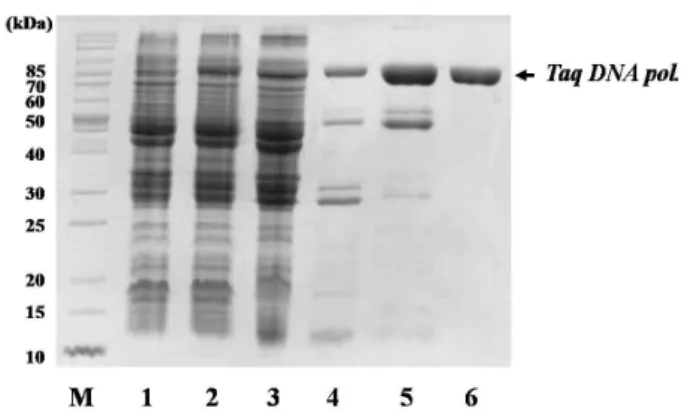
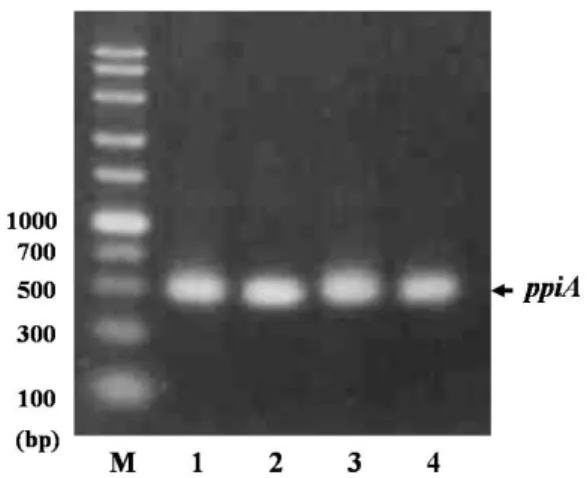
Production of DNA polymerase from Thermus aquaticus in recombinant Escherichia coli
Sung-Gun Kim
1, Jong-Tae Park
2*
1
Department of Biomedical Science, Youngdong University, Chungbuk 370-701, Korea
2
전체 글
1
2
수치

관련 문서
Measurements items between the maxillary second premolar and the first molar: A, The occlusal plane and reference lines which were above 4, 6, 8 mm from cementoenamel junction
In addition, Wallace and Froum 20) reported that the bone formation rate was superior in the cases using a barrier membrane in comparison with the
We determined the nucleotide sequences of the mitochondrial DNA (mtDNA) control region using cloning and sequencing, and obtained the complete sequence from the cattle bones
The results show that the green seed extracts are only shown as antioxidants, but the others are shown as a prooxidants against DNA damage and Escherichia coli
KAERI tested FSW between 10 mm thick CSC Cu coating and normal cu plate to get a sealing method of a disposal canister. Before the FSW test, Bead-On test was done on the
The desalted crude is then sent to another series of heat exchangers (6 10) for heating to 427 o F then sent to exchangers (6-10) for heating to 427 o F, then, sent
tricuspidata on the production of proinflammatory cytokines in TNFα+IFNγ-stimulated HaCaT cells ...15 Fig.5: The cell viability of sub-fractions from 70% EtOH
Blunt ends DNA containing restriction enzyme
