Vol. 19, No. 2, August, 2011
□ 원 저 □Prader-Willi 증후군과 Angelman 증후군의 임상양상 및 유전형에 대한 고찰
울산대학교 의과대학 서울아산병원 소아청소년병원 소아청소년과학교실
*경희대학교 의과대학 소아청소년과학교실
†, 상계백병원 뇌경련센터 소아청소년과
‡김주현*ㆍ염미선*ㆍ최해원*ㆍ이은혜*, †ㆍ유수정‡ㆍ고태성*ㆍ유한욱*
= Abstract =
Clinical and Genetic Characteristics of Prader-Willi Syndrome and Angelman Syndrome
Ju-Hyun Kim, M.D.
*, Mi-Sun Yum, M.D.
*, Hae-Won Choi, M.D.
*Eun-Hye Lee, M.D.
*, †, Su Jeong You, M.D.
‡, Tae-Sung Ko, M.D.
*and Han-Wook Yoo, M.D.
*Department of Pediatrics*, Asan Medical Center, University of Ulsan College of Medicine Department of Pediatrics†, College of Medicine, Kyung Hee University
Department of Pediatrics and Epilepsy Center‡ Sanggye Paik Hospital, Inje University College of Medicine
Purpose : Two different disorders, Prader-Willi syndrome (PWS) and Angelman syndrome (AS) are caused by the deletion of 15q11-13 or the maternal/paternal uniparental disomy of chromosome 15 (mUPD(15)/pUPD(15)) through the genomic imprinting phenomenon. We studied the clinical manifestations of both diseases and genotype-phenotype correlations in PWS.
Methods : We retrospectively analyzed medical records of patients who had been genetically confirmed as PWS or AS from December 1998 to March 2010 at Asan Medical Center. Clinical characteristics at diagnosis and genetic causes were re- viewed. In PWS, clinical characteristics of the patients with microdeletions were compared with those with mUPD(15).
Results : During the study period, we found 90 patients with PWS and 30 with AS.
In cases of PWS, the male to female ratio was 1.65:1 and the mean age at initial diagnosis was 41 months. Symptoms at first diagnosis were hypotonia (70 cases) and developmental delay (66 cases). More hypopigmentation and eye abnormalities occurred in the microdeletion group (n=62) than in the mUPD(15) group (n=21).
In AS, the male to female ratio was 1.3:1 and the mean age at initial diagnosis was 23 months. Distinguishing symptoms were speech impairment, seizure, and behavioral uniqueness. Microdeletion by FISH was detected in 19 patients among 20 patients and one of the non-deletion patient showed pUPD(15) on a DNA methylation test.
Conclusion : PWS and AS, two distinct neurogenetic disorders with different clinical presentations were the first known examples of human diseases involving imprinted genes. This study about clinical characteristics and genetic analysis of PWS and AS may help our understanding of these diseases and thus, assist in making correct diagnoses.
1)
접수 : 2011년 6월 29일, 수정 : 2011년 8월 5일, 승인 : 2011년 8월 6일 책임저자 : 고태성, 서울아산병원 소아청소년병원 소아청소년과
Tel : 02)3010-3386, Fax : 02)473-3725, E-mail : [email protected]
Key Words : Prader-Willi syndrome, Angelman syndrome, Clinical characteristics, Genetic causes, FISH, DNA methylation test
서 론
Prader-Willi 증후군(PWS)과 Angelman 증후 군(AS)은 15번 염색체의 동일 부위(15q11-q13) 에 이상을 보이면서 임상적으로는 구분이 되는 질환 이다. 이 염색체의 특정 영역이 부계와 모계로부터 각각 하나씩 유래하지 않고 어느 한쪽 유전자의 발 현만이 나타나는 것이 그 병인으로 알려져 있다1).
