies were observed by phase-contrast microscopy. Bars, 20 μm. Cells were treated with Ler and/or Btz (for SNU-668 cells, 4 nM Btz and/or 10 μM Ler; for NCI-H460 cells, 15 nM Btz and/or 15 μM Ler; for BxPC-3 and SNU-475 cells, 20 nM Btz and/or 10 μM Ler) for 24 h.
38
Fig. 14. The morphological changes in breast cancer cells or normal breast cells treated with Ler/Cfz or Ler/Ixz.
Cells were treated with 10 μM Ler and/or PIs (20 nM Cfz or 100 nM Ixz) for 24 h and cellular morphologies were observed by phase-contrast microscopy. Bars, 20 μm.
39
Fig. 15. Ler/Btz-induced vacuolization and cell death is not associated with apoptosis, necroptosis, or autophagic cell death.
(A) MDA-MB 435S cells were pretreated with 20 μM z-VAD, 20 μM Nec-1, 0.5 mM 3-MA, or 20 nM Bafilo and further treated with 10 μM and 4 nM Btz for 12 h. Cells were observed by phase-contrast microscopy. Bars, 20 μm. (B) MDA-MB 435S cell were pretreated with the indicated concentrations of z-VAD-fmk (z-VAD), necrostatin-1 (Nec-1), 3-methyladenine (3-MA), or bafilomycin A1 (Baf), and further treated with Ler/Btz for 24 h. Cellular viability was assessed using IncuCyte. Data represent the means ± S.D. (n=7). One-way ANOVA and Bonferroni’s post hoc test. *p <0.001 vs. untreated cells, control. (C and D) Cells were treated with 10 μM Ler and/or 4 nM Btz, or 5 μg/ml doxorubicin (Doxo.) for 24 h. Western blotting of the indicated proteins was performed with β-actin used as a loading control.
40 4. Combination of Ler and Btz induces paraptosis
We investigated whether Ler/Btz-induced vacuolation is derived from the organelles, including the endoplasmic reticulum (ER) and/or mitochondria. To observe these structures, we performed confocal microscopy in YFP-ER cells (MDA-MB 435S sublines stably transfected with the YFP-ER plasmid) after stained Mitotracker-Red (MTR). In untreated cells, we observed filamentous mitochondria and reticular morphology of the ER (Figure 16), and in treated with 4 nM Btz did slightly reduce the mitochondrial length (Figure 17). Although in treated with 10 μM Ler, we did not observe morphological change in the ER, mitochondrial length was shortened at 8 h and mitochondrial dilation was slightly increased at 16 h. But, at 24 h, filamentous morphology of mitochondria was restored (Figure 17). Interestingly, combined treatment with Ler/Btz-treated cells exhibited a slight dilation of mitochondria at 8 h, and gradually increased mitochondrial swelling and the size of enlarged mitochondria at 16 h (Figure 16). Since the fluorescence of MTR was weakened by Ler/Btz treatment at 16 h, we tested whether combined treatment with Ler/Btz induces the loss of mitochondrial membrane potential (MMP). After Ler/Btz treatment for 24 h, we observed the initiation of death-mediated cellular detachment. To further study the relationship between mitochondrial morphology and MMP after Ler/Btz treatment, we performed confocal microscopy in YFP-Mito cells treated with Ler and/or Btz then stained with tetramethylrhodamine methyl ester (TMRM) (Figure 18 and Figure 19). We found that mitochondrial dilation showed a peak at 16 h of Ler/Btz treatment
41
(Figure 19). Interestingly, many expanded mitochondria lost MMPs (green mitochondria, blue arrows), although some of them still retained MMPs (yellow mitochondria, white arrow heads). At 24 h of Ler/Btz treatment, most mitochondria were showed irregularly fragmented morphology and weaker TMRM fluorescence, indicating the MMP loss. In adddtion, dilation of the ER was observed in Ler/Btz-treated cells at 12 h (Figure 16). The size of extended the ER was increased from 16 h and it was maintained 24 h.
