https://doi.org/10.14734/PN.2021.32.2.85 pISSN 2508-4887•eISSN 2508-4895
Donghoon Joo, MD, Mun Hui Jeong, MD, Seong Hee Jeong, MD, Narae Lee, MD, Mi Hye Bae, MD, Young Mi Han, MD, PhD, Kyung Hee Park, MD, PhD, Shin Yun Byun, MD, PhD Department of Pediatrics, Pusan National University School of Medicine, Yangsan, Korea
Microcephaly can occur with autosomal dominant or autosomal recessive inherited pattern in a family, but more commonly it is a form of many syndromes that lead to intellectual impairment.
Several reports of ocular anomalies accompanied with microcephaly vary from microphthalmia, nystagmus, cataracts, falciform retinal folds, retinal dysplasia, optic disc coloboma and optic atrophy to chorioretinal degeneration have been documented. In this report, the patient hospitalized for intrauterine growth restriction and microcephaly was diagnosed with congenital retinal detachment on fundus examination incidentally and whose result of chromosomal microarray analysis revealed about 1.4 Mb of duplication at Yq11.23 that is irrelevant with clinical symptom. The result of whole exome sequencing was not significant, either. So by enrolling the patient’s genetic information obtained through whole genome sequencing and clinical feature to the national bio big data, we plan to research on genetic disease in comparison with genetic information of similar cases continuously. The authors experienced a case of congenital retinal detachment accompanied by microcephaly without family history and regardless of congenital viral infection, this has not been reported until now.
Key Words: Microcephaly, Detachment of retina, Congenital, Genetics
서론
소두증은 상염색체우성 또는 상염색체열성유전 양식을 따르는 가족성으로 발생할 수 있 으나 조금 더 흔하게는 지능 장애를 일으키는 많은 증후군들의 한 형태로 잘 나타난다.1 전형 적인 유전 양식을 따르는 소두증에 동반된 소안구증, 안진, 백내장, 낫모양망막주름, 망막이 형성증, 시신경유두결손, 시신경위축, 맥락망막변성 등과 같은 다양한 안구 이상이 보고되 어 있다.2 그러나 이들은 각각 분리된 임상적 범주로서 알려져 왔고, 안구 이상의 산발적인 임상적 특성과 명확한 유전 양식의 부재로 인해 유전적 이질성을 가지는 것으로 간주되었 다.3-6 본 저자들은 가족력이 전혀 없고 선천 바이러스 감염과도 상관없이 산발적으로 발생 한 소두증에 동반된 선천 망막박리를 경험하였고, 현재까지 이러한 증례가 보고된 적이 없 기에, 보고하는 바이다.
증례
환아는 산전 초음파에서 확인된 태아성장지연과 소뇌증 평가를 위해 유도 분만으로 본원 에서 재태기간 38주 1일, 출생체중 2,350 g (3-10 백분위수), 신장 43 cm (<3 백분위수), 두 위는 28.5 cm (<3 백분위수)로 출생하였다. 출생 시 Apgar 점수는 1분 6점, 5분 8점이었다.
산모는 임신 중 갑상선 기능저하증으로 Levothyroxine을 복용하며 갑상선 기능이 정상으 로 유지된 것 외에 특이 병력은 없었으며, 감염이나 방사선에 노출된 적은 없었다. 환아는 34 Received: 19 September 2020
Revised: 1 December 2020 Accepted: 24 December 2020 Correspondence to Shin Yun Byun, MD, PhD Department of Pediatrics, Pusan National University Children’s Hospital, 20 Geumo-ro, Mulgeum-eup, Yangsan 50612, Korea
Tel: +82-55-360-2180 Fax: +82-55-360-2181 E-mail: [email protected] Copyright© 2021 by The Korean Society of Perinatology
This is an Open Access article distributed under the terms of the Creative Com- mons Attribution Non-Commercial License (http://creativecommons.org/
license/by-nc/4.0/), which permits unrestricted non-commercial use, distribution, and reproduction in any
A Case of Microcephaly with Congenital
Retinal Detachment
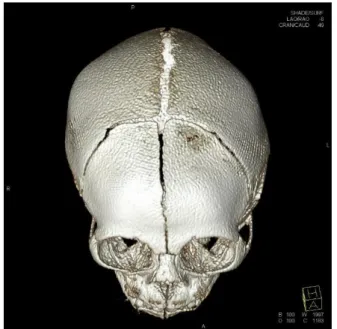
출생시 활력징후는 맥박 150회/분, 호흡 48회/분, 체온 37.2°C, 혈압은 77/42 mmHg였다. 두경부 진찰에서 소두증, 소하악 증, 소안구증 소견을 보였고, 대천문이 만져지지 않았으나 그 외 콧등 높이, 귀 모양과 위치 등에는 특이 소견이 없었다(Fig.
