Water for Future
이산화탄소(CO 2 ) 연료화를 위한 메커니즘 이해 및 고효율 촉매 개발
Ⅰ. 서 론
전 세계적으로 미래 에너지 공급에 대한 문제 해결과 환경오염 및 공해를 방지할 수 있는 친환 경 에너지에 관한 연구는 최근 전 인류가 당면한 가장 큰 과제중의 하나이다. 이에 대한 해결 대안 으로서 주목 받고 있는 기술들 중의 하나는, 석탄 화력 발전소 같은 오염원에서 배출되는 지구온난 화의 주요 원인 물질인 이산화탄소(CO2)[1-3]를 포집하여 유용한 에너지로 전환 시킬 수 있는 기 술이다. 이는 오염원으로부터 직접 CO2를 유용한 에너지로 전환시킴으로써 별도의 CO2 저장 공간 이 필요치 않을 뿐 아니라 지구온난화 방지에 큰 기여를 할 수 있다는 장점이 있다.
CO2를 감축하기 위해 용매를 이용한 흡수법, 고 체 흡수제를 이용한 흡수·흡착법 등 여러 가지 기술들이 연구되어왔다. 그 중 이산화탄소의 포집 및 저장기술은 화석연료를 사용하면서 온실가스의 농도를 안정화시키는 방법 중 하나이다[4,5]. 또한
CO2를 연료물질로 변환시키는 연료화 기술은 이 산화탄소의 양을 줄이는 동시에 에너지자원으로써 활용이 가능한 생성물질을 얻을 수 있어 일석이조 의 효과를 얻을 수 있는 방법이다. 하지만 열역학 적으로 안정한 화합물인 이산화탄소는 이용 가능 한 탄화수소화합물로 전환시키기 위해서는 추가적 인 에너지가 요구되는 반응이다. 따라서 이산화탄 소 연료화를 효과적으로 사용하기 위한 촉매 개발 연구가 필요하다.
CO2의 전기화학적 환원(electrochemical reduction)이란 외부로부터 직접 전기에너지를 공 급 받아 이산화탄소를 환원하는 방법으로 CH4, COOH, CO 등과 같은 탄화수소 화합물과 중간 생 성물들이 생성되는 반응이다. 전기화학적 방법을 통한 CO2의 환원반응은 수용액상에서 전극을 통 해 직접적으로 이루어지는데, 추가적인 외부에너 지가 필요하며 수소발생반응(hydrogen evolution reaction, HER)과 경쟁 관계에 있다. 따라서 수소 발생반응을 억제하며 원하는 생성물이 발생하는 촉매를 사용하여야하며 이를 위한 촉매 연구가 활 발히 수행중이며[6-8], Hori et al.은 다양한 촉 매를 전극으로 사용하여 전기화학적 반응으로 다 양한 유기물(e.g., H2, CO, HCOOH, CH4 etc.)로 전환시키는 연구를 발표하였으며[9,10] 그 외에도 이산화탄소 전환률을 높이기 위한 연구가 진행 중 이다[11,12]. 하지만 다양한 연구에도 불구하고, 임 동 희 ●●●
충북대학교 환경공학과 교수 [email protected]
Water for Future
이산화탄소의 탄화수소 전환은 상대적으로 높은 오버포텐션(overpotential)을 필요로 한다는 문제 점이 있다[13,14]. 이에 이산화탄소를 탄화수소로 효율적으로 전환하기 위해서는 다양한 촉매 표면 에서 CO2의 전기화학적 환원 메커니즘에 대한 이 해가 필요할 것이다. 이를 바탕으로 전환율을 향 상시킬 수 있는 주요 인자를 찾고 이를 이용해 새 로운 촉매물질을 설계하는 것이 이산화탄소를 유 용한 에너지원으로서 활용하는 일에서 중요한 부 분일 것이다.
본 학술기사에서는 밀도범함수이론(density functional theory, DFT) 계산화학 모델링 툴을 활용한 CO2 환원반응 메커니즘 연구에 관해 저자 가 최근 수행했던 연구를 소개하고자 한다. 먼저, 기존 구리, 금, 백금 촉매 조건에서 CO2의 환원 메커니즘을 DFT 계산을 통하여 비교 분석하였다.