Since the vacuolation of the ER and mitochondria is the morphological feature of paraptosis (Sperandio S et al., 2000; Sperandio S et al., 2004; Lee D et al., 2016), and paraptosis is known to require de novo protein syntyesis (Sperandio S et al., 2000; Sperandio S et al., 2004), we examined whether pretreatment with protein synthesis blocker, cycloheximide (CHX), affects the Ler/Btz-induced vacuolation and cell death in these cells. We found that CHX pretreatment significantly blocked the cell death (Figure 20A) and vacuolization (Figure 20B) induced by Ler/Btz in MDA-MB 435S cells. In addition, pretreatment with CHX effectively suppressed the expansion of mitochondria and the ER in YFP-ER and YFP-Mito cells treated with Ler/Btz for 12 h (Figure 20C). Collectively, these results suggest that combined treatment with Ler and Btz eliminates various cancer cells by induction of paraptosis.
42
Figure 16. Combined treatment with Ler/Btz induces the ER and mitochondrial dilation in cancer cells.
YFP-ER cells were treated with 10 μM Ler and 4 nM Btz for indicated time durations and then stained with MTR. Cells were observed by confocal microscopy. Bars, 20 μm.
43
Figure 17. Morphological changes in cells treated with Btz or Ler.
YFP-ER cells treated with 4 nM Btz or 10 μM Ler for the indicated time durations and then stained with MTR. Cells were observed by confocal microscopy. Bars, 20 μm.
44
Figure 18. Morphologies and membrane potential of mitochondria in cells treated with Btz or Ler alone.
YFP-Mito cells treated with 4 nM Btz or 10 μM Ler for the indicated time durations and then stained with TMRM. Cells were observed by confocal microscopy. Bars, 20 μm.
45
Figure 19. Combined treatment with Ler and Btz induces mitochondrial vacuolization and MMP loss in cancer cells.
YFP-Mito cells were treated with 10 μM Ler and 4 nM Btz for the indicated time durations and then stained with TMRM. Cells were observed by confocal microscopy. Bars, 20 μm.
46
Figure 20. Pretreatment with CHX inhibits Ler/Btz-induced cytoplasmic vacuolation and cell death in cancer cells.
(A) MDA-MB 435S cells were pretreated with CHX and then further treated with Ler and/or Btz at the indicated concentrations for 24 h. Cellular viability was assessed using IncuCyte system. Data represent the means ± S.D. (n=7). One-way ANOVA and Bonferroni’s post hoc test. *p <0.001 vs. untreated cells; #p <0.05 vs. Ler/Btz-treated cells. (B) Cellular morphologies were observed by phase-contrast microscopy. Bars, 20 μm. (C) YFP-ER and YFP-Mito cells were pretreated with 2 μM CHX and further treated with 10 μM Ler plus 4 nM Btz for 12 h. Cells were observed by confocal microscopy. Bars, 20 μm.
47
5. Combined treatment with Ler and Btz enhances ubiquitinated protein accumulation and ER stress
It is well known that major mechanism of PI-mediated cell death involves the accumulation of toxic poly-ubiquitinated proteins and misfolded protein aggregates, which induce ER stress (Ding WX et al., 2007). Therefore, we investigated whether the underlying mechanism of Ler/Btz-mediated anticancer effect was also related to ER stress. Western blotting showed that Ler enhances the Btz-mediated accumulation of poly-ubiquitinated protein (Figure 21A). Immunocytochemistry of ubiquitin revealed that while treatment with Btz formed aggresome-like structures in MDA-MB 435S cells, combined treatment with Ler and Btz induced more scattered expression of ubiquitinated protein aggregates for 16 h (Figure 21B). In addition, Ler increased the Btz-induced phosphorylation of PERK and eIF2α, and up-regulation of ATF4 and CHOP protein levels (Figure 21). Interestingly, levels of the ER stress marker proteins was much higher in breast cancer cells treated with Ler/Btz, compared to those in normal breast cells (Figure 22). These results suggest that Ler can sensitize cancer cells to Btz by enhancing Btz-mediated ER stress.
48
Figure 21. Ler enhances Btz-mediated ubiquitinated protein and ER stress.
(A) MDA-MB 435S cells were treated with 10 μM Ler and/or 4 nM Btz for indicated time durations. Western blotting of the indicated proteins was performed with β-actin used as a loading control. (B) MDA-MB 435S cells were treated with 10 μM Ler and/or 4 nM Btz for 16 h. Immunocytochemistry of Ub was performed. Cells were observed by confocal microscopy. Bars, 20 μm.
49
Figure 22. The expression levels of ER stress marker proteins in Ler/Btz-treated breast cancer cells or normal breast cells.
MDA-MB 435S or MCF-10A cells were treated with 10 μM Ler and/or 4 nM Btz for 8 h. Western blotting of the indicated proteins was performed with β-actin used as a loading control.