PWS은 Prader 등에 의해 처음 보고된 질환2)으 로 그 임상양상이 매우 다양하다. 발달지연, 근력 저 하, 정신 지체 등의 신경학적 이상과 섭식과다에 따 른 비만, 저신장, 성선기능저하 등의 내분비계의 이 상, 작은 손과 발, 수면장애, 눈의 이상, 통증에 대한 역치 증가 등이 흔한 증상이다1, 3, 4). PWS 환자의 유전학적인 이상은 미세결실, 어머니로부터의 이체 성(mUPD(15))이 대부분을 차지하고 드물게 각인 센터(imprinting center)의 돌연변이가 그 원인이 된다5). 또한 이러한 유전형에 따른 표현형의 차이가 존재한다고 알려져 있는데, 저색소증은 미세결실형 에서 더 흔한 반면에 산모의 고연령 및 조산아, 출생 당시 저체중은 mUPD(15)형에서 더 흔하다6-9).
AS는 PWS 보다 늦은 1965년 Angelman 의해 처음 기술되었으며10) 발달지연, 언어 장애, 돌발적 인 웃음이나 행복감, 주의력 결핍과 같은 행동 장애 와 소두증, 경련이 잘 동반된다고 알려져 있다1, 3, 11). 특히 PWS과 비교해 EEG의 이상과 경련이 흔하다
12, 13). 환자의 약 70-75%는 미세결실에 의해 발생
하며, UBE3A의 점 돌연변이나 아버지로부터의 이 체성(pUPD(15))이 그 원인이 된다11, 14). 이전 연구 들에서 AS 역시 유전형에 따른 표현형의 차이가 존 재하며, 미세결실형에서 발달 및 신경학적 예후가 불량하다고 알려져 있다15-17).
PWS와 AS 모두 비특이적인 증상들로 인하여 진 단이 되지 않거나 정신지체, 심한 비만이나 조절되 지 않는 경련으로 내원하여 뒤늦게 진단이 되는 경
우가 대부분이었다. 그러나 최근에는 분자세포유전 학의 발달로 조기 진단이 가능해지면서 매년 진단되 는 환자 수가 증가하는 추세에 있다. 이에 본 연구에 서 저자들은 1998년 12월부터 2010년 3월까지 서 울아산병원 소아청소년과 및 의학유전학 클리닉에서 PWS과 AS으로 확진이 된 환아들의 의무기록을 토 대로 임상양상과 유전형을 분석하였다. 또한 진단 시 연령에 따른 표현형의 차이를 관찰하고, 유전형 에 따른 표현형의 차이가 있는지 여부를 조사하였다.
저자들은 본 연구 결과를 통하여 PWS와 AS 환자들 의 조기 진단과 치료에 도움이 되고자 하였다.
대상 및 방법
1. 대 상
본 연구는 기관윤리심의위원회의 허가를 받아 시 행되었다. 1998년 12월부터 2010년 3월까지 울산 의대 서울아산병원 소아청소년과에서 PWS과 AS으 로 진단받은 환자들을 대상으로 후향적 병록지 기록 조회를 통해 진단시의 연령, 성별, 임상양상과 분자 세포유전학적 소견에 대해 분석하였다.
2. 방 법 1) 임상양상
연구 기간 동안 PWS과 AS으로 진단된 환자들의 증상에 대해 분석하였고, 분자세포유전학적 검사의 시행 결과를 조사하였다. PWS 환자들에 대한 임상 적 진단은 Holm 등에 의해 제시된 scoring system 에 따랐다18). AS 환자들에 대한 임상적 진단은 Williams 등에 의해 2005년에 수정된 내용에 근거 하였다19).