1). 청진상 폐음과 심음에 이상 없었으며 복부 팽만은 없고 장 음은 정상이었으며 간, 비장은 만져지지 않았다. 신경학적 검 사에서 모로반사 및 파악 반사는 정상이었다. 입원 당시 시행 한 혈액 검사상 백혈구 14,890/mm3, 혈색소 17.8 g/dL, 혈소판 268,000/mm3였고, C-반응단백질 0.01 mg/dL로 혈액 검사에 서 감염을 시사하는 소견은 없었다. 혈청 생화학 검사, 선천성 대사 이상 검사, 갑상선 기능 검사에서 특이 소견은 없었고 선 천성 감염과 관련된 Toxoplasma immunoglobulin M, Rubella virus immunoglobulin M, Cytomegalovirus polymerase chain reaction (PCR), Herpes simplex virus type 1, 2 PCR 검사 결 과는 모두 음성이었다.
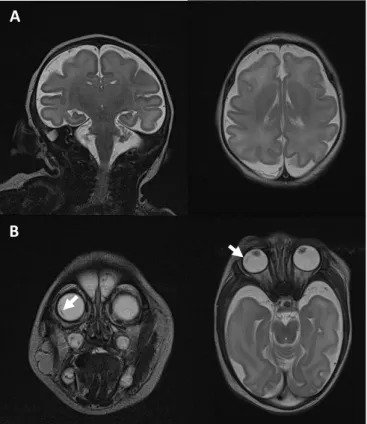
흉부 X-선에서 이상 소견은 보이지 않았고, 대천문이 만져지 지 않아 두개골 조기 유합증을 감별하기 위해 시행한 3차원 안 면골 전산화단층촬영에서 양측 이마뼈, 마루뼈가 겹쳐지고 관 상 봉합이 상대적으로 좁아진 조기 부분 두개골유합증 및 사두 증 소견이 확인되었다(Fig. 2). 출생 9일째 시행한 뇌자기공명 촬영에서는 뇌실질의 전반적인 부피 감소 및 뇌척수액 공간이 현저하게 넓어진 소견이 관찰되어 소두증에 합당한 소견이었 고, 뇌고랑 및 뇌이랑이 정상보다 덜 발달한 뇌이랑없음증 소 견이 보였다(Fig. 3A). 또한 우안에 현저하게 접힌 막, 좌안에
접힌 것으로 의심되는 막이 각각 관찰되어 망막박리 소견이 의 심되었다. 동반 기형 여부를 확인하기 위해 시행한 경흉부 심 장 초음파에서는 난원공 개존증 외에는 특이 소견은 없었고, 신 장 도플러 초음파에서도 특이 소견은 없었다. 출생 이후 아기 가 잘 울지 않고 대부분 자고 있는 것처럼 눈 뜨는 것이 관찰 되지 않아 안과적 평가를 의뢰하였고, 예상했던 소안구증 외에 각막과 수정체혼탁 등 전안부에서의 이상은 관찰되지 않았으 나 안저 검사에서 뜻밖에 양안의 일차 유리체증식증으로 인한 황반끌림 및 시신경 기울어짐 소견과 견인 망막박리가 확인되 어 뇌자기공명촬영과 일치하는 소견을 보여주었다. 안저 검사 소견에서 Norrie병이 의심되어 염색체와 마이크로어레이 분 석을 시행하였고 염색체는 정상 남아로 확인되었으나 마이크 로어레이 분석에서 Yq11.23 부위에 약 1.4 Mb의 중복이 관찰 되었다(Fig. 4). 그러나 Yq11.23은 정자발생과 관련된 유전자 좌로서 해당 부위의 복제수 변이는 환아의 임상 증상과는 명확 한 연관성이 보고된 바가 없다. 이에 따라 추가로 whole exome sequencing을 시행하였으나 특이 소견이 관찰되지 않아 유전 적 연관성을 발견하지 못하였다. 생후 6일째 시행한 자동화 이 음향 방사 검사에서 2회 연속 비정상으로 나와 타각적 청력역 치 측정 검사를 시행하였고, 우측 40 dB, 좌측 60 dB로 좌측 감 각신경성 난청 소견이 확인되었다. 입원 기간 동안 호흡곤란이 나 청색증 등 다른 주산기 문제는 없었고, 경구 수유 또한 안정 적으로 진행되었다. 퇴원 이후 선천 망막박리에 대해 안과 외래 에서 추적 관찰하고 있으며 우안은 황반끌림이 지속적으로 보
Fig. 2. Computed tomography 3-dimension facial bone revealed relatively narrowing of both coronal sutures with overlapping of both frontal and parietal bones.
