DOI 10.17480/psk.2020.64.2.124
의약품 품목갱신제도 개선에 대한 제약업계의 인식 조사 - 유익성-위해성 평가를 중심으로 -
박양수#·오하나·여현석·홍진태·한상배
충북대학교 약학대학원
Pharmaceutical Industry’s Perception on the Improvement of Drug Renewal System Focused on Benefit-risk Assessment
Yangsu Park#, Ha-Na Oh, Hyun Seok Yeo, Jin Tae Hong, and Sang-Bae Han
Graduate School of Pharmacy, Chungbuk National University
(Received January 14, 2020; Revised March 5, 2020; Accepted March 5, 2020)
Abstract Drug renewal system is one of the effective tools for the re-evaluation of benefit-risk for the approved drug periodically. This study was purposed to investigate pharmaceutical industry’s perception on the improvement of drug renewal system and benefit-risk assessment. Comparative analysis was conducted on domestic and foreign pharmaceutical companies, as it is expected that the survey results would be affected by differences in product types, retention level of data and infrastructure. We conducted both on-line and off-line survey, which results in a total of 77 out of 255 staffs responded from December 2019 to January 2020; most of them were working at the department of regulatory affair or pharmacovigilance. The questionnaire consisted of 3 major parts; appropriateness of the current document requirements, introduction of the benefit-risk assessment, and pharmaceutical company’s status. Sixty-five percent of the respondents said that the current document requirements for drug renewal were appropriate, though 77% responded that the current system cannot assess the benefit-risk. For the introduction of benefit-risk assessment, 57% responded that it was unnecessary. The main reason was that it was inappropriate for the domestic situation. Only 22% responded that they had a workforce for benefits-risk assessment in the company. The overall response tendency was similar between domestic and foreign companies. However, for the benefit-risk assessment method, foreign companies had a higher recognition than domestic companies (61% vs 35%). In conclusion, it was thought that the pharmaceutical industry still had a great reluctance to introduce benefit-risk assessment due to the lack of expertise and the burden of preparing documents. Therefore, it is needed that infrastructure construction and sufficient training should precede for the improvement of the drug renewal system and the introduction of a benefit-risk assessment.
Keywords Drug renewal system, benefit-risk assessment, perception, pharmaceutical industry
서 론(Introduction)
국내에서는 의약품 사후관리를 위한 여러 제도들이 이미 오 랫동안 시행되고 있었는데, 대표적인 것이 재평가, 재심사 및 안 전성 정보 관리이다.
1),2)그러나 이러한 제도들이 존재함에도 불 구하고, 그 동안의 사후관리제도를 통해 품목이 취소되거나 자 발적으로 취하 또는 처분을 받은 품목의 수는 극히 적었는데, 그 이유는 해당 제도들의 제출자료의 요건과 수준을 살펴보면
잘 알 수 있다. 먼저, 재평가에 있어서 제출자료의 핵심은 해외 의 사용례(의약품집 수재 현황 등)를 수집하는 것이었는데, 이 들 자료는 업체들이 공동으로 준비할 수 있어 제출에 큰 어려 움이 없었다. 설령 일부 자료가 미비한 경우에도 다른 회사가 제출한 자료를 근거로 동일한 성분의 모든 제품들이 함께 지위 를 인정받을 수 있었기 때문에 특별한 경우를 제외하고는 대부 분의 품목들이 재평가를 무사히 통과할 수 있었다.
3)재심사의 경 우에도, 신약의 경우 3,000례, 자료제출의약품의 경우에는 600례 의 시판 후 조사를 시행하지만,
4)이는 시판 전 임상시험과는 달 리 일상 진료 하에서 이루어지는 조사활동이기 때문에 안전성 유효성의 특별한 차이를 발견하기는 쉽지 않아 재심사를 통한 품목 취소 사례 역시 거의 없는 것이 사실이었다. 그리고 그동 안 국내에는 품목 허가에 대한 유효기간이 없었기 때문에 한번 허가를 받은 제품은 평생토록 그 권리를 누릴 수 있었고, 회사
#
Corresponding author
Yangsu Park, Graduate School of Pharmacy, Chungbuk National Uni- versity, 194-21, Osongsaengmyeong 1-ro, Osong-eup, Heungdeok- gu, Cheongju-si, Chungcheongbuk-do, Korea
Tel: +82-70-4609-7135, Fax: +82-2-6952-1357
E-mail: [email protected]
들 역시 특별한 사유가 없는 한 품목에 대한 허가권을 지속적 으로 보존하기 위하여 많은 노력을 기울였다.
이러한 국내 품목허가 및 사후관리에 대한 인식의 변화를 가 져온 것이 품목갱신제도의 도입이었다.
5),6)품목갱신제도란 의약 품의 품목허가 시 일정한 유효기간을 부여하고, 유효기간 만료 시기가 도래하면 해당 시판 기간 동안 수집된 자료를 제출하여 갱신의 적절성을 평가한 후 유효기간을 연장(갱신)하는 제도이 다. 국내 품목갱신제도는 2013년 처음 시행되었고, 갱신의 주기 는 5년, 갱신의 대상은 일부(원료의약품, 수출용의약품, 희귀의 약품 등)를 제외한 시판되는 모든 의약품을 대상으로 하였다. 이 에 따라 2017년에 갱신을 위한 자료가 처음 제출되었고, 2018 년도부터 첫 갱신이 진행되었다.
품목갱신제도의 도입 취지는 크게 두가지로 볼 수 있다. 첫째 는 5년마다 시판 여부를 확인하여 실제 생산되지 않는 품목을 정비함으로써 불필요한 행정 낭비를 막고자 하는 것이고, 둘째 는 갱신 심사를 통해 정기적으로 시판약에 대한 유익성과 위해 성을 평가하고자 하는 것이었다. 최근 식약처 보도자료를 통해 품목 갱신이 진행된 지난 2년간의 실적을 살펴보면, 전체 8,232 개의 품목 중 5,546개 품목(67%)이 갱신되었고, 나머지 2,686개 품목은 품목 취하 또는 미신청 사유로 갱신이 되지 않았다고 밝 혔다.
7)갱신이 되지 않은 주요 이유는 생산·수입 실적이 없는 품목이 72%였는데, 이러한 사실로 볼 때, 미생산 품목에 대한 정리의 목적은 어느정도 달성한 것으로 보여진다. 그러나, 반대 로 식약처가 평가를 통해 퇴출시킨 제품이 한 개도 없다는 사 실은, 유익성-위해성 평가에 대한 부분에서는 아직까지 그렇지 못한 듯 하다. 이에 대한 가장 큰 이유는 품목갱신 제출자료에 서 찾을 수 있다. 현재 품목갱신을 위하여 제출하여야 하는 자 료는 총 6가지로서 ‘1. 유효기간 동안 수집된 안전관리에 관한 자료 및 조치계획, 2. 외국에서의 사용현황 및 안전성 관련 조 치에 관한 자료, 3. 유효기간 동안 수집된 품질관리에 관한 자 료, 4. 표시기재에 관한 사항, 5. 유효기간 동안의 제조·수입 실적에 관한 자료, 6. 유효한 제조판매·수입 품목허가증 또는 품목신고증’이 필요하다. 그러나 자료 목록에서 볼 수 있듯이 이 들 자료에서는 실제 유익성-위해성 평가를 위한 내용이 포함되 어 있지 않으므로 평가 역시 불가능한 것이다.