50
6. Combination of Ler and Btz disrupts Ca2+ homeostasis via mitochondrial Ca2+ overload
We previously reported that Ca2+ homeostasis, particularly mitochondrial Ca2+ overload, plays an important role in the paraptosis induced by curcumin (Yoon MJ
et al., 2012) or celastrol (Yoon MJ et al., 2014). In addition, Ler is known to inhibit
L-type Ca2+ channels (Burnier M et al., 2009; Bang LM et al., 2003), and Therefore, we tested whether Ler and/or Btz altered intracellular Ca2+ levels. Fluorescence microscopy (Figure 23A) and flow cytometry (Figure 23B) using Fluo-3 showed that combination of Ler and Btz progressively and markedly increased cytosolic Ca2+ levels. In contrast, Btz or Ler alone triggered very slight increase of intracellular Ca2+ at 16 h (Figure 23A and 23B). Next, when we assessed the changes in mitochondrial Ca2+ levels using Rhod-2, Ler/Btz markedly increased mitochondrial Ca2+ levels from 4 h with a peak at 12 h (Figure 24). Thus, these results indicate that the increase in mitochondrial Ca2+ preceded the increase in cytoplasmic Ca2+ levels in response to Ler/Btz.
We next investigated the functional importance of increased cytosolic Ca2+ in Ler/Btz-induced paraptosis using BAPTA-AM, a scavenger of intracellular Ca2+. Since Ca2+ is mainly taken up into mitochondria through the mitochondrial Ca2+ uniporter (MCU) (De Stefani D and Rizzuto R, 2014; De Stefani D et al., 2015), we examined the effect of MCU inhibitors, such as Ru360 or ruthenium red (RR), in Ler/Btz-induced paraptosis. We found that pretreatment with Ru360, RR, or
51
BAPTA-AM dose-dependently inhibited Ler/Btz-induced cell death, although the blocking effect of Ru360 or RR was greater than that of BAPTA-AM (Figure 25A). We also investigated the effects of Ru360 or BAPTA-AM on the Ler/Btz-induced vacuolization of mitochondria and the ER. Interestingly, pretreatment with Ru360 effectively blocked the dilation of both mitochondria and the ER by Ler/Btz treatment. In contrast, BAPTA-AM partially inhibited mitochondrial dilation, whereas it potently inhibited the expansion of the ER (Figure 25B). Next, since Ca2+ was shown to be imported across the outer mitochondrial membrane (OMM) through VDAC (Rizzuto R et al., 2009), we investigated whether pretreatment with DIDS, a VDAC inhibitor (Benítez-Rangel E et al., 2015), blocks the Ler/Btz-induced cell death. In contrast to the effect of Ru360, DIDS did not affect this cell death (Figure 26A). Also, we investigated whether increased Ca2+ originates from the ER or extracellular environment following treatment with Ler/Btz. Pretreatment with 2-APB, an IP3 receptor inhibitor (Maruyama T et al., 1997), did
not block the Ler/Btz-induced cell death. Moreover, high concentration of 2-APB (about 80 μM), which is shown to inhibit full name (SOCE) (DeHaven WI et al., 2008), did not block the Ler/Btz-induced cell death. Taken together, these results suggest that initially the increase in mitochondrial Ca2+ may critically contribute to Ler/Btz-induced paraptosis, while later increase in cytosolic Ca2+ may be important for the ER dilation triggered by Ler/Btz.
52
Figure 23. Combined treatment with Ler and Btz markedly increases cytosolic Ca2+ levels.
MDA-MB 435S cells were treated with 10 μM Ler and/or 4 nM Btz for the indicated time points. Treated cells were stained with Fluo-3 and subjected to fluorescence microscopy (A) and flow cytometry (B). Bars, 20 μm.
53
Figure 24. Combined treatment with Ler and Btz significantly increases mitochondrial Ca2+ levels.
MDA-MB 435S cells were treated with 10 μM Ler and/or 4 nM Btz for the indicated time points. Treated cells were stained with Rhod-2 and subjected to confocal microscopy (A) and flow cytometry (B). Bars, 20 μm.
54
Figure 25. Pretreatment with the MCU inhibitors significantly suppresses Ler/Btz-induced cell death and dilation of mitochondria and the ER.