Fig. 1. Lateral view of our patient shows the microcephaly.
이나 견인 망막박리는 다소 호전되었고, 좌안은 유리체 끌림이 끊어지며 견인 망막박리가 소실되었다. 퇴원 이후 눈맞춤이 잘 되지 않고 양안의 수평 고속 안진이 동반되어 평가 진행하였고, 주시반응이 거의 형성되어 있지 않은 상태로 지속적인 추적 관 찰 중이다. 5개월에 시행한 대동작 운동기능 평가(Gross motor function measurement)에서 눕기와 뒤집기 영역에서 49.02%
(25/51), 앉기 영역에서 16.67% (10/60) 확인되어 Gross Motor Function Classification System level 4에 해당하는 발달지연 소견으로 소아재활의학클리닉 외래를 통하여 재활치료 유지 중 이고, 11개월에 시행한 베일리 영유아 발달검사(Bayley scales of infant development)에서도 인지영역 6개월, 수용성 언어영 역 2개월 10일, 표현성 언어영역 3개월 20일, 소근육 운동영역 8개월, 대근육 운동영역 9개월로 전반적인 발달지연이 확인되 었다. 그러나 근긴장도와 근력 자체에는 특이 소견이 없고, 생 후 4개월에 다시 시행한 타각적 청력역치 측정 검사에서도 우 측 30 dB, 좌측 40 dB로 정상 범위로 호전되었다. 현재 환아 는 14개월로 신체계측에서 체중 7 kg, 신장 68 cm, 두위 35 cm 로 모두 3백분위수 미만이다.
고찰
소뇌증과 선천 망막박리가 동반될 수 있는 질환에는 노리병 (Norrie disease)과 신경원이행이상(neuronal migration disor- der)을 동반하는 질환군인 후쿠야마형 선천성 근이영양증
A
B
Fig. 3. Magnetic resonance brain of a microcephaly with congenital retinal detachment. (A) Both coronal and axial T2 weighted image show decreased volume of brain parenchyme and prominent cere- brospinal fluid space (microcephaly), also show decreased number of sulci and gyri without cortical thickening (mild lissencephaly). (B) Coronal and axial T2 weighted image of orbit. White arrows show folded membrane at periphery of the right orbit (retinal detachment).
Fig. 4. Chromosomal microarray revealed approximately 1.4 Mb of duplication at Yq11.23.
성 및 뇌량이 관찰되지 않는 등의 신경학적 증상을 보이는 상염 색체 열성질환이다.15 후쿠야마형 선천성 근이영양증과 동일하 게 알파-디스트로글라이칸의 당화(glycosylation)에 관여하는 단백질 생산과 관련된 유전자인 POMT1, POMT2, POMGNT1, LARGE, FKTN, FKRP 유전자의 변이로 인해 발생하며 전체 환 자의 50% 미만에서 이들 유전자가 발견된다.16,17 이 질환 또한 환아와는 임상 증상이 맞지 않았다.
근육-눈-뇌 질환(Muscle-Eye-Brain disease)은 근력 저 하, 발달지연, 지능장애와 함께 진행성 고도근시, 시신경위 축, 망막변성 등의 눈 이상을 동반하는 상염색체 열성질환이 다.15 1p32-p34에 위치하는 22개의 엑손(exon)으로 이루어진 POMGNT1 유전자의 점돌연변이로 발생하는 것으로 알려져 있
다.18,19 뇌자기공명촬영 소견은 전반적 피질 비후와 큰뇌이랑증
(pachygyria)을 보이는 것이 특징적이고, 시신경위축, 다리뇌 (pons)와 소뇌벌레(cerebellar vermis)의 형성 부전 등도 동반 될 수 있다.7 진단은 특징적인 magnetic resonance imaging 소 견과 눈 이상, 혈중 크레아틴 키나아제(creatine kinase) 상승, 근 생검 소견 및 POMGNT1의 변이를 통해 할 수 있다.15 환아는 전 반적인 발달지연이 동반되었으나 근력 저하는 보이지 않는 등 근육-눈-뇌 질환과 임상 증상이 맞지 않았다. 본 환아는 태아성 장지연과 소두증으로 입원하여 평가를 시행하였고, 부모를 닮지 않은 외형과 눈을 잘 뜨지 않아 시행한 안저 검사에서 우연히 선 천 망막박리가 진단된 경우로 선천 감염 및 유전력도 없어 원인 을 확인할 수 없었다.