국내에 품목갱신제도를 도입할 당시 식약처와 업체 간의 가장 큰 의견 차이를 보였던 부분은 안전성평가 관련 자료의 제출 범 위였다. 국내의 품목갱신제도는 도입 검토 초기에 EU와 FDA의 갱신제도를 상당부분 참고하였는데, EU와 FDA 에서도 이미 오 래전부터 Renewal 또는 Annual report 와 같은 방식으로 품목갱 신을 운영하고 있었고, 갱신 신청시에 유익성-위해성에 관한 자 료를 함께 제출하고 있었다
8). 국내에서도 이러한 부분을 함께 반 영하고자 하였으나, 최종적으로 “외국의 사용현황”만을 제출하는 것으로 조정이 되었다. 이는 국내 산업의 특성을 감안한 조치라 는 점에서 제약업계는 충분히 반기는 입장이었으나, 한편으로는 사후관리의 가장 핵심이라고 할 수 있는 유익성과 위해성에 대 한 평가가 해외의 사례에 의존하게 된다는 점과 기존 운영해 오
던 재평가 제도와의 차별성이 없게 되었다는 점에서 제도 도입 취지가 상실되었다는 비판을 낳았다. 아울러, 제출자료에 대한 평 가를 어떤 방식으로 하겠다는 부분이 명시되어 있지 않아, 단지 생산(수입) 실적만 확인되고 해외에서의 사용 현황만 확인되면 해당 품목은 또다시 5년간의 유효기간 연장이 가능한, 품목갱신 의 목적이 상실된 형식적인 제도로 운영되는 상황이 되었다.
한편, 현재 US FDA (Food and Drug Administration)와 EMA (European Medicines Agency) 에서 유익성-위해성 평가를 위하 여 사용하고 있는 대표적인 평가기법으로서 BRF (Benefit-Risk Framework)
10),11)와 ET (Effect table)
12),13)가 있다. FDA BRF는 유 익성-위해성 평가를 위한 기본 틀로서 ‘질병 분석(Analysis of Condition), 현재 사용 가능한 치료제(Current Treatment Options), 유익성(Benefit), 위해성(Risk) 및 위해 관리(Risk Management)’
의 총 5가지 항목을 포함하고 있고, 이들 개별 요소들에 대하여 규제 의사 결정을 위한 ‘근거자료와 불확실성(Evidence and Uncertainties)’ 및 ‘결론과 이에 대한 근거(conclusions and Rea- sons)’를 추가하여 최종적인 평가틀이 구성된다. EMA ET 역시 핵심 유익성과 위해성을 요약하는 도구표로서, 각 유익성과 위 해성에 대하여 ‘효과(Effect), 요약 설명(Short description), 단위 (Unit), 신청약(Treatment), 비교약(Control), 자료의 불확실성/강점 (Uncertainties/Strength of Evidence), 인용 자료(Reference)’의 내 용을 포함하고 있다. 위에서 살펴본 품목갱신제도의 운영상의 문제점과 유익성-위해성 평가 부재에 대한 문제점을 해결하기 위한 일환으로서 해외의 유익성-위해성 평가기법 도입이 좋은 대안이 될 수 있다.
9)그러나 해외에서는 활발히 사용중인 평가 기법들임에도 불구하고, 아직까지 국내에서는 이를 이용한 자료 제출 사례가 거의 없고, 전문가 역시 부족한 상황으로서 도입을 위해서는 평가기법에 대한 소개 및 업계 담당자들의 인지도 수 준을 파악하는 것부터 필요한 실정이다.
한편, 국내에서 의약품허가를 보유하고 있는 회사는 크게 국 내제약사와 외국계제약사로 나눌 수 있는데, 이들 두 제약사들 은 품목의 구성 및 자료의 보유측면에서 많은 차이점이 있다.
먼저, 외국계제약사가 보유하고 있는 품목군은 신약 또는 개 량 제품들이 많아 품목허가를 위한 의무사항으로 대부분 임상 자료를 보유한 경우가 많고, 허가 시에도 6년 또는 4년간의 재 심사를 부여받도록 되어 있어 시판 후에도 다양한 임상 자료를 꾸준히 수집할 수 있다. 또한, 외국계제약사의 경우에는 규모가 큰 제약사가 많아 꾸준한 연구 투자와 시험을 진행하여 안전성 및 유효성에 대한 정보들을 지속적으로 수집하고 있다. 그리고, 국내에 진출해 있는 외국계제약사는 미국, 유럽 등 대부분 의약 개발선진국가들이 많아 국내보다 이미 빠른 규제기준을 적용하 고 있어, 국내 규제기관의 자료 요청이 있는 경우 본사가 이미 해당 자료를 보유하고 있거나, 회사가 이미 보유하고 있는 자료 와 전문가의 지원을 통해 신속하게 자료를 준비하게 된다.
반면, 국내제약사의 경우 보유하고 있는 품목군의 대부분이
제네릭의약품으로서 품목허가 시 단지 생물학적동등성 시험을
실시하거나 일부 품목의 경우에는 이마저도 없이 허가를 받은
품목도 있다. 아울러, 시판을 하더라도 안전성에 관련한 정기보 고 이외에는 어떠한 사후관리에 대한 의무사항도 없기 때문에 허가 이후 추가적으로 수집되는 자료는 거의 없는 상황이다.
따라서, 기본적으로는 이러한 국내제약사와 외국계제약사의 환경과 보유 인프라의 차이로 인해 유익성-위해성 평가라고 하 는 새로운 규제를 바라보는 입장에 차이가 있을 것으로 생각하 였으며, 아울러 단순히 자료의 보유와 전문성의 수준 이외에 또 어떤 요소들을 제약사에서는 주요하게 생각하는지에 대해서도 연구가 필요할 것으로 판단하였다.
품목갱신제도 도입단계에서 식약처와 제약업계는 많은 이견 이 있었음에도 불구하고, 제도 시행 이전은 물론 시행 이후에도 업계에서 느끼는 문제점 및 개선사항 등의 현황에 대해서는 한 번도 조사가 이루어진 적이 없다. 유익성-위해성에 대한 연구 역 시 해외의 제도에 대한 비교연구와 제약사 역량 에 대한 조사 연구 자료가 단편적으로 있었으나 품목갱신제도와 유익성-위해 성 평가의 관계 속에서 두가지를 연구한 사례는 아직까지 없다.
따라서, 본 연구에서는 품목갱신제도 개선 및 유익성-위해성 평 가 도입과 관련한 본격적인 연구에 앞서, 설문조사를 통하여 업 계의 현황을 파악하고, 품목갱신제도의 개선을 위해 어떤 부분 에 중점을 두어야 하는지에 대해 살펴보고자 하였다.
연구방법(Research Methods)
설문지 개발
설문지는 품목갱신제도 운영과 관련하여 제출자료의 적절성 및 갱신제도가 유익성-위해성 평가라고 하는 본래의 취지에 부 합하게 운영되고 있는지, 개선이 필요한 부분은 어떤 점이 있는 지에 대하여 제약사의 인식을 우선 조사한 후, 이를 보완하기 위한 방법으로서 유익성-위해성 평가방법을 도입하는 것에 대한 입장(필요성, 도입방법 및 제약사 현황)을 조사하는 것으로 순 서를 정하고, 이에 따라 설문항목을 구성하였다. 설문 항목 중 품목갱신과 관련한 내용들은 자체 개발하였고, 설문 참가자 현 황, 유익성-위해성 평가와 관련한 내용 및 업계 현황 항목은 기 존의 국내 제약회사를 대상으로 실시된 역량분석 선행연구
14)를 참고하였다. 세부적으로는 설문 참가자 현황 및 제약업계 현황 부분의 응답자에 대한 정보, 보유 인력 및 전문성, 식약처 준비 요구사항에 대한 내용은 선행 연구 항목과 유사하게 인용하였 고, 유익성-위해성 평가방법인 FDA Benefit-Risk Framework 및 EMA Effect Table 에 대한 인지 여부에 대해서는 동일하게 질문 하되(있다/없다), 적용 품목 및 도입 방식에 대해서는 질문과 답 변을 일부 변형 및 추가하여 작성하였다. 개발된 설문지는 업계 전문가 2인의 감수 및 의견을 반영하여 최종 완성하였다.