또한, CO2 환원반응을 촉진하기 위한 새로운 물질 개발연구로서 그래핀에 흡착된 구리나노입자 표 면에서의 CO2 에너지 전환 효율 및 메커니즘 분석 결과를 소개한다.
Ⅱ. 이론적 계산 방법
Ⅱ-1. 밀도범함수이론(Density function theory, DFT) 배경
밀도범함수이론(DFT)은 물질, 분자 내부에 전 자가 들어있는 모양과 그 에너지를 양자 역학으 로 계산하기 위한 이론의 하나이다. 이를 통해 어 떤 분자가 세상에 존재할 수 있는지 없는지의 여 부, 특성 분자의 모양과 성질 등등을 예측할 수 있 다. 컴퓨터를 사용하는 과학 계산들 중에서, 가 장 널리 쓰이는 양자 역학 계산 분야 중 하나이다.
Kohn과 Sham[15]은 전자밀도가 어떤 시스템에 서 에너지 등 모든 것을 대표할 수 있는 변수라는 이론을 정립하였다. 전자밀도 ρ(r)를 파동함수의 대표변수로 지정함으로써 파동함수가 3n (n=전 자의 개수) 좌표의 함수이지만 전자밀도로써 단지 3D 좌표 x, y, z의 함수로 결정할 수 있으며, 이에 따라 시스템의 바닥상태 에너지값을 효율적으로 계산할 수 있게 되었다.
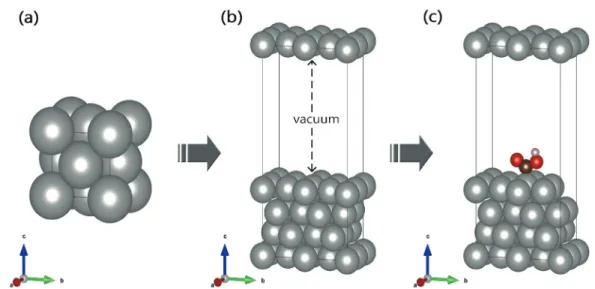
Fig. 1. (a) Structure of bulk for Face centered cubic (FCC). (b) surface of (2×2) slab model. (c) Image of adsorbate adsorbed on the surface.
Water for Future
Ⅱ-2. DFT 계산방법
밀도범함수이론(Density functional theory, DFT) 계산은 Vienna ab initio simulation package (VASP)[16-19]를 이용하여 projector- augmented wave (PAW)[20,21]방법으로 써 원자와 전자의 구조와 에너지를 계산하였 다. 전자 교환-상관성 범함수 (exchange- correlation functional)는 generalized gradient approximation (GGA)를 기반으로 한 Perdew, Burke, Ernzerhof (PBE) 모델 방법으로 계산되 었다[22]. Kinetic energy cutoff는 400 eV로 설 정하였고, Brillouin zone의 적분은 각 촉매의 벌 크구조에서 16×16×16, (2×2) 표면에서 8×8×1 Monkhorst-Pack[23] 방법을 사용하였다. 각 계 산은 (111)-(2×2)표면에서 계산하였으며 전체 4 층 중 아래 2층을 고정하였다(Fig. 1). 각 단계마 다 흡착물질은 Top, Bridge, Hollow site에 흡착 하였다. 또한, 반응이 일어나는 전기화학적 수용
액 상태를 모사하기 위하여 Nørskov et al.에 의 해 고안된 computational hydrogen electrode (CHE) model을 사용하였다[24,25]. CHE model 은 수용액상에서 양성자-전자쌍(H++e-)의 화학적 포텐셜(μ)은 가스 상태 수소(H2)의 화학적 포텐셜 (μ)의 절반과 같다고 가정하였다.