현재 환아는 유전자 이상 및 유전자 관련 배경이 강력히 의심 되어 전장 유전체 분석(whole genome sequencing)을 통해 국 가 바이오 빅데이터에 등록된 상태로 추후 유사한 임상 양상을 보이는 증례와의 유전 정보 비교 분석을 통해 유전질환에 대한 연구를 이어갈 예정이다. 저자들은 재태기간 38주 1일, 출생 체 중 2,350 g의 남아에서 원인을 모르는 소두증과 동반된 선천 망 막박리 1례를 경험하였기에 문헌고찰과 함께 보고하는 바이다.
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Ethics statement
The patient consented to the use of these photographs.
(Fukuyama congenital muscular dystrophy), 워커-워버그 증 후군(Walker-Warburg syndrome), 근육-눈-뇌 질환(Muscle- Eye-Brain disease) 등이 알려져 있다.7
노리병(Norrie disease)은 불완전한 망막의 혈관화로 인한 망 막박리, 망막주름, 일차 유리체증식증, 유리체출혈, 홍채위축, 각막혼탁 등의 안구증상과 진행성 감각신경성 난청 및 정신지 체, 발달지연, 간질 등의 신경학적 증상을 동반하는 성염색체 열성유전질환이다.8 X염색체의 단완(Xp11.4-p11.3)에 위치하 여 노린(norrin) 단백질을 코딩하는 NPD 유전자의 변이에 의해 발생하며, 노린 단백질은 안구의 분화와 발달, 망막, 내이(inner ear)의 혈관 발달에 중요한 역할을 한다.9 소수의 비전형적 노리 병 환자에게 안구 이상과 더불어 소뇌증, 성장 지연, 정신지체, 간질 등의 신경학적 증상이 동반되기도 한다.10 진단은 대개 전 형적인 임상 소견과 가족력을 통해 할 수 있으며, NPD 유전자의 변이가 확인된 경우 확진할 수 있다.11 환아는 이 질환이 강력히 의심되어 마이크로어레이 검사를 시행하였으나 노리병과 관련 된 유전자 변이는 발견되지 않았으며, 환아는 감각신경성 난청 도 오히려 호전을 보여 진행성 감각신경성 난청과는 임상 경과 가 일치하지 않았다.
후쿠야마형 선천성 근이영양증(Fukuyama congenital mus- cular dystrophy)은 골격근의 퇴행성 변화로 인한 근긴장저하, 신경원이행이상으로 인한 뇌겉질 형성이상(cerebral cortical dysplasia), 정신지체, 발달지연, 뇌전증 등의 신경학적 증상 및 망막박리를 포함하는 망막 이상 등의 특징적인 관련 증상을 보 이는 상염색체 열성질환이다.12 염색체 9q31-q33에 위치하는 FKTN 유전자의 변이에 의해 fukutin과 frutin related protein 생성이 결핍되며 이로 인해 세포외바탕질(extracellular matrix) 형성에 필수적인 알파-디스트로글라이칸(α-dystroglycan)의 불완전한 생성이 일어나 근육, 중추신경에서 비정상적인 세포 외 바탕질의 분해가 발생하여 관련된 증상이 나타난다.13 진단 은 임상양상과 혈중 크레아틴 키나아제(creatine kinase) 상승, 뇌자기공명촬영에서의 조약돌뇌회결손, 소뇌의 다미세이랑증 (polymicrogyria) 등의 특징적 소견과 근육 생검, 근전도 검사 및 FKTN 유전자의 변이 확인을 통해 할 수 있다.14 환아는 출생 당시 시행한 신경학적 검사에서 특이 소견이 없었고, 성장하면 서 근긴장 저하가 진행되는 퇴행성 변화가 관찰되지 않는 등 임 상 증상과 유전자 변이 소견이 후쿠야마형 선천성 근이영양증 과는 맞지 않았다.