설문지는 총 5개 파트(A~E), 15개 문항으로 구성되었다. 수집 정보는 크게 설문참가자에 대한 기본정보, 품목갱신 제출자료에 대한 적절성, 품목갱신에 대한 유익성-위해성 평가 도입 필요성, 유익성-위해성 평가방법에 대한 인지도, 업계 현황으로 구성하 였다. 설문 형식은 단답형과 복수 선택 가능한 형태로 각 질문
의 내용에 맞게 적절히 선택하였다.
파트 A는 설문 참가자의 기본정보로서, 회사 구분(국내 제약 회사, 외국계 제약회사, 기타), 매출액 규모, 연령, 근무경력, 담 당 업무
(*
), 전공을 확인하였다.
파트 B는 품목갱신 제출자료의 적절성을 조사하는 부분으로 서, 현재의 제출자료가 적절하다고 생각하는지, 부적절하거나 개 선이 필요하다면 어떤 부분인지
(*
), 품목갱신에서 가장 중요시되 어야 하는 항목
(*
)과 현재의 품목갱신제도를 통해 유익성-위해성 평가가 가능하다고 생각하는지, 아니라고 생각한다면 그 이유
(*
)는 무엇인지 물었다.
파트 C에서는 품목갱신에 대한 유익성-위해성 평가의 도입 필 요성과 불필요하다고 생각하는 경우에는 그 이유
(*
)를 물었고, 평가를 위해 선행되어야 하는 사항
(*
), 평가가 필요한 품목과 불 필요하다고 생각하는 품목
(*
), 유익성-위해성 평가 도입을 위해 가장 중요시 되어야 하는 부분
(*
)이 무엇인지에 대해서도 조사하였다.
파트 D는 유익성-위해성 평가방법에 대한 인지도를 조사하는 부분으로서, FDA와 EMA에서 사용하는 Benefit-Risk Framework 및 Effect table에 대한 인지도를 조사하였다. 아울러, 평가방법 을 어떤 식으로 도입하는 것이 적절한지
(*
)에 대해서도 질문하였다.
마지막 파트 E에서는 유익성-위해성 평가를 위한 제약회사의 인력 현황과 전문성을 조사하고, 평가방법 도입 시 식약처가 우 선적으로 준비해주기를 바라는 사항
(*
)에 대해 설문을 진행하였다.
대상자 및 설문조사 실시
품목갱신제도의 시행에 따라 가장 영향을 많이 받는 곳은 품 목허가를 보유한 제약회사이며 그 중에서도 갱신자료를 직접 준 비해야 하는 인허가부서 및 약물감시 부서의 부담이 제일 크기 때문에, 설문조사는 “Regulatory affairs (인허가)” 또는 “Pharma- covigilance ( 약물감시)” 업무 경험이 2년 이상 있거나, 품목갱신 업무 경험이 있는 제약회사 또는 관련 종사자들만을 대상으로 진행하였다. 대상자는 국내사, 외국계 회사 또는 기타 회사(컨설 팅업체 등) 종사자를 불문하고, 업무와의 관련성이 많은 대상자 를 최대한 모집하기 위하여 인허가 담당자가 다수 가입되어 있 고 제약관련 교육활동을 활발히 수행하고 있는 한국제약산업연 구회, 의약품규제과학센터 및 제약개발업무종사자 커뮤니티의 협조를 얻어 대상자 정보를 수집하였고, 총 255명에 대하여 직 접 이메일을 발송하였다. 이들 중 이메일을 통하여 수신된 설문 지는 총 63건이었다. 한 명의 메일로부터 2개 이상의 설문지를 수신한 경우도 있었으며, 이는 동일 회사의 동료로부터 수집된 설문지임을 확인하였다. 아울러 이메일을 수령하였더라도 대면 조사에 동의한 경우에는 직접 방문을 통한 설문을 진행하였고, 이를 통한 총 14건의 설문지를 합하여 전체 수집된 설문지는 총 77 건이었다. 자료는 2019년 12월 11일부터 2020년 1월 10일까 지 수집하였다. 본 연구는 충북대학교 생명윤리심의위원회의 정 식 심의를 통하여 승인받았다(CBNU-201912-SB-969-01).
(*): 복수 답변 가능 항목
자료 분석
수집된 정보는 Microsoft Excel 2016을 이용하여 통합하였다.
각 설문 항목에 대한 특성 비교를 위하여 응답자 수와 백분율 을 제시하였다. 설문 답변은 회사별로 보유한 자료 및 인프라의 수준 등에 따라 차이가 있을 것으로 예상되어 국내제약사, 외국 계제약사, 기타로 각각 응답 결과를 구분하여 분석하였으며, ‘기 타’ 회사 종사자의 응답은 소수(4명, 5.2%)로서 분석은 진행하 였으나 회사별 고찰에서는 제외하였다. 설문 문항은 대부분 독 립적 설문내용 형식으로 구성되어 있고, 명목형 설문에 해당하 여 별도의 신뢰도 분석은 진행하지 않았다.
또한, 현재의 품목갱신 제출자료가 적절하지 않다고 답변하였 거나, 유익성-위해성 평가제도를 도입하여야 한다고 답변한 응 답자들의 현황을 파악하기 위하여 회사, 성별, 매출액, 연령, 근 무경력, 담당업무, 전공에 대하여 Fisher’s exact test를 이용한 분 석을 진행하였고(모든 항목에서 기대 빈도가 5 이하인 셀이 25%
이상에 해당), 품목갱신제출자료의 적절성 유무 답변에 대한 응 답자별로 유익성-위해성 평가 도입의 필요성과 유익성-위해성 평 가 가능성에 대하여 답변 비율 차이에 대해서도 비율검정을 통 하여 유의성을 검증하였다. 모든 자료는 SAS ver. 9.4를 이용하 여 통계분석을 진행하였고, 유의수준은 0.05를 적용하였다.
결 과(Results)
응답자의 특성
본 설문조사는 대상자 255명 중 총 77명이 설문에 응답하여 30.2% 의 응답률을 나타냈다. 설문에 참여한 77명의 대상자에 대 한 회사 구분, 매출액 규모, 연령, 근무경력, 담당 업무, 전공에 대한 현황은 Table 1에 정리하였다. 회사별로는 국내 제약회사 근무자가 55명(71.4%)으로 가장 많았고, 외국계 제약회사 근무 자가 18명(23.4%), 기타 4명(5.2%) 순이었다. 회사의 매출액은 3000억 이상이 29.9%로 가장 많았으나, 1,000억 미만, 1,000억
~2,000 억 미만, 2,000억~3,000억 미만 회사의 비율도 24.7, 22.1, 22.1%로 대체로 비슷하였다. 응답자의 연령대는 30대와 40대가 각각 36.4, 46.8%로 가장 많았다. 근무경력은 10년 이상이라고 응답한 대상자가 47명(61.0%)으로서 과반수를 넘었다. 응답자의 84.4% 는 의약품 인허가와 관련한 업무를 하고 있었으며, 48.1%
는 신제품개발 업무와 관련이 있었다. 응답자의 전공은 약학이 53명(68.8%)으로 가장 많았으며, 다음으로 생화학 16명(20.8%), 간호학 2명(2.6%), 경제/경영학 2명(2.6%) 순이었다. 담당 업무 및 전공에 대하여는 복수 답변을 허용하였고, 응답자 중 38명은 회사에서 1가지 이상의 업무를 담당하고 있었으며, 전공이 2개 인 응답자도 1명이었다.