Ⅲ. 결과 및 고찰
Ⅲ-1. 벌크(bulk) 구조 최적화
연구를 수행하기 위한 기초단계로 먼저 구리, 금, 백금의 벌크구조들을 최적화 하였다. 세 금속 모두 면심 입방 구조(face centered cubic, FCC)구 조로 unit cell이 4개의 원자로 구성되어있다(Fig.
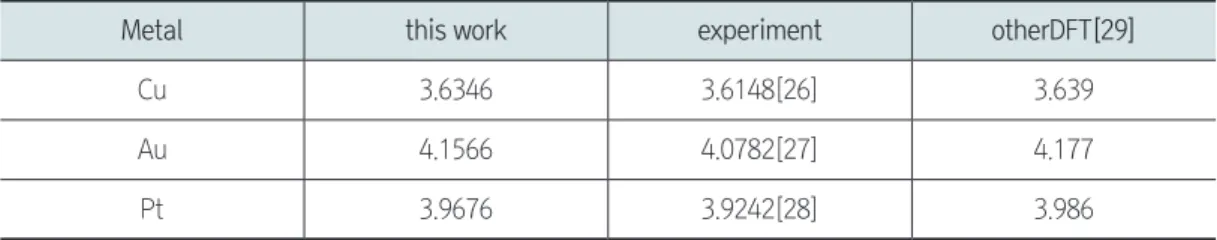
1). Birch-Murnaghan 상태방정식을 이용하여 Cu, Au, Pt 의 격자상수를 결정하였다(Table 1).
Table 1. DFT-calculated lattice constants compared with experimental and other DFT data.
Metal this work experiment otherDFT[29]
Cu 3.6346 3.6148[26] 3.639
Au 4.1566 4.0782[27] 4.177
Pt 3.9676 3.9242[28] 3.986
Ⅲ-2. Cu, Au 촉매의 CO2 환원반응 에너지 계산
이산화탄소가 메탄으로 환원되기 위해서는 총 9 단계를 거치게 된다. 여러 단계의 반응 중 반응제 한단계가 존재하는데, 반응제한단계란 다음 단계 로 넘어갈 때 필요한 에너지 중 가장 큰 값을 말한 다. Peterson et al.은 DFT를 이용하여 Cu(211) 표면에서 일어나는 다양한 반응경로를 조사하여
가장 안정한 경로를 제안했다[22]. 구리 표면에 흡 착된 CO2에 수소가 첨가되면서 COOH* → CO*
→ CHO* → CH2O* → CH3O* → O* → OH* (*
은 표면, *이 붙은 것은 표면에 흡착된 상태임을 나타냄) 로 전환된다. 이 중 반응제한단계는 CO*
→ CHO* 단계라고 발표했다. 본 연구는 이를 참 고하여 전제 경로 중 앞의 4단계에서 반응제한단 계가 나타날 것이라 가정하였다.
Water for Future
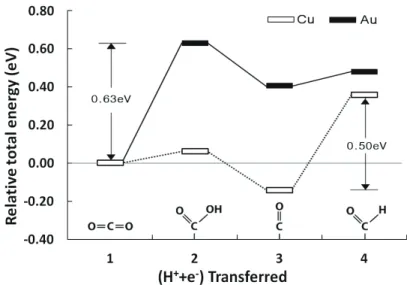
Fig. 2에 나타난 것처럼, 상대적 에너지 값을 비 교했을 때 앞 단계의 상대적 에너지 값보다 그 다 음 단계의 상대적 에너지 값이 크면 반응이 진행 될 때 오버포텐셜 만큼의 에너지가 필요함을 의미 한다. Cu와 Au 표면에서 이산화탄소 환원반응 4 단계 중 CO2 → COOH* 단계와 CO* → CHO*
단계에서 상대적 에너지가 높아졌다. 이 두 단계 는 추가적인 에너지가 주어져야 반응이 진행된다.