2) 유전학적 진단
말초 혈액에서 림프구를 채취하고 phytohemag- glutinin으로 배양시켜 세포 분열 중기의 염색체를 얻은 후 G-banding을 이용하여 염색체를 분석하였
Table 1. Clinical Manifestations in Subjects with PWS and AS at Initial Diagnosis
PWS AS
Clinical manifestations
No. of patients
(%)
Average age (yr)
Clinical manifestations
No. of patients
(%)
Average age (yr) Neonata and infantile
hypotonia
Developmental delay
Feeding problem in infancy Obesity
Hypogonadism
Hypopigmentation
Short stature
Eye abnormalities (esotropia, myopia)
70 (77.8)
66 (73.63)
52 (57.7)
51 (56.6)
41 (45.5)
34 (37.7)
30 (33.3)
23 (25.5)
0.7 (0.1-13)
1.9 (0.1-15)
0.6 (0.1-15)
6.7 (0.8-20)
4.3 (0.6-13)
5.4 (0.1-11)
6.8 (0.6-14)
3.3 (0.8-12)
Developmental delay
Speech impairment
Hypopigmentation
Seizure
Behavioral uniqueness
Microcephaly
Strabismus
Scoliosis
29 (96.7)
22 (73.3)
16 (53.3)
15 (50.0)
15 (50.0)
12 (40.0)
9 (30.0)
3 (10.0)
1.2 (0.6-7.5)
1.7 (0.6-7.5)
4.6 (0.6-7.5)
2.5 (0.6-7.3)
5.3 (1.1-7.5)
1.7 (0.6-7.5)
2.2 (0.8-3.4)
3.1 (0.6-7.3) Abbreviations : PWS, Prader-Willi syndrome; AS, Angelman syndrome; yr, years
다. 다음으로 15q11-13 영역에 있는 small nuclear ribonucleoprotein polypeptide N (SNRPN)을 소 식자로 하여 fluorescence in situ hybridization (FISH) 방법으로 미세결실의 유무를 확인하였다.
이 때, 미세결실이 발견되지 않은 환아에 대해서는 SNPRN 유전자의 5’에 모계와 부계가 서로 다르게 메틸화되어 있는 CpG island에 대해 메틸화 특이 PCR을 이용해 maternal uniparental disomy (mUPD) (15), paternal uniparental disomy (pUPD)(15)형 을 진단하였다.
3) 통 계
본 연구는 분석프로그램인 SPSS 12.0 (SPSS, an IBM Company, USA)을 사용하여 통계분석을 하였 다. PWS에서 유전형에 따른 표현형의 차이 비교에 는 독립표본 T 검정을 이용하였다. P값은 0.05 미 만(
P
-value<0.05)인 경우 통계적으로 유의성이 있 는 것으로 판단하였다.결 과
1. Prader-Willi 증후군 1) 연령 및 성별 분포
연구기간 동안 PWS으로 진단이 된 환아는 총 90 명이었다. 남아는 56명, 여아가 34명으로 남녀 비는 1.65:1로 조사되었다. 진단 당시의 연령은 생후 1개 월에서 20세로 평균 41개월이었다.
2) 내원 시 증상
첫 내원 시 증상은 신생아기와 영아기 저긴장증이 70례(77.7%)로 가장 많았으며, 발달지연으로 내원 해 진단된 경우가 66례(73.3%)로 뒤를 이었다. 만 3세 미만의 어린 연령에서는 수유곤란, 저긴장증, 발 달지연을 주소로 내원해 진단되는 경우가 많았고, 연령이 증가하면서 성선기능저하, 저신장, 비만, 행 동장애 및 수면장애 등이 상대적으로 높은 빈도로 나타났다(Table 1).