품목갱신 제출자료의 적절성 조사
품목갱신 제출자료의 적절성에 대한 설문에서 참여한 77명의 응답자 중 현재의 품목갱신을 위한 제출자료가 ‘적절하다’고 답 변한 비율은 64.9% (50/77)이었다. 국내 제약회사와 외국계 제
약회사에 근무하는 응답자 모두에서 ‘적절하다’라고 답변한 비 율이 67.3% (37/55), 61.1% (11/18)로서 ‘적절하지 않다’라고 답 Table 1. Respondent’s characteristics
Characteristic n(%)
Total No. of respondents 77
Company
Domestic company 55(71.4) International company 18(23.4)
Others 4(5.2)
Missing -
Sales (in 2018) (bil. KRW)
<50 12(15.6)
≥50 to <100 7(9.1)
≥100 to <200 17(22.1)
≥200 to <300 17(22.1)
≥300 23(29.9)
Missing 1(1.3)
Age
20-29 10(13)
30-39 28(36.4)
40-49 36(46.8)
50-59 3(3.9)
60-69 0(0)
≥70 0(0)
Missing -
Career (years)
≥2 to <3 10(13.0)
≥3 to <5 7(9.1)
≥5 to <8 10(13.0)
≥8 to <10 3(3.9)
≥10 47(61.0)
Missing -
Work
(*
)Regulatory affairs 65(84.4) Pharmacovigilance 14(18.2) Business development 37(48.1) Clinical research 11(14.3) Academic work 9(11.7)
Others 4(5.2)
Missing -
Major
(*
)Medicine 0(0)
Pharmacy 53(68.8)
Nursing 2(2.6)
Economics/Management 2(2.6) Biochemistry 16(20.8)
Statistics 0(0)
Others 5(6.5)
Missing -
(*): Multiple answers available
변한 비율보다 높았다. 제출항목 중 ‘부적절하거나 개선’되어야 하는 항목으로서는, ‘외국에서의 사용현황 및 안전성 관련 조치 에 관한 자료’(59.3%), ‘유효기간 동안의 제조·수입 실적에 관 한 자료’(25.9%), ‘표시기재에 관한 자료’(22.2%), ‘유효기간 동 안 수집된 품질관리에 관한 자료’(18.5%) 순으로 나타났다. 품 목갱신에 있어 가장 중요시되어야 한다고 생각하는 부분은 ‘안 전성 관련자료’라고 응답한 비율이 80.5%로서 가장 높았으며, 다음으로 ‘유익성-위해성 균형 평가’(44.2%), ‘유효성 관련자료’
(39.0%) 순이었다. 한편, 현재의 품목갱신제도를 통하여 유익성 - 위해성을 충분히 평가할 수 있다고 생각하는지에 대한 질문에 서는 ‘그렇지 않다’라고 응답한 비율이 76.6%로 조사되어, 국내 제약회사(70.9%), 외국계 제약회사(88.9%) 모두 부정적인 의견
이 다수였다. 유익성-위해성 평가가 어려운 이유로는 ‘평가방법 이 구체적으로 제시되어 있지 않다’(62.7%) 의견이 가장 많았으 며, ‘평가를 위한 제출자료의 수준이 적절하지 않다’(47.5%), ‘평 가를 위한 전문가가 없다’(39.0%)의 응답도 높은 수준으로 나타 났다(Table 2).
품목갱신에 대한 유익성-위해성 평가방법 도입 조사
품목갱신제출자료로서 유익성-위해성 평가방법의 도입이 필요 한지에 대한 조사에서는 총 77명의 응답자 중 ‘아니오’라고 응 답한 비율이 57.1% (44/77)로서 ‘예’라고 응답한 비율(42.9% (33/
77)) 보다 다소 높게 나타났다. 도입이 필요하다고 응답한 비율은 외국계 제약회사(55.6% (10/18))가 국내 제약회사(38.2% (21/55)) Table 2. Adequacy of submission data requirement for drug renewal
Answer Total Domestic International Others
n(%) n(%) n(%) n(%)
Total No. of respondents 77 55 18 4
Do you think the current submission data requirement for drug renewal
is adequate?
Yes 50(64.9) 37(67.3) 11(61.1) 2(50.0)
No 27(35.1) 18(32.7) 7(38.9) 2(50.0)
Missing - - - -
Sub-total No. of respondents 27 18 7 2
Which of the following items is inappropriate or should be
improved?
(*)Collected Safety data and action plan during validity period 2(7.4) 1(5.6) 0(0) 1(50.0) The Status of foreign use and Safety related action 16(59.3) 12(66.7) 4(57.1) 0(0) Collected Quality control data during validity period 5(18.5) 3(16.7) 2(28.6) 0(0)
Labeling 6(22.2) 5(27.8) 1(14.3) 0(0)
Manufacturing and importing record during validity period 7(25.9) 3(16.7) 3(42.9) 1(50.0) Valid manufacturing/ importing approval certificate 2(7.4) 1(5.6) 1(14.3) 0(0)
Missing 1(3.7) 1(5.6) - -
What is most important thing for drug renewal?
(*
)Safety data 62(80.5) 42(76.4) 17(94.4) 3(75.0)
Efficacy data 30(39.0) 20(36.4) 9(50.0) 1(25.0)
Benefit-risk balance assessment 34(44.2) 23(41.8) 8(44.4) 3(75.0) Assessment by Medical Specialist 3(3.9) 3(5.5) 0(0) 0(0)
User assessment 2(2.6) 2(3.6) 0(0) 0(0)
Economic evaluation 3(3.9) 1(1.8) 1(5.6) 1(25.0)
Missing Can the current drug renewal
system fully assess the benefit-risk of the product?
Yes 18(23.4) 16(29.1) 2(11.1) 0(0)
No 59(76.6) 39(70.9) 16(88.9) 4(100.0)
Missing - - - -
Sub-total No. of respondents 59 39 16 4
Why do you think that the current drug renewal system is inadequate for benefit-risk
assessment?
(*
)The required data is not appropriate for assessment 28(47.5) 16(41.0) 7(43.8) 4(100.0) The assessment method is not specified 37(62.7) 21(53.8) 12(75.0) 4(100.0) Lack of expert for assessment 23(39.0) 13(33.3) 9(56.3) 1(25.0) Limitation of data collection 18(30.5) 14(35.9) 4(25.0) 0(0)
Others 1(1.7) 1(2.6) 0(0) 0(0)
Missing - - - -
(*): Multiple answers available
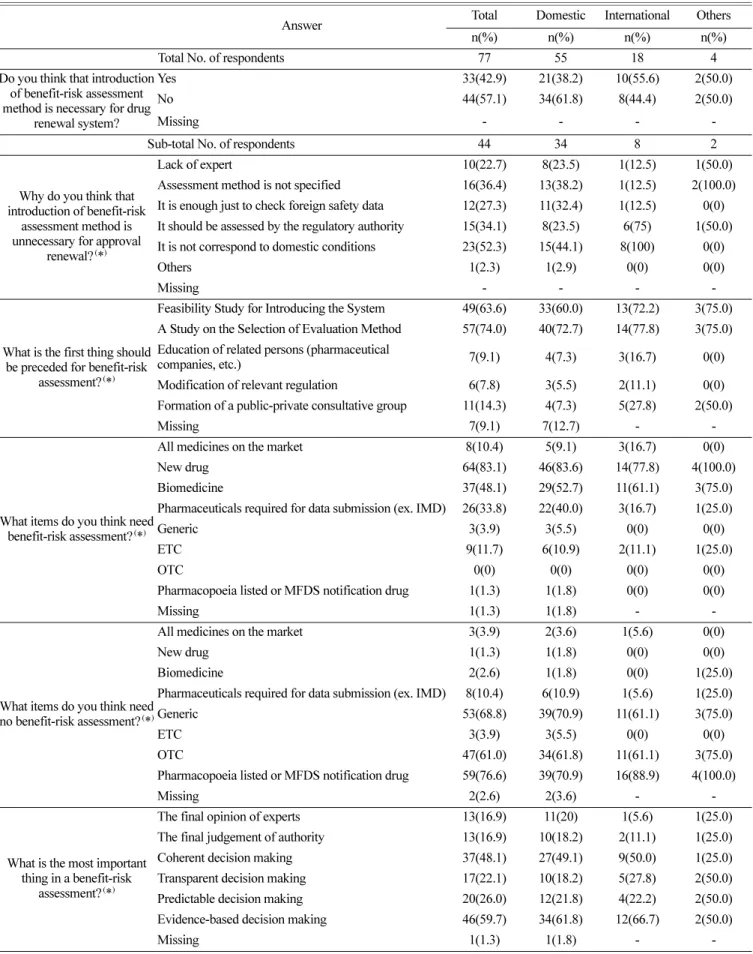
Table 3. Introduction of benefit-risk assessment method for drug renewal system
Answer Total Domestic International Others
n(%) n(%) n(%) n(%)
Total No. of respondents 77 55 18 4
Do you think that introduction of benefit-risk assessment method is necessary for drug
renewal system?