이 두 단계 중 상대적 에너지 값 차이가 큰 단계 가 반응제한단계이며 이 값의 차이는 에너지 벽이 다. Cu의 경우 CO* → CHO* 단계, Au의 경우 CO2 → COOH* 단계가 반응제한단계이며 각 에 너지 벽은 0.50 eV, 0.63 eV로 나타났다. 반응제 한단계의 에너지 벽이 높을수록 오버포텐셜이 높 아지므로 반응이 진행되기 어려운데 Cu와 Au의 에너지 벽을 비교했을 때 Au(Ebarrier=0.63eV) 보다 Cu(Ebarrier=0.50eV)의 에너지 벽이 낮다.
이는 금보다는 구리 촉매에서 이산화탄소 환원 반 응이 더 잘 진행될 수 있음을 알 수 있다. 또한 이 결과는 다양한 촉매에서 전기화학적 환원반응을
연구한 Hori et al.의 결과와도 일치한다[10].
Hori et al.은 Au 촉매를 사용했을 때 메탄이 생성되지 않고 대부분 CO가 생성된다고 발표했 다[10]. 단계에서 추가적인 에너지가 필요하며 이 값이 적을수록 반응이 잘 진행되는데 본 연구에 서 Cu와 Au의 CO* → CHO* 단계의 오버포텐 셜을 비교하면 Au가 더 낮음을 확인할 수 있다 (Fig. 2). CO2가 CH4로 환원되기 위해서는 표면 에 흡착된 CO에 수소가 추가되어 CHO 단계로 진 행되어야한다. 반면에 수소가 추가되지 않고 CO 가 표면에서 탈착되어 가스 물질서 중간생성물이 될 수도 있다. CO* → CO(g) 의 상대적 에너지 를 비교했을 때 Au는 발열반응(exothermic, Δ ECO*→CO(g) = -1.79eV)이고 Cu는 흡열반응 (endothermic, ΔECO*→CO(g) = 0.02eV)으로 나타났다. Au 표면에서 CO*는 추가 에너지를 요 구하는 CHO* 단계로 진행되기보다 CO(g)로 탈착 되는 반응이 더 잘 일어나며 메탄이 생성되지 않고 CO(g)가 생성되는 결과와 일치한다.
Fig. 2. Relative total energy diagrams of the electrochemical reduction of CO2 on the Cu and Au surfaces (Reference:
Lee et al., Chungbuk National University (2016)).
Water for Future
Ⅲ-3. 백금 촉매의 CO2 환원반응과 수 소생성반응 에너지 계산
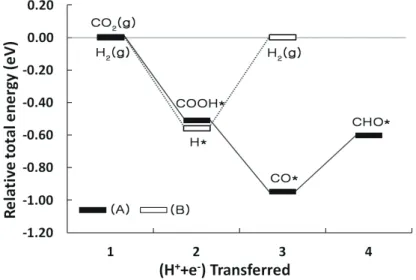
Pt 표면 위에서도 Cu, Au와 같은 계산을 진행하 였다. Pt는 CO* → CHO* 단계가 반응제한단계이 며 에너지 벽은 0.35eV이다. Cu와 Au와 달리 Pt 는 CO2 → COOH 단계의 상대적 에너지 값 차이가 음수로 나타났다. 이를 종합해보면 Cu와 Au, Pt 중 반응제한단계의 에너지 벽이 가장 낮은 Pt가 이 산화탄소를 메탄으로 가장 잘 전환하는 촉매라고 할 수 있다. 하지만 실험 결과와 비교했을 때 Pt는 메탄보다 수소를 잘 생성한다는 결과와 일치하지 않는다. 이는 전기화학적 환원반응에서 메탄 생성 반응과 경쟁관계에 있는 수소 생성 반응(hydrogen evolution reaction, HER)이 관여했기 때문이라고
사료되어 추가 계산을 진행하였다.
수소생성반응은 촉매 표면에 수용액 상의 수 소 원자가 흡착되었다가 수소 가스로 전환되는 반 응이다. 수소생성반응의 상대적 에너지를 구하여 Table 2.에 나타내었다. 또한 이산화탄소 환원반 응과 수소생성반응의 상대적 에너지 차이를 비교 했다(Fig. 3). 전체 반응이 진행되기 위해서 첫 단 계가 진행이 되어야 다음 단계가 진행되므로 각 반응의 첫 단계의 에너지 차이를 비교했다. CO2
환원 반응은 -0.51eV, 수소생성반응은 -0.56eV 값을 나타냈다. 상대적 에너지 값의 차이가 클수 록 반응이 일어나기 어려운 반면 그 값이 낮을수 록 반응이 쉽게 일어난다. Pt 표면에서는 상대적 으로 더 낮은 값을 나타내는 수소생성반응이 우세 하며 경쟁관계인 CO2 환원 반응은 제한된다.