Table 2. Genetic Test Abnormalities in Subjects with PWS and AS
PWS AS
Test methods No. of patients (%) Test methods No. of patients (%) FISH (deletion)
UPD study (mUPD) Unclassified
62/83 (74.6) 21/83 (25.3)
7
FISH (deletion) UPD study (pUPD) Unclassified
19/20 (95.0) 1/20 ( 5.0)
10
Abbreviations : FISH, fluorescence in situ hybridization; mUPD, maternal uniparental disomy; pUPD, parental uniparental disomy
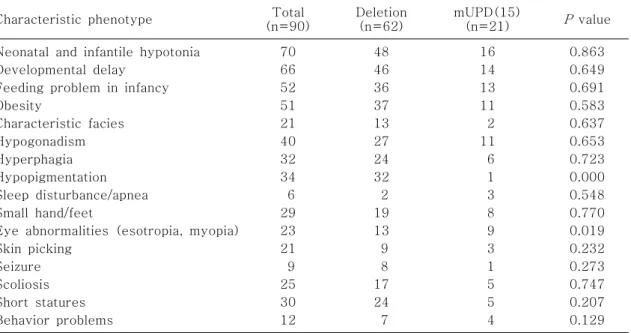
Table 3. Comparative Phenotypes in PWS Patients with or Without Deletions
Characteristic phenotype Total
(n=90)
Deletion (n=62)
mUPD(15)
(n=21) P value Neonatal and infantile hypotonia
Developmental delay Feeding problem in infancy Obesity
Characteristic facies Hypogonadism Hyperphagia Hypopigmentation Sleep disturbance/apnea Small hand/feet
Eye abnormalities (esotropia, myopia) Skin picking
Seizure Scoliosis Short statures Behavior problems
70 66 52 51 21 40 32 34 6 29 23 21 9 25 30 12
48 46 36 37 13 27 24 32 2 19 13 9 8 17 24 7
16 14 13 11 2 11 6 1 3 8 9 3 1 5 5 4
0.863 0.649 0.691 0.583 0.637 0.653 0.723 0.000 0.548 0.770 0.019 0.232 0.273 0.747 0.207 0.129 3) 분자세포유전학적 검사
대상 환아 90명 중 FISH 검사는 83명에서 시행 되었으며 15q11-13 부위의 미세결실이 확인된 경 우는 62명(74.6%)이었다. FISH 검사 결과상 미세 결실이 확인되지 않은 21명(25.3%)은 DNA 메틸화 분석 검사에서 모두 비정상 메틸화 양상, 즉 mUPD (15)을 확인할 수 있었다. DNA 메틸화 분석 검사만 을 시행한 환자는 7명이었고, 이 경우에는 유전적인 원인이 미세결실에 의한 것인지 mUPD(15)에 의한 것인지 여부를 알 수 없었다(Table 2).
4) 유전형에 따른 임상적 특징 비교
FISH 검사와 DNA 메틸화 검사를 모두 시행하여 유전적인 원인을 확인할 수 있었던 83명의 환아를
대상으로 미세결실형과 mUPD(15)형으로 나누어 두 군의 임상양상을 비교하였다. 저색소증과 근시, 내사시와 같은 눈의 이상은 미세결실형에서 통계적 으로도 유의(
P
-value<0.05)하게 높은 빈도를 나타 냈으나, 이외의 임상 증상은 두 군 사이에 유의한 차 이가 없었다(Table 3).2. Angelman 증후군 1) 연령 및 성별 분포
연구기간 동안 AS으로 진단이 된 환아는 총 30명 이었다. 남아가 17명, 여아가 13명으로 남녀 비는 1.3:1로 조사되었다. 진단 당시의 연령은 생후 7개 월에서 7세 6개월로 평균 23개월이었다.
2) 내원 시 증상
대부분 내원 시 발달지연(29례, 96.7%)을 보였 으며, 특히 언어 장애(22례, 73.3%)가 두드러졌다.
피부 및 체모의 탈색소화가 16례(53.3%), 경련이 15례(50.0%)에서 나타났고 과다웃음, 과다행동과 같은 특징적인 행동양상이 15례(50.0%)에서 동반 되었다(Table 1).