Yes 33(42.9) 21(38.2) 10(55.6) 2(50.0)
No 44(57.1) 34(61.8) 8(44.4) 2(50.0)
Missing - - - -
Sub-total No. of respondents 44 34 8 2
Why do you think that introduction of benefit-risk
assessment method is unnecessary for approval
renewal?
(*
)Lack of expert 10(22.7) 8(23.5) 1(12.5) 1(50.0)
Assessment method is not specified 16(36.4) 13(38.2) 1(12.5) 2(100.0) It is enough just to check foreign safety data 12(27.3) 11(32.4) 1(12.5) 0(0) It should be assessed by the regulatory authority 15(34.1) 8(23.5) 6(75) 1(50.0) It is not correspond to domestic conditions 23(52.3) 15(44.1) 8(100) 0(0)
Others 1(2.3) 1(2.9) 0(0) 0(0)
Missing - - - -
What is the first thing should be preceded for benefit-risk
assessment?
(*
)Feasibility Study for Introducing the System 49(63.6) 33(60.0) 13(72.2) 3(75.0) A Study on the Selection of Evaluation Method 57(74.0) 40(72.7) 14(77.8) 3(75.0) Education of related persons (pharmaceutical
companies, etc.) 7(9.1) 4(7.3) 3(16.7) 0(0)
Modification of relevant regulation 6(7.8) 3(5.5) 2(11.1) 0(0) Formation of a public-private consultative group 11(14.3) 4(7.3) 5(27.8) 2(50.0)
Missing 7(9.1) 7(12.7) - -
What items do you think need benefit-risk assessment?
(*
)All medicines on the market 8(10.4) 5(9.1) 3(16.7) 0(0)
New drug 64(83.1) 46(83.6) 14(77.8) 4(100.0)
Biomedicine 37(48.1) 29(52.7) 11(61.1) 3(75.0)
Pharmaceuticals required for data submission (ex. IMD) 26(33.8) 22(40.0) 3(16.7) 1(25.0)
Generic 3(3.9) 3(5.5) 0(0) 0(0)
ETC 9(11.7) 6(10.9) 2(11.1) 1(25.0)
OTC 0(0) 0(0) 0(0) 0(0)
Pharmacopoeia listed or MFDS notification drug 1(1.3) 1(1.8) 0(0) 0(0)
Missing 1(1.3) 1(1.8) - -
What items do you think need no benefit-risk assessment?
(*
)All medicines on the market 3(3.9) 2(3.6) 1(5.6) 0(0)
New drug 1(1.3) 1(1.8) 0(0) 0(0)
Biomedicine 2(2.6) 1(1.8) 0(0) 1(25.0)
Pharmaceuticals required for data submission (ex. IMD) 8(10.4) 6(10.9) 1(5.6) 1(25.0)
Generic 53(68.8) 39(70.9) 11(61.1) 3(75.0)
ETC 3(3.9) 3(5.5) 0(0) 0(0)
OTC 47(61.0) 34(61.8) 11(61.1) 3(75.0)
Pharmacopoeia listed or MFDS notification drug 59(76.6) 39(70.9) 16(88.9) 4(100.0)
Missing 2(2.6) 2(3.6) - -
What is the most important thing in a benefit-risk
assessment?
(*
)The final opinion of experts 13(16.9) 11(20) 1(5.6) 1(25.0) The final judgement of authority 13(16.9) 10(18.2) 2(11.1) 1(25.0) Coherent decision making 37(48.1) 27(49.1) 9(50.0) 1(25.0) Transparent decision making 17(22.1) 10(18.2) 5(27.8) 2(50.0) Predictable decision making 20(26.0) 12(21.8) 4(22.2) 2(50.0) Evidence-based decision making 46(59.7) 34(61.8) 12(66.7) 2(50.0)
Missing 1(1.3) 1(1.8) - -
(*): Multiple answers available
Table 4. Awareness of benefit-risk assessment method
Answer Total Domestic International Others
n(%) n(%) n(%) n(%)
Total No. of respondents 77 55 18 4
Have you ever heard of FDA benefit-risk Framework or
EMA Effects Table?
Yes 32(41.6) 19(34.5) 11(61.1) 2(50.0)
No 45(58.4) 36(65.5) 7(38.9) 2(50.0)
Missing - - - -
Sub-total No. of respondents 32 19 11 2
If you’ve ever heard of it, What’s your level of
understanding?
I know very well 0(0) 0(0) 0(0) 0(0)
I know well 2(6.3) 2(10.5) 0(0) 0(0)
I know 20(62.5) 11(57.9) 7(63.6) 2(100.0)
I don’t know. 10(31.3) 6(31.6) 4(36.4) 0(0)
I don’t know at all 0(0) 0(0) 0(0) 0(0)
missing - - - -
Which of the above two methods do you think is appropriate to introduce?
(*
)FDA Benefit-Risk Framework 9(11.7) 7(12.7) 1(5.6) 1(25.0)
EMA Effects Table 7(9.1) 6(10.9) 1(5.6) 0(0)
Mixed method 12(15.6) 8(14.5) 3(16.7) 1(25.0)
Modified method to suit domestic conditions 54(70.1) 38(69.1) 12(66.7) 4(100.0)
No matter 9(11.7) 6(10.9) 3(16.7) 0(0)
Missing 3(3.9) 2(3.6) 1(5.6) -
(*): Multiple answers available
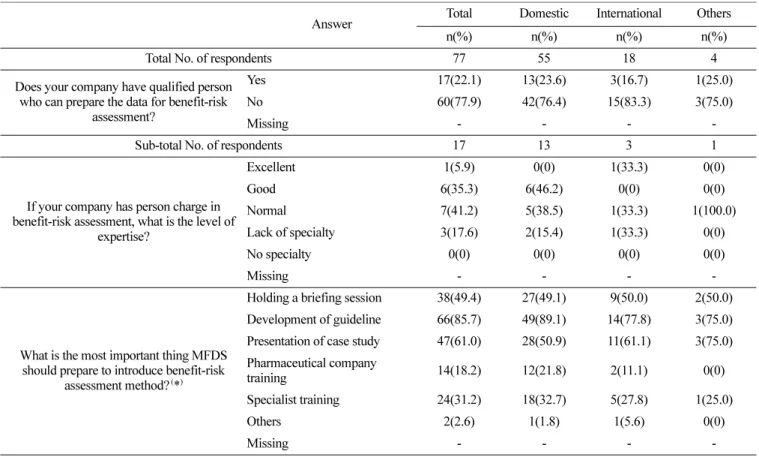
Table 5. Current status of pharmaceutical industry
Answer Total Domestic International Others
n(%) n(%) n(%) n(%)
Total No. of respondents 77 55 18 4
Does your company have qualified person who can prepare the data for benefit-risk
assessment?
Yes 17(22.1) 13(23.6) 3(16.7) 1(25.0)
No 60(77.9) 42(76.4) 15(83.3) 3(75.0)
Missing - - - -
Sub-total No. of respondents 17 13 3 1
If your company has person charge in benefit-risk assessment, what is the level of
expertise?
Excellent 1(5.9) 0(0) 1(33.3) 0(0)
Good 6(35.3) 6(46.2) 0(0) 0(0)
Normal 7(41.2) 5(38.5) 1(33.3) 1(100.0)
Lack of specialty 3(17.6) 2(15.4) 1(33.3) 0(0)
No specialty 0(0) 0(0) 0(0) 0(0)
Missing - - - -
What is the most important thing MFDS should prepare to introduce benefit-risk
assessment method?