Fig. 3. Relative total energy diagrams of (A) the reduction of CO2 and (B) the hydrogen evolution reaction on Pt (Reference: Lee et al., Chungbuk National University (2016)).
Ⅲ-4. 그 래 핀 - 구 리 나 노 입 자 활 용 한 CO2 환원반응 연구
CO2 환원반응에 대한 DFT 모델링을 위해 최적
구조의 구리나노입자에 대한 구조적, 물리적 특 성 분석이 필요하게 된다. 가능성 있는 구리나노 입자 모형으로서 icosahedron, cuboctahedron, 그리고 D4h symmetry 구조등이 있을 수 있으며,
Water for Future
icosahedron의 구조일 경우 원자의 개수는 55개 로써 구성될 수 있다. 그리고 그래핀 구조로서 그 래핀에 탄소 원자 공백(vacancy)을 형성하여 구 리나노입자가 더욱 효과적으로 그래핀 표면에 흡 착될 수 있으며, 또한 촉매 활성도의 향상을 기대
할 수 있다. Fig. 4는 구리나노입자(icosahedral Cu55)가 한 개의 탄소 공백을 가진 그래핀에 흡착 되어 있는 최적화된 시스템(구리나노입자+그래핀 시스템) 분자 모형을 나타내고 있다.
Fig. 4. (A) Side view of a Cu55 nanoparticle supported on defective graphene. (B) Top view of a 5-8-5 vacancy site of defective graphene with two missing C atoms. (C) Icosahedral Cu55 nanoparticle. Brown and yellow colors represent Cu and C, respectively. (Reference: Lim et al. Nanoscale, 6, 5087 (2014)).
Fig. 5(A)는 CO2환원반응 중간 생성물들의 그 래핀-구리나노입자 표면에서의 흡착 특성 분석 을 수행하고, 이로써 전체적인 CO2 환원 반응 메 커니즘을 보여준다. 그래핀-구리나노입자의 특 성을 기존의 벌크구리(Cu(111))와 비교하기 위해 동일한 CO2 환원반응경로에 대해 두 물질에 대해 비교 분석하였다. CO2 환원반응의 중간생성물은 CO2(gas) → COOH* → CO* → CHO* → CH2O*
→ CH3O* → O* → OH* → H2O(gas)로 나타난 다(* 표시는 중간생성물이 구리나노입자 표면에 흡착되었다는 의미). Fig. 5(B)는 CO2 환원반응 각 단계 중 전체반응을 제한하는 단계가 CO* →
CHO* 단계(Step 2→3)임을 보여준다. 그래핀과 구리나노입자에서의 반응제한단계의 에너지 벽은 0.68 eV임에 반해, 기존의 벌크구리(Cu(111))의 에너지 벽은 0.97 eV임을 보여준다. 이로써, 그 래핀-구리나노입자를 활용함으로써 CO2가 메탄 으로 전환되는 환원반응이 더 효율적으로 잘 일어 날 수 있음을 알 수 있다. Fig. 5(C)는 전기화학적 환경에서 이산화탄소 환원반응이 일어 날 수 있는 포텐셜을 나타내는데, 이는 CO* → CHO* 단계 의 에너지 벽을 제거하는데 필요한 포텐셜로서 나 타낼 수 있다.
Water for Future
Ⅳ. 결론
본 학술기사에서는 밀도범함수이론(density functional theory, DFT) 계산화학 모델링 툴을 활용한 CO2 환원반응 메커니즘 연구에 관해 저자 가 최근 수행했던 연구를 소개하였다.