(A) MDA-MB 435S cells were pretreated with the indicated calcium antagonists, and further treated with 10 μM Ler and/or 4 nM Btz for 24 h. Cellular viability was assessed using IncuCyte. Data represent the means ± S.D. (n=7) One-way ANOVA and Bonferroni’s post hoc test. *p <0.001 vs. untreated cells; #p <0.05 vs. Ler/Btz-treated cells. (B) YFP-ER and YFP-Mito cells were pretreated with 10 μM BAPTA-AM or 20 μM Ru360, and further treated with 10 μM Ler plus 4 nM Btz for 12 h. Cells were observed under the confocal microscope. Bars, 20 μm.
55
Figure 26. The effects of pretreatment with DIDS or 2-APB on Ler/Btz-induced cell death in MDA-MB 435S cells.
(A and B) MDA-MB 435S cells were pretreated with the indicated inhibitors and further treated with 10 μM Ler and/or 4 nM Btz for 24 h. Cellular viability was assessed using IncuCyte. Data represent the means ± S.D. (n=7) One-way ANOVA and Bonferroni’s post hoc test. *p <0.001 vs. untreated cells; #p <0.05 vs. Ler/Btz-treated cells.
56
7. Mitochondrial Ca2+ uptake critically contributes to Ler/Btz-induced paraptotic cell death
To further understand the roles of mitochondrial Ca2+ overload and ER stress in Ler/Btz-induced paraptosis, we compared the effects of Ru360 and CHX on the major signals of Ler/Btz-induced paraptosis. We found that pretreatment with Ru360 or CHX significantly suppressed Ler/Btz-induced mitochondrial Ca2+ accumulation (Figure 27A and B) and MMP loss (Figure 27C). In addition, Ru360 or CHX effectively blocked the Ler/Btz-induced increase of intracellular Ca2+ levels (Figure 27D). When we investigated the effect of Ru360 on Ler/Btz-induced ER stress, pretreatment with Ru360 partially inhibited Ler/Btz-induced accumulation of ubiquitinated proteins, ATF, and CHOP by. In contrast, CHX completely blocked it (Figure 28). In contrast, pretreatment with BAPTA-AM did not inhibit Ler/Btz-induced ER stress. These results suggest that mitochondrial Ca2+ overload (which is important for mitochondrial vacuolation) may act downstream of signals associated with ER stress but upstream of increase in the cytoplasmic Ca2+ levels (which is important for ER dilation) in Ler/Btz-induced paraptotic signals. In addition, there may be some cross-talk between ER stress and mitochondrial Ca2+ overload. In summary, our novel results show that the combination of Ler and Btz demonstrates the anticancer effect by inducing paraptosis, where ER stress and disruption of Ca2+ homeostasis (particularly mitochondrial Ca2+ overload) play important roles.
57
Figure 27. Mitochondrial Ca2+ overload is critical for Ler/Btz-induced paraptosis.
(A and B) YFP-Mito or MDA-MB 435S cells were pretreated with 20 μM Ru360 or 2 μM CHX and then treated with 10 μM Ler plus 4 nM Btz for 8 h. Cells were stained with Rhod-2 and subjected to confocal microscopy (A) or flow cytometry (B). Bars, 2 μm. (C and D) YFP-Mito cells or MDA-MB 435S cells were pretreatment with Ru360 or CHX and then treated with Ler/Btz for 24 h. (C) Cells were stained with TMRM and observed under the confocal microscope. Bars, 2 μm. (D) Cells were stained with Fluo-3 and subjected to flow cytometry.
58
Figure 28. Mitochondrial overload and the increase in cytosolic Ca2+ may localize downstream of ER stress in Ler/Btz-induced signals.
MDA-MB 435S cells were pretreated with Ru360, CHX, or BAPTA-AM and then treated with Ler/Btz for 8 h. Western blotting of the indicated proteins was performed, with β-actin used a loading control.
59
IV. DISCUSSION
Although surgical resection, radiotherapy, and chemotherapy have been used to treat tumors, cancer recurrence and acquirement of cancer resistance to further treatments lead to the failure in cancer therapies. Therefore, it is necessary to identify novel therapeutics to overcome to improve the effects of cancer therapy. Drug repositioning, sometimes referred to as ‘drug repurposing or drug recycling’, is a promising approach to overcome barrier to discovering and developing new drugs by identifying new therapeutic applications of known drugs (Turanli B et al., 2018).