(*
)Holding a briefing session 38(49.4) 27(49.1) 9(50.0) 2(50.0) Development of guideline 66(85.7) 49(89.1) 14(77.8) 3(75.0) Presentation of case study 47(61.0) 28(50.9) 11(61.1) 3(75.0) Pharmaceutical company
training 14(18.2) 12(21.8) 2(11.1) 0(0)
Specialist training 24(31.2) 18(32.7) 5(27.8) 1(25.0)
Others 2(2.6) 1(1.8) 1(5.6) 0(0)
Missing - - - -
(*): Multiple answers available
보다 높았다. 유익성-위해성 평가 도입이 불필요한 이유는 ‘국 내 제약 여건과 부합하지 않다’는 응답이 52.3%로 가장 높았으 며, 이외에 ‘평가방법의 불확실성’(36.4%), ‘규제기관이 판단할 사항’(34.1%), ‘해외안전관리 조치 확인으로 충분하다고 판단’
(27.3), ‘전문가 부재’(22.7%)라는 의견 역시 고르게 나타났다. 유 익성-위해성 평가를 위해 가장 먼저 선행되어야 하는 것은 ‘평 가방법 선정을 위한 연구’(74.0%)와 ‘제도 도입 타당성 연구’
(63.6%) 로 조사되었으며, 신약(83.1%), 바이오의약품(48.1%), 자 Table 6. Characteristics of the respondents who are negative for the current submission data requirement
Characteristic total Current submission data requirement is
not appropriate, n(%) p-value
Company
Domestic company 55 18(32.7)
0.6959
International company 18 7(38.9)
Others 4 2(50.0)
Missing - -
Sales (in 2018) (bil. KRW)
<50 12 3(25.0)
0.2551
≥50 to <100 7 3(42.9)
≥100 to <200 17 9(52.9)
≥200 to <300 17 3(17.6)
≥300 23 8(34.8)
Missing 1 -
Age
20-29 10 2(20.0)
0.0741
30-39 28 7(25.0)
40-49 36 18(50.0)
50-59 3 0(0)
60-69 0 0(0)
≥70 0 0(0)
Missing - -
Career (years)
≥2 to <3 10 2(20.0)
0.7924
≥3 to <5 7 2(28.6)
≥5 to <8 10 3(30.0)
≥8 to <10 3 1(33.3)
≥10 47 19(40.4)
Missing -
Work
Regulatory affairs 65 24(36.9)
0.9571
Pharmacovigilance 14 4(28.6)
Business development 37 13(35.1)
Clinical research 11 4(36.4)
Academic work 9 4(44.4)
Others 4 2(50)
Missing - -
Major
Medicine 0 0(0)
0.8360
Pharmacy 53 18(34.0)
Nursing 2 0(0)
Economics/Management 2 1(50.0)
Biochemistry 16 7(43.8)
Statistics 0 0(0)
Others 5 2(40.0)
Missing - -
료제출의약품(33.8%) 순으로 우선 적용해야 한다는 의견이 다수 였다. 반면, 공정서 및 고시 해당 품목(76.6%), 제네릭의약품 (68.8%), 일반의약품(61.0%)의 경우에는 유익성-위해성 평가가 불필요하다고 생각하는 응답이 많았다. 아울러, 유익성-위해성
평가에 있어 가장 중요시되어야 하는 부분은 ‘근거에 기반한 의 사결정’과 ‘일관성 있는 의사결정’이라고 응답한 비율이 59.7, 48.1%로 다수였다(Table 3).
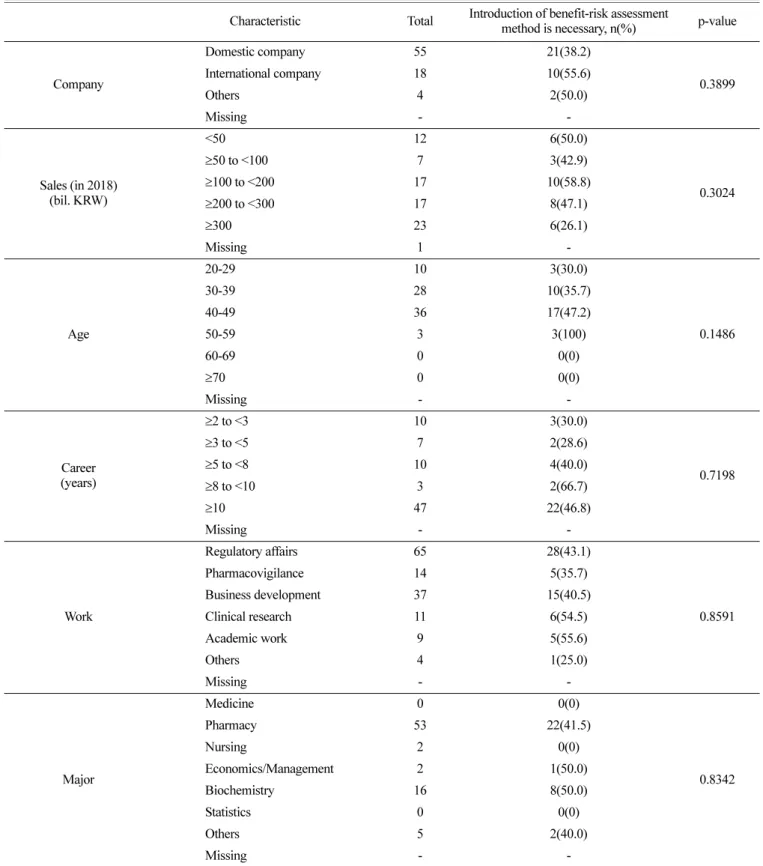
Table 7. Characteristics of the respondents who are positive for the introduction of benefit-risk assessment method Characteristic Total Introduction of benefit-risk assessment
method is necessary, n(%) p-value
Company
Domestic company 55 21(38.2)
0.3899 International company 18 10(55.6)
Others 4 2(50.0)
Missing - -
Sales (in 2018) (bil. KRW)
<50 12 6(50.0)
0.3024
≥50 to <100 7 3(42.9)
≥100 to <200 17 10(58.8)
≥200 to <300 17 8(47.1)
≥300 23 6(26.1)
Missing 1 -
Age
20-29 10 3(30.0)
0.1486
30-39 28 10(35.7)
40-49 36 17(47.2)
50-59 3 3(100)
60-69 0 0(0)
≥70 0 0(0)
Missing - -
Career (years)
≥2 to <3 10 3(30.0)
0.7198
≥3 to <5 7 2(28.6)
≥5 to <8 10 4(40.0)
≥8 to <10 3 2(66.7)
≥10 47 22(46.8)
Missing - -
Work
Regulatory affairs 65 28(43.1)
0.8591
Pharmacovigilance 14 5(35.7)
Business development 37 15(40.5)
Clinical research 11 6(54.5)
Academic work 9 5(55.6)
Others 4 1(25.0)
Missing - -
Major
Medicine 0 0(0)
0.8342
Pharmacy 53 22(41.5)
Nursing 2 0(0)
Economics/Management 2 1(50.0)
Biochemistry 16 8(50.0)
Statistics 0 0(0)
Others 5 2(40.0)
Missing - -
유익성-위해성 평가방법에 대한 인지도 조사
유익성-위해성 평가방법인 FDA Benefit-Risk Framework 또는 EMA Effects Table에 대해 들어본 적이 있는지에 대한 조사에 서는 전체 77명의 응답자 중 41.6% (32/77)만이 ‘들어본 적이 있다’고 응답하여 과반수에 미치지 못하였다. 회사별로는 외국 계 제약회사에 근무하는 응답자의 61.1% (11/18)가 ‘들어본 적 이 있다’고 답한 반면, 국내 제약회사 근무자의 경우에는 34.5%
(19/55)만이 ‘들어본 적 있다’라고 대답하여 차이를 보였다. ‘들 어본 적이 있다’ 라고 응답한 경우에도, ‘매우 잘 알고 있다’ 라 고 응답한 사람은 한 명도 없었으며, 대부분 ‘보통’(62.5%) 또는
‘ 잘 모른다’(31.3%)라고 대답하였다. FDA와 EMA 방법을 도입 하고자 할 경우에도 ‘연구사업을 통하여 국내 실정에 맞게 수정 된 방법으로 도입하여야 한다’는 의견이 70.1%로 다수였다(Table 4).