요약하면, 각 단일금속 촉매(Cu, Au, Pt)의 표 면과 그래핀과 구리나노입자 표면에서 CO2의 환 원 메커니즘을 분석함으로써, 각 촉매물질의 CO2
환원 특성을 비교하였다. 각 촉매 단일 금속에서 의 CO2 환원반응 생성물 생성의 상대적인 비교에 있어서는 기존 실험데이터 경향성을 잘 재현할 수 있었다. 또한, 그래핀과 나노입자 시스템을 활용 함으로써 CO2 환원반응에서 CH4 생산이 더 효율 적으로 될 수 있음을 보였다. 이로써, 향후 CO2환 원반응에서의 효율적인 연료물질(CO, HCOOH, CH4 등)생성을 위한 새로운 촉매 개발 연구에 기 초자료로서 활용할 수 있을 것으로 기대한다.
추가적으로, 계산화학 모델링 툴을 활용함으로 써 아직 실험을 통해 연구되지 않은 다양한 물질 들의 반응성을 비교적 빠른 시간 내에 비교 조사 할 수 있고, 이를 통해 에너지 생산 및 저장 효율 성을 높일 수 있는 새로운 물질들을 연구 개발하 는데 큰 역할을 할 수 있을 것으로 기대한다.
사사
본 학술기사는 본 저자의 두 편의 게재된 논문 을 재구성하여 작성되었음: 1) Dong-Hee Lim et al., Nanoscale, 6, 5087 (2014), 2) Chang- Mi Lee et al., Research Institute of Industrial Science & Technology, Chungbuk National University (2016).
Fig. 5. (a) The lowest energy pathways of CO2 reduction on the Cu55-graphene. (B and C) Relative free energy diagrams without (B) and with (C) applied potential. (Reference: Lim et al. Nanoscale, 6, 5087 (2014)).
Water for Future
참고문헌
1. IPCC (Intergovernmental Panel on Climate Change), 2013, 5th Assessment Report
2. IPCC (Intergovernmental Panel on Climate Change), 2007, 4th Synthesis Report
3. Bernstein, L.; Bosch, P.; Canziani, O.; Chen, Z.; Christ, R.; Davidson, O.;
Hare, W.; Huq, S.; Karoly, D.; Kattsov, V., Climate change 2007: synthesis report. Intergovernmental Panel on Climate Change 2007, 20, 2011.
4. Figueroa, J. D.; Fout, T.; Plasynski, S.; McIlvried, H.; Srivastava, R. D., Advances in CO2 capture technology-The US Department of Energy's carbon sequestration program. International Journal of Greenhouse Gas Control 2008, 2, (1), 9-20.
5. Pevida, C.; Plaza, M.; Arias, B.; Fermoso, J.; Rubiera, F.; Pis, J., Surface modification of activated carbons for CO2 capture. Applied Surface Science 2008, 254, (22), 7165-7172.
6. Azuma, M., Hashimoto, K., Hiramoto, M., Electrochemical Reduction of Carbon Dioxide on Various Metal Electrodes in Low-Temperature Aqueous KHCO3 Media, Journal of The Electrochemical Society , vol. 137, 1990, pp.1772-1778
7. Lee, Eun Y.; Hong, D.; Park, Han W.; Suh, Myunghyun P., Synthesis, properties, and reactions of trinuclear macrocyclic nickel(II) and nickel(I) complexes: Electrocatalytic reduction of CO2 by nickel(II) complex. European Journal of Inorganic Chemistry 2003, 2003, (17), 3242-3249.
8. De Jesus-Cardona, H.; del Moral, C.; Cabrera, C. R., Voltammetric study of CO2 reduction at Cu electrodes under different KHCO3 concentrations, temperatures and CO2 pressures. Journal of Electroanalytical Chemistry 2001, 513, (1), 45-51.
9. Yoshio Hori, Hidetoshi Wakebe, Toshio Tsukamoto and Osamu Koga,
“Electrocatalytic process of CO selectivity in electrochemical reduction of CO2
at metal electrodes in aqueous media,” Electrochimica Acta, vol. 39, 1994, pp.1833-1839.