워커-워버그 증후군(Walker-Warburg syndrome)은 구순열 등의 얼굴기형을 비롯하여 눈 이상으로 소안구증, 전방유착, 백 내장, 망막기형, 시신경형성부전 등이 있으며 뇌자기공명촬영 에서 심한 뇌이랑없음증, 교뇌와 소뇌의 기형과 심한 백질의 변
Acknowledgemants
This study was supported by clinical research grant in 2019 from Pusan National University Yangsan Hospital.
References
1) Mitchell AL. Congenital anomalies. In: Martin RJ, Fanaroff AA, Walsh MC, editors. Fanaroff and Martin's neonatal-perinatal medicine. 11th ed.
New York: Elsevier; 2020. p.497.
2) Atchaneeyasakul LO, Linck L, Weleber RG. Microcephaly with chorio- retinal degeneration. Ophthalmic Genet 1998;19:39-48.
3) Mukhopadhyay A, Kramer JM, Merkx G, Lugtenberg D, Smeets DF, Oortveld MA, et al. CDK19 is disrupted in a female patient with bilateral congenital retinal folds, microcephaly and mild mental retardation.
Hum Genet 2010;128:281-91.
4) Warburg M. Heterogeneity of congenital retinal non-attachment, falciform folds and retinal dysplasia. A guide to genetic counselling.
Hum Hered 1976;26:137-48.
5) Tenconi R, Clementi M, Moschini GB, Casara G, Baccichetti C. Chorio- retinal dysplasia, microcephaly and mental retardation. An autosomal dominant syndrome. Clin Genet 1981;20:347-51.
6) Warburg M, Heuer HE. Chorioretinal dysplasia-microcephaly-mental retardation syndrome. Am J Med Genet 1994;52:117.
7) Valanne L, Pihko H, Katevuo K, Karttunen P, Somer H, Santavuori P. MRI of the brain in muscle-eye-brain (MEB) disease. Neuroradiology 1994;
36:473-6.
8) Drenser KA, Fecko A, Dailey W, Trese MT. A characteristic phenotypic retinal appearance in Norrie disease. Retina 2007;27:243-6.
9) Black G, Redmond RM. The molecular biology of Norrie's disease. Eye (Lond) 1994;8(Pt 5):491-6.
10) Rodriguez-Revenga L, Madrigal I, Alkhalidi LS, Armengol L, González E, Badenas C, et al. Contiguous deletion of the NDP, MAOA, MAOB, and EFHC2 genes in a patient with Norrie disease, severe psychomotor retardation and myoclonic epilepsy. Am J Med Genet A 2007;143A:916- 20.
11) Wu LH, Chen LH, Xie H, Xie YJ. Prenatal diagnosis of a case of norrie disease with late development of bilateral ocular malformation. Fetal Pediatr Pathol 2017;36:240-5.
12) Yoshioka M, Kuroki S, Kondo T. Ocular manifestations in fukuyama type congenital muscular dystrophy. Brain Dev 1990;12:423-6.
13) Martin PT. Mechanisms of disease: congenital muscular dystrophies- glycosylation takes center stage. Nat Clin Pract Neurol 2006;2:222-30.
14) Aida N. Fukuyama congenital muscular dystrophy: a neuroradiologic review. J Magn Reson Imaging 1998;8:317-26.
15) Fahnehjelm KT, Ygge J, Engman ML, Mosskin M, Santavuori P, Malm G. A child with muscle-eye-brain disease. Ophthalmological and neurologi- cal characteristics. Acta Ophthalmol Scand 2001;79:72-5.
16) Beltrán-Valero de Bernabé D, Currier S, Steinbrecher A, Celli J, van Beusekom E, van der Zwaag B, et al. Mutations in the O-mannosyltrans- ferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 2002;71:1033-43.
17) Vajsar J, Schachter H. Walker-Warburg syndrome. Orphanet J Rare Dis 2006;1:29.
18) Cormand B, Avela K, Pihko H, Santavuori P, Talim B, Topaloglu H, et al.
Assignment of the muscle-eye-brain disease gene to 1p32-p34 by linkage analysis and homozygosity mapping. Am J Hum Genet 1999;
64:126-35.
19) Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, et al.
Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 2001;1:717-24.