제약업계 현황 조사
전체 응답자 77명 중 회사내에 유익성-위해성 자료를 준비하 여 제출할 수 있는 인력이 ‘있다’고 대답한 사람은 17명(22.1%) 에 불과하였다. 이들 해당 인력의 전문성에 대해서는 ‘매우 우 수’(1명, 5.9%), ‘우수’(6명, 35.3%), ‘보통’(7명, 41.2%), ‘부족’(3 명, 17.6%)으로 응답하였다. 한편, 제약업계는 유익성-위해성 평 가 도입에 앞서 식약처가 ‘제출자료 및 평가방법에 대한 가이드 라인 개발’(85.7%), ‘유익성-위해성 평가 사례발표’(61.0%), ‘제도 도입을 위한 설명회 개최’(49.4%)를 선행해야 한다고 생각하는 것으로 조사되었다(Table 5).
품목갱신제출자료의 적절성 및 유익성-위해성 평가방법 도입 답 변에 대한 응답자의 현황
현재의 품목갱신을 위한 제출자료가 ‘적절하지 않다’고 답변 한 27명(35.1%)에 대한 특성을 회사(국내, 외국계), 매출액, 나이, 경력, 담당업무 및 전공별로 각각 분석해 보았으나 유의성 있는 차이를 확인할 수 없었다(Table 6). 아울러, 유익성-위해성 평가 방법을 ‘도입해야 한다’고 답변한 30명(45.5%) 역시 각 항목별 로 유의적인 차이를 나타내지 않았다(Table 7).
또한, 품목갱신제출자료에 대해 ‘적절하다’와 ‘그렇지 않다’라 고 답변한 각각의 응답자를 대상으로 ‘유익성-위해성 평가방법 도입이 필요하다고 생각하는지’와 ‘현재의 갱신제도로서 유익성
- 위해성 평가가 가능하다고 생각하는지’에 대한 응답 비율을 분 석하였다. 그 결과, 품목갱신자료가 ‘적절하다’고 생각하는 응답 자의 64.0% (32/50)는 유익성-위해성 평가방법 도입 역시 ‘불필 요하다’고 생각하고 있었고, 제출자료가 ‘적절하지 않다’고 생각 하는 응답자의 55.6% (15/27)는 유익성-위해성 평가가 ‘필요하다’
고 생각하였다. 한편, 품목갱신제출자료가 ‘적절하다’고 생각하 는 응답자 중에서 현재의 제출자료로서 유익성-위해성 평가 가 능하다고 답변한 응답자는 28% (14/50)에 불과하였고, 두 응답 군 모두에서 ‘불가능하다’는 답변이 72.0% (36/50), 85.2% (23/
27) 로 높게 나타났다(Table 8).
고 찰(Discussion)
본 연구는 현재의 품목갱신제출자료 및 평가의 적절성에 대 하여 유익성-위해성 관점에서 제약회사 및 관련 종사자를 대상 으로 의견을 조사하여, 운영 현황 파악 및 제도 본래의 목적과 취지를 달성하기 위하여 어떠한 개선점이 필요한지 분석해 보 고자 하였다. 또한, 유럽에서는 품목갱신시 안전성정기보고(Periodic Safety Update Report; PSUR) 자료와 비임상 및 임상에 관한 자 료를 취합하여 종합적인 검토서를 제출하도록 하고 있고,
15)미 국의 경우에도 연례보고서(Annual Report)를 1년 주기로 제출하 며 매번 비임상 연구, 임상시험, 소아에 대한 자료, FDA 요구 사항, 이행 약속 현황 등을 보고하게 함으로써 유익성-위해성을 평가하고 있는데 반해,
16)아직까지 국내 품목갱신자료에는 이러 한 평가의 내용이 포함되어 있지 않으므로, 유익성-위해성 평가 에 대한 도입 의견과 현황을 함께 파악할 필요가 있었다. 설문 조사 실시에 앞서, 품목갱신제도와 유익성-위해성 평가는 해외 에서는 이미 오래전부터 시행이 되고 있던 제도들인 만큼, 외국 계제약사들의 경우 많은 경험과 충분한 인프라를 기반으로 하 여 본 제도를 받아들이는데 대한 거부감이 국내제약사 대비하 여 훨씬 덜할 것으로 예상하고 조사를 시작하였다.
먼저, 국내의 제약업계 종사자들은 국내 제약회사 또는 외국 계 제약회사를 불문하고 현재의 품목갱신제도가 적절하다고 생 각하다는 비율이 그렇지 않다고 생각하는 비율보다 높았고, 유 익성-위해성 평가방법 도입 역시 불필요하다고 생각하는 사람의 비율이 높았다. 그러나 현재의 품목갱신제도로서 유익성-위해성 에 대한 평가가 가능한지에 대한 질문에서는 ‘그렇지 않다’고 Table 8. Correlation of the response between adequacy of submission data requirement for drug renewal and introduction of benefit- risk Assessment Method
Do you think that introduction of benefit-risk assessment method is necessary for drug renewal system?
n (%)
Is the current drug renewal system possible to fully assess the benefit-risk of the product?
n(%)
Do you think the current submission data requirement for drug renewal is adequate?
Yes (50)
Necessary 18(36.0)
p=0.2342
Possible 14(28.0)
p<0.001 Unnecessary 32(64.0) Impossible 36(72.0)
No (27)
Necessary 15(55.6) Possible 4(14.8)
Unnecessary 12(44.4) Impossible 23(85.2)
응답한 비율이 상대적으로 높았고(76.6%), 현재의 품목갱신 제 출자료가 ‘적절하다’라고 답변한 응답자 중에서도 현재의 자료 로서는 유익성-위해성 평가가 ‘불가능’하다고 답변한 비율이 72.0%로서 ‘가능’하다고 답변한 비율보다 더 높게 나타나, 앞의 응답에 대한 예측되는 답변과는 상반된 결과를 보였다. 이처럼 유익성-위해성 평가가 불가능함에도 불구하고, 현재의 제도가 적 절하고, 새로운 평가기법을 도입하지 않아도 된다고 답변을 한 이유는 응답자의 특성 및 업계의 현황을 통하여 유추해 볼 수 있으며, 주된 원인은 제도의 변화에 대한 두려움과 부담감에서 기인하는 것으로 판단된다. 응답자의 대부분이 실제 품목갱신과 직접적으로 관련이 있는 담당자로서 제도가 변화되거나 제출자 료가 늘어나는 것에 대해 현실적으로 많은 우려가 있는 것으로 생각되고, 이러한 사실은 실제 제약회사의 유익성-위해성 평가 를 위한 전문 인력이 거의 없다고 응답한 설문 결과를 통해서 도 이를 확인할 수 있었다.
품목갱신 제출자료에서 제일 먼저 개선되어야 할 항목으로 ‘외 국의 사용 현황과 안전성 관련 조치 부분’(59.3%)이 꼽혔고, 유 익성-위해성 평가 도입이 불필요하다고 생각하는 이유 중에서도
‘ 국내 제약 여건과 부합하지 않는다’(52.3%)는 답변이 가장 많 았는데, 이는 국산 신약 또는 개량신약의 비율이 약 2.5% 에 불 과하고(2017년 제약산업분석 보고서, 한국보건산업진흥원), 상대 적으로 의약품 개발 및 평가에 대한 전문가가 부족한 국내 제 약 현실에서 해외의 자료에만 의존해야 하는 기업들의 어려움 이 반영된 결과라고 보여지는 대목이다. 아울러, 유익성-위해성 평가방법 도입 시에도 연구사업을 통해 국내실정에 맞게 수정 된 방법으로 진행하는 것이 좋겠다는 의견은 식약처가 항상 염 두에 두고 있어야 할 부분으로 생각된다.