3) 분자세포유전학적 분석
총 30명의 환자 중 FISH 검사는 20명에서 시행 되었고, 미세결실이 확인된 경우는 19명(95.0%)이 었다. FISH 검사상 미세결실이 없었던 1명(5.0%) 은 메틸화 분석검사상 비정상 메틸화 양상, 즉 pUPD (15)이 확인되었다(Table 2). 8명의 환아는 FISH 검사를 시행하지 않았고, 메틸화 분석검사만이 시행 되었기 때문에 유전학적 원인이 미세결실에 의한 것 인지 pUPD(15)에 의한 것인지 여부를 확인할 수 없었다. 2명의 환아들은 타원에서 진단 후 본원으로 전원이 되어 유전자 검사 결과가 누락되어 있었다.
4) 유전형에 따른 임상적 특징 비교
FISH 검사와 메틸화 분석이 모두 시행이 된 증례 가 적어서 유전형에 대한 자료가 불충분했기 때문에 유전형에 따른 임상 양상의 차이를 분석할 수 없었 다.
고 찰
PWS은 1956년 Prader 등에 의해 발표되어2) 연 간 1/15,000-1/30,000 발생 빈도를 나타낸다4). 유전학적 진단을 시행해야 하는 환자들을 선별할 목 적과 이 증후군에 대한 경험이 부족한 임상가들에게 정보를 줄 목적으로 임상적 진단 기준의 필요성이 제기되었으나, 임상양상이 다양하고 비특이적이며 연령에 따른 임상양상의 변이성 때문에 진단 기준을 마련하는데 어려움이 있었다20). 이후 임상 보고가 축적되면서 1993년 Holm 등이 113명의 PWS 환자 들을 대상으로 한 연구를 통해 임상 진단 기준을 제 안하였고 이후 수정을 거쳐 사용하게 되었다18).
PWS 환자들은 태아기에 약한 태동, 출생 시 저체 중, 영유아기에 저긴장증 및 수유곤란이 나타나고
나이가 들면서 섭식 과다, 비만 등이 나타나 연령에 따른 임상양상의 차이가 존재한다는 점이 특징적이 다. 연령에 따른 임상양상의 변화는 임상진단 기준 에서 3세 이하와 그 이상을 구분하는 근거가 된다3,
4, 18). 이외에도 작은 손과 발, 특징적인 얼굴모양(긴
얼굴, 좁은 미간, 아몬드 모양의 눈, 얇은 윗입술, 작 은 입, 쳐진 입꼬리)과 공격적 행동, 강박적 충동적 행동과 같은 행동 문제를 보이기도 한다3, 18, 21). 저 자들의 연구에서도 이전 연구들과 유사한 결과를 나 타냈다. PWS으로 진단된 총 90명의 환자들 중 저긴 장증이 70례(77.8%)로 가장 높은 빈도를 보였고, 이어 발달지연이 66례(73.3%)로 많았다. 이는 진단 시기와도 관련이 있을 수 있는데 각각의 임상양상과 발현 시기를 분석했을 때, 3세 미만에서는 수유곤란, 저긴장증, 발달지연이 흔한 증상이었으며, 연령이 증 가하면서 내사시 또는 근시와 같은 눈의 이상, 성선 기능저하, 비만 등이 두드러지게 나타났다. 특히나 섭 식 과다에 따른 조기 소아비만은 당뇨, 고혈압, 뇌혈 관 질환 및 수면장애 등의 합병증을 초래하기 때문에 이환률과 사망률을 증가시키는 주요한 원인이라는 점에서 중요성을 갖는다22). 본 연구에서는 PWS 으로 진단된 환아 중 51명(56.6%)에서 비만이 동반되었 다. 이 중 1례에서는 외래 추적관찰 실패 후, 식이조 절을 중단하면서 급격한 체중의 증가로 인하여 고도 비만(BMI>40)에 의한 폐쇄성 무호흡이 발생해 기 계호흡치료 중 합병증으로 사망했던 사례도 있었다.
이에 비만에 대한 보다 적극적인 치료와 추적 관찰 이 필요하며 조기에 행동요법, 약물치료, 수술적 치 료가 모두 고려되어야 할 것으로 생각된다23, 24).