Herein, we show that combination of various dihydropyridine calcium channel blockers (DHP-CCB), anti-hypertensive drugs, and bortezomib (Btz) synergistically killed MDA-MB 435S cells. Interestingly, while amlodipine, nicardipine, niguldipine, or felodipine plus bortezomib demonstrated a slight cytotoxicity in normal breast (MCF-10A) cells, combination of lercanidipine (Ler) and Btz did not induce cell death in these cells. Ler belongs to the third-generation of DHP-CCB, it shows greater vascular selectivity than other DHPs, including Amlo, Nigul, Nicar, and Felo (Meredith PA, 1999; Angelico P et al., 1999). Beneficial efficacy and safety of Ler has made it a flexible choice for the treatment of hypertension in a wide range of patients (Angelico P
et al., 1999; Burnier M and Gasser UE, 2007). Here we have found that Ler effectively
enhances the anticancer effects of various PIs, including Btz, Cfz, and Ixz, and conserves normal breast cells in breast cancer cells. The synergistic effects of Ler/Btz were also observed in other types of cancer cells, including gastric, lung, pancreatic, and
60
liver cancer cells. Our observation of cellular morphology dramatically triggered cellular vacuolation and subsequent cell death by combination of Ler and Btz, although the sub-lethal dose of Ler or Btz did not induce notable morphological changes. These results suggest that combination therapy with Ler and PIs can provide a safe and effective treatment strategy to improve the effectiveness of PI-based cancer treatment. The vacuoles found prior to paraptosis were derived from mitochondria and the ER by Ler/Btz-induced cell death in cancer cells. In response to Ler/Btz, mitochondrial swelling was first observed at 8 h and peaked at 16 h, thereafter that mitochondria became contracted. In contrast, dilation of the ER first appeared after 12 h in Ler/Btz treated cells and remained until death-related cell detachment. The vacuolation of mitochondria and the ER, and finally cell death were effectively suppressed by CHX treatment, suggesting that new protein synthesis is required for this cell death. When we investigated that underlying mechanism by which Ler sensizes cancer cells to Btz-mediated cell death, we found that co-treatment with Ler and Btz significantly elevated accumulation of poly-ubiquitinated protein, phosphorylation of PERK and eIF2α, and increases of ATF and CHOP protein levels by Btz treatment. When we compared the expression of ER stress marker proteins in breast cancer cells and normal breast cells, the expression levels of ER stress proteins in normal cells were lower than those in cancer cells. These results suggest that Ler enhances the low-dose Btz-mediated ER stress selectively in cancer cells. We have previously shown that variety natural products, including curcumin (Yoon MJ et al., 2012; Yoon MJ et al., 2014a) and celastrol (Yoon MJ et al., 2014b), effectively kill cancer cells by inducing paraptosis. In
61
that study, we showed that proteasome inhibition is necessary but not sufficient for paraptosis, and perturbation of Ca2+ homeostasis, particularly mitochondrial Ca2+ influx, critically contributes to the efficacious inducing paraptosis. In this study, Ler/Btz induced accumulation of Ca2+ in mitochondria and resultant increase cytoplasmic Ca2+ levels. Inhibition of MCU-mediated mitochondrial Ca2+ influx blocked Ler/Btz-induced mitochondrial swelling, the ER dilation, and subsequent cell death. Interestingly, pretreatment with MCU inhibitors blocked the Ler/Btz-induced MMP loss and increase of cytosolic Ca2+ levels. Scavenging of intracellular Ca2+ levels using BAPTA-AM effectively inhibited Ler/Btz-induced the ER dilation but not mitochondrial dilation. These results indicate that MCU-mediated mitochondrial Ca2+ overload has a critical role in Ler/Btz-induced mitochondria dilation, subsequent ER dilation, and cell death, whereas increased cytoplasmic Ca2+ may play an important role in Ler/Btz-induced ER dilation at the late phase of Ler/Btz treatment.
VDAC (voltage-dependent anion channel) is a Ca2+ transport channel which locates in the outer membrane of mitochondria (OMM) and highly permeable to Ca2+ (Liao Y et
al., 2015). Thus, we tested whether treatment with DIDS, a VDAC inhibitor
(Benítez-Rangel E et al., 2015), affects the cell death induced by combined treatment with Ler and Btz. In contrast to the effect of Ru360, DIDS did not affect this cell death. Ruthenium red (RR) or Ru360 was shown to induce VDAC1 channel closure in a time-dependent manner and stabilize the channel in a low conduction state (Shoshan-Barmatz V et al., 2017). Therefore, this result shows that DIDS may suppress only VDAC, but Ru360 may block both VDAC and MCU, resulting in the greater blocking effect of