품목갱신에 있어서 응답자들은 ‘안전성’ 부분이 가장 중요하 다고 생각하고 있었고, 다음이 ‘유익성-위해성’이라고 답변하였 다. 유익성-위해성 평가가 안전성을 포함하는 보다 확장된 개념
17)인 것을 고려해 보면 아직까지 유익성-위해성 평가에 대한 충분 한 인식과 공감대가 형성되지 않은 것으로 생각된다. 아직까지 국내에는 FDA의 BRF 또는 EMA의 ET와 같은 평가기법이 생 소하고, 전문가가 부족하다는 답변 결과를 고려해 보더라도 제 도 도입에 앞서 가장 먼저 필요한 부분은 인프라 구축인 것으 로 판단되었다. 실제 해외에서도 유익성-위해성 평가방법에 대 한 연구가 국제의약품규제조화위원회(ICH)를 비롯해 미국과 유 럽에서 이미 2009년부터 진행되어 왔음에도 불구하고,
18~20)아직 까지 국제적으로 PhRMA BRAT, IMI-PROTECT 와 같은 민관 협의체를 통한 사례연구와 평가체계 개발이 지속적으로 진행되 는 것을 보면,
10),11),21~24)우리나라 역시 국내 상황에 맞는 제도 수 립과 인프라 구축에는 상당한 기간이 필요할 것임에는 명백하다.
아울러, 평가기법에 대한 인지도 조사 결과를 살펴보면, 2014 년에 실시되었던 설문조사
14)결과에서도 BRF 및 ET 에 대해 ‘들 어본 적 없다’라고 응답한 비율이 61.5, 84.6%였는데, 5년이 지 난 현재에도 크게 달라진 점이 없다는 점은 그 동안 식약처 및 제약업체에서 이 부분에 대해 얼마나 무관심했는지 다시 한번
되짚어 볼 필요가 있는 부분이다. 그동안 국내에서도 2010년 11 월에 “의약품 유익성/위험성 정량평가 방법”에 대한 보고서를 발간하였고,
25)2013년도에는 심혈관계 약물 중 3가지 Statins (Atorvastatin, Simvastatin, Cerivastatin),
26)2014 년도에는 근골격계 질환에 사용되는 3가지 Cox-2 inhibitors (Celecoxib, Etoricoxib, Rofecoxib),
27)그리고 2017년도에는 백신에 대한 유익성-위해성 평가연구
28)를 시행한 바 있지만, 이러한 연구들은 제약사의 참 여나 의견 수렴이 전혀 없이 몇몇 해당 품목에 한해서만 실시 된 방법론적 용역 연구였던 만큼 제약업계의 인지도 증대에는 한계가 있었다. 따라서, 향후의 연구에서는 업계 전반이 함께 참 여할 수 있는 방식으로 연구계획을 수립하는 것이 필요할 것이다.
한편, 유익성-위해성 평가 도입에 있어서는 국내의 제약업계 상황을 고려한 신중한 접근을 고려해 달라는 응답이 많았는데, 대부분 식약처의 협조와 교육지원을 바라는 내용들이었다. 도입 에 앞서 도입 타당성 연구와 평가방법 선정을 위한 연구를 선 행하고, 설명회 개최, 평가사례 발표, 가이드라인 등을 사전에 진행해 주도록 식약처에 요청하였다. 또한, 도입을 하더라도 신 약 및 바이오의약품에 한하여 우선 도입하기를 희망하였고, 공 정서 및 고시 해당품목, 제네릭의약품에 대해서는 유익성-위해 성 평가 도입이 불필요하다는 응답이 많아, 품목군 선정에서도 다시 한번 검토가 필요함을 제안하였다.
본 설문조사에서 품목갱신 제출자료의 적절성 및 유익성-위해 성 평가방법 도입에 대한 긍정과 부정 응답자 현황을 회사(국내 제약회사 또는 외국계 제약회사), 매출액 규모, 근무경력, 업무, 전공별로 분석해 보았으나 각 항목별로 차이를 확인할 수 없었 으며, 이를 통해 국내에 소재하고 영업을 하고 있는 제약관련 종사자들의 의견은 전반적으로 비슷한 것임을 알 수 있었다.
결과적으로, 본 설문조사를 통하여 국내 제약관련 업계 종사 자들은 제도 개선에 대한 필요성을 인식하고 있음에도 불구하 고, 전문인력의 부족과 제출자료 준비에 대한 부담감으로 인하 여 새로운 제도의 도입이나 변화를 받아들이는데 많은 어려움 을 느끼고 있으며, 이러한 생각은 외국계제약사와 국내제약사의 차이가 없음을 확인할 수 있었다. 따라서, 제도의 성공적인 정 착을 위해서는 업계의 인식개선이 동반되어야 할 것으로 판단 되고, 규제기관 역시 업계의 이러한 어려움을 이해하고, 전문가 육성 등의 지원과 노력이 충분히 이루어져야 할 것으로 사료된다.
본 연구는 품목갱신제도의 본격적인 시행 이후에 실시된 최 초의 설문조사로서 제출자료의 적절성과 현행 평가의 한계점을 되짚어 보고, 이와 더불어 새로운 평가기법 도입에 대한 업계의 의견을 조사하여 복합적으로 평가하였다는 점에서 의미가 있으 며, 향후 품목갱신제도의 개선 및 유익성-위해성 평가 도입을 위 한 기초자료로서 활용될 수 있을 것으로 사료된다.
그럼에도 불구하고, 본 연구의 한계점으로는 첫째, 조사 대상
업체를 각 조건별로 사전 지정하지 않고 협회 또는 교육센터 등
의 회원사가 보유하고 있는 정보만을 기반으로 진행함에 따라
설문대상자와 응답자의 대표성에 한계가 있을 수 있다는 점이
다. 설문조사의 특성상 응답 역시 개인의 자발적 회신에 의존함
에 따라 응답률 및 총 응답자 수에 한계가 있었고, 이로 인해 국내 제약회사 및 외국계 제약회사에 대한 응답자수의 불균형 이 발생하였다. 특히 이메일을 통한 응답자의 수가 적었던 외국 계 제약회사에 대해서는 추가적으로 직접 대면을 통한 설문을 진행하였으나, 여전히 국내 제약회사 응답자 대비하여 상대적으 로 응답자수가 부족하였다. 이러한 대표성의 한계로 인해 본 연 구의 결과를 일반화하기에는 다소 어려움이 있으므로, 해당 응 답자의 범위로 한정하여 연구 결과를 해석함이 바람직할 것으 로 사료된다. 둘째, 본 연구는 국내 제약단체의 의견이 반영되 지 않은 업계 종사자 개인별 설문조사로 진행된 만큼, 각 단체 의 공식 의견 및 다수에 대한 설문조사는 별도 연구로 이루어 져야 할 것이다.
결 론(Conclusion)
본 연구를 통하여 제약업계에서도 유익성-위해성 평가가 품목 갱신제도에 필요하다는 것을 인식하고 있으며 다만, 그 평가방 법의 어려움과 인력부족으로 인해 도입을 꺼려한다는 것을 확 인할 수 있었다. 따라서 품목갱신제도의 당초 역할을 위해 식품 의약품안전처 또는 관련부처는 유익성-위해성 평가 도입에 대한 업계와의 공동 연구를 통해 국내 제약 실정에 적합한 가이드라 인 개발과 전문가 양성 등을 선행하여야 할 것이며, 이를 통해 업계에서도 충분히 새로운 제도를 도입할 수 있도록 기반을 구 축할 필요가 있다.
Conflict of Interest
본 연구와 관련하여 모든 저자는 이해상충을 가지고 있지 않 음을 알려드립니다.
References