임상적으로 PWS이 의심되는 경우에 분자세포유 전학적 검사를 시행하여 확진한다. PWS의 전형적인 증상을 보이는 환자의 약 75-80%에서 15번 염색 체 장완 근위부(15q11-13)의 미세결실, 약 20%는 어머니로부터의 이체성(mUPD), 약 5% 미만에서는 각인센터의 돌연변이가 원인이 되어 발생한다고 알 려져 있다4, 5). 미세결실이 원인인 경우에는 FISH 검사만으로 확진이 가능한데, 본 연구에 포함된 PWS 환아 90명중 83명에서 FISH 검사가 시행되 었다. 이중 62명(74.6%)에서 결실이 확인되었으며,
결실이 없었던 21명(25.3%)의 환아들은 DNA 메틸 화 분석 결과 모두 비정상 메틸화 양상(maternal methylation)을 확인함으로써 PWS으로 확진할 수 있었다(Table 2). 분자세포유전학적 검사가 보편적 으로 시행된 이후에는 미세결실형과 mUPD(15)형 에서 유전형과 표현형 사이의 상관 관계를 밝히려는 연구들이 시도된 바 있다. 지금까지 알려진 것에 의 하면 저색소증은 미세결실형에서 더 높은 빈도로 나 타나며, 산모의 고연령과 조산아, 출생시 저체중은 mUPD(15)형에서 더 높은 빈도를 보이지만 이외의 임상양상은 뚜렷한 차이가 없다6-9). 본 연구에서도 미세결실군과 mUPD(15)군으로 나누어 유전형에 따른 임상양상의 차이가 있는지 여부를 분석하였다.
이전 연구에서와 같이 미세결실형에서 저색소증이 더 높은 빈도로 나타났고, 본 연구에서는 특징적으 로 눈의 이상(근시, 내사시) 또한 통계적으로 의미 (
P
value<0.05) 있게 더 많은 빈도로 나타났다.Maria 등26)의 연구에서와 같이 두 군의 표현형의 차 이는 중추신경계와 연관된 지표에서 나타나는 것으 로 생각된다.
AS의 유병률은 1/10,000-1/20,000 정도로 PWS 보다 약간 더 높다1, 11). AS으로 진단된 거의 모든 환아에서 심한 발달지연을 보이고 실조성 보행과 같 은 운동장애 혹은 균형 장애, 특징적인 행동 양상이 나타난다고 알려져 있다. 이외에도 언어 장애, 특이 한 얼굴모양과 소두증, 경련, 돌발적인 웃음, 과다행 동 등이 동반된다3, 11). 특히 PWS과 비교했을 때 경 련이 흔한데, 3세 이전에 환자의 85%에서 조절하기 힘든 심한 경련이 동반되며 간질이 진단되기 이전에 종종 열성 경련이 선행한다12). 저자들의 조사에서는 전체 환자 30명 중 29명(96.6%)에서 발달지연이 관찰되었고, 22명(73.3%)에서 언어 장애가 나타났 다. 알려진 것보다 언어 장애의 빈도가 상대적으로 적은 것은 언어 발달의 지연 여부를 판단하기 힘든 연령인 15개월 미만의 환아들이 환자군에 포함되어 있었기 때문으로 생각된다. 또한 본 연구에 포함된 PWS 환자 중 9례(10%)에서만 경련을 보인 반면에 AS에서는 15례(50%)에서 경련이 동반되어 발달지 연, 언어 장애, 탈색소화 다음으로 흔한 증상임을 알
수 있었다. 그러나 다른 연구들에서 보다 본 연구에 포함된 AS 환자군에서 경련의 빈도가 상대적으로 낮은 것은 경련이 발현되기 전 환아들의 장기적인 추적 관찰 실패에 기인하는 것으로 생각된다.