10. Takahashi, I.; Koga, O.; Hoshi, N.; Hori, Y., Electrochemical reduction of CO2 at copper single crystal Cu (S)-[n(111)×(111)] and Cu (S)-[n(110)×(100)]
electrodes. Journal of Electroanalytical Chemistry 2002, 533, (1), 135-143.
11. Shin, D. Y.; Jo, J. H.; Lee, J.-Y.; Lim, D.-H., Understanding mechanisms
Water for Future
of carbon dioxide conversion into methane for designing enhanced catalysts from first- principles. Computational and Theoretical Chemistry, vol. 1083, 2016, pp.31-37.
12. Hirunsit, P., Electroreduction of Carbon Dioxide to Methane on Copper, Copper-Silver, and Copper-Gold Catalysts: A DFT Study, THE JOURNAL OF PHYSICAL CHEMISTRY C , vol.
117, 2013, pp.8262-8268
13. Yoshio Hori, Akira Murata and Ryutaro Takahashi, “Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution,”
Journal of the Chemical Society, Faraday Transactions 1, vol. 85(8), 1989, pp.2309-2326.
14. Yin-Jia Zhang, Vijay Sethuraman, Ronald Michalsky, and Andrew A. Peterson, Competition between CO2 Reduction and H2 Evolution on Transition-Metal Electrocatalysts, ACS Catalysis, vol. 4(10), 2014, pp.3742-3748.
15. Kohn, W. and Sham, L. J., Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev., vol. 140 (4A), 1965, pp.A1133-A1138.
16. Kresse, G. and Hafner, J., Ab initio molecular dynamics for liquid metals, Physical Review B, vol. 47(1), 1993, pp.558-561.
17. Kresse, G. and Hafner, J., Ab initio molecular-dynamics simulation of the liquid-metal amorphous-semiconductor transition in germanium, Phys. Rev. B, vol. 49(20), 1994, pp.14251-14269.
18. Kresse, G. and Furthmuller, J., Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B, vol. 54(16), 1996 pp.11169-11186.
19. Kresse, G. and Furthmuller, J., Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., vol. 6(1), 1996, pp.15-50.
20. Bloochl, P. E., Projector augmented-wave method, Physical Review B, vol. 50(24), 1994 pp.17953-17979.
21. Kresse, G. and Joubert, D., From ultrasoft pseudopotentials to the projector augmented- wave method, Phys. Rev. B, vol. 59 (3), 1999 pp.1758-1775.
22. Perdew, J. P., Burke, K. and Ernzerhof, M., Generalized Gradient Approximation Made Simple, Physical Review Letters, vol. 77, 1996, pp.3865-3868.
23. Monkhorst, H. J. and Pack, J. D., Special points for Brillouin-zone integrations, Physical Review B, vol. 13, 1976, pp.5188-5192.
24. Nørskov, J. K., Kitchin, J. R., Bligaard, T., Jonsson, H., 2004, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, THE JOURNAL OF PHYSICAL CHEMISTRY B ,vol. 108, pp.17886-17892
Water for Future
25. Peterson, A. A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J. K., How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy &
Environmental Science 2010, 3, (9), 1311-1315.
26. M. E. Straumanis and L. S. Yu, Lattice parameters, densities, expansion coefficients and perfection of structure of Cu and of Cu-In α phase Acta Crystallographica, vol. A25. 1969, pp.676-682.
27. A. Maeland and T. B. Flanagan, Lattice spacings of gold-palladium alloys, Canadian Journal of Physics, vol. 42(11), 1964, pp.2364-2366
28. Y. Waseda, K. Hirata and M. Ohtani, High-temperature thermal expansion of platinum, tantalum, molybdenum, and tungsten measured by x-ray diffraction, High Temperatures- High Pressures, vol. 7(2), 1975 pp.221-226.
29. Annapaola Migani, Carmen Sousa and Francesc Illas, Chemisorption of atomic chlorine on metal surfaces and the interpretation of the induced work function changes, Surface Science, vol. 574, 2005, pp.297-305.