AS의 유전학적 이상은 15번 염색체 장완 부위의 결실(15q11-13)이 약 70-75%, UBE3A의 점 돌 연변이가 5-11%, 아버지로부터의 이체성(pUPD) 이 3-7%를 차지한다5, 11). 본 연구에 포함된 30명 의 환자 중 20명에서 FISH 검사가 시행되었고, 미 세결실이 확인된 경우는 19명(95.0%)이었다. 미세 결실이 없었던 1명(5.0%)의 환아는 메틸화 분석검 사상 비정상 메틸화 양상, 즉 pUPD(15)이 확인되 었다. 8명의 환아는 FISH 검사는 시행되지 않았고, 메틸화 분석검사만이 시행되어 명확한 유전학적 원 인을 확인할 수 없었다. AS에서도 유전형에 따른 표 현형의 차이를 보이는지 여부를 밝히려는 연구들이 시도된바 있으나16, 17) 본 연구에서는 유전형에 대한 검사 결과가 불충분하여 분석에 제한이 있었다.
또한 알려진 바에 의하면 PWS 보다 AS의 빈도 가 높은데, 실제 본 연구에 포함된 환자의 경우 PWS 으로 진단된 환자가 더 많았다. 이는 AS 환자들이 주로 정신발달지연 등으로 내원하기 때문에 제대로 된 유전학적 진단을 받지 못했을 가능성을 고려해 볼 수 있겠고, 또한 AS의 경우 UBE3A의 점 돌연변 이가 전체 유전학적 원인의 5-11% 정도로 알려져
있는데5, 11) 본 연구에 포함된 환자들에서는 이에 대
한 검사를 시행하지 않았기 때문에 상당수의 환자가 대상에서 제외되었을 가능성을 생각해 볼 수 있겠다.
저자들은 1998년 12월부터 2010년 3월까지 울 산의대 서울아산병원 소아청소년과에서 PWS 또는 AS으로 진단된 환아 120명을 대상으로 병록지 조 회를 통해 후향적인 방법으로 임상양상과, 유전형에 대한 분석을 시행하였다. 두 질환의 임상양상에 관 한 국내 연구가 발표된 바 있으나20, 21) 본 연구는 국 내에서 발표된 PWS과 AS에 관한 연구 가운데 가장 많은 수의 환자를 대상으로 하였으며 각각의 임상양 상을 비교하고 유전형과 표현형의 차이를 분석했다 는 점에서 의의가 있을 것으로 생각된다. 살펴본 바 와 같이 PWS과 AS 환아들에서 임상 소견이 다른
질환들과 뚜렷이 구분되지 않고 비특이적인 증상25) 이 많아 조기 진단에 어려움이 있다. 유전학적 검사 기법이 발전하고 있으나, 다양한 유전 방식과 비용 문제로 인하여 실제 임상에서 이를 모두 적용해 시 행하는 데에는 제한이 있다. 따라서 자세한 병력 청 취 및 신체 검진을 통해서 이를 의심해 보는 것이 무 엇보다 중요하다고 할 수 있겠다. 특히 저긴장증과 수유곤란(PWS) 또는 언어 장애와 함께 심한 발달 지연(AS)이 있는 경우에는 두 질환을 한번쯤 의심 해야 할 필요가 있다고 생각된다. 환자들의 정확한 임상양상을 숙지함으로써 이를 의심하고 FISH 검사 나 메틸화 분석검사를 통해 조기에 진단을 한다면 치료적 접근 및 2차적 예방 역시 조기에 이루질 수 있을 것으로 기대된다.
요 약
목 적 : Prader-Willi 증후군(PWS)과 Angelman 증후군(AS)은 동일한 15번 염색체의 장완 근위부 (15q11-13)의 미세결실이 주원인이나 서로 구분 되는 질환이다. 저자들은 단일병원에서 진단된 PWS 과 AS의 임상양상 및 분자세포유전학적 검사 결과 에 대해 비교, 분석함으로써 향후 두 질환의 조기 진 단과 치료에 도움이 되고자 하였다.
방 법 : 1998년 12월부터 2010년 3월까지 울산 의대 서울아산병원 소아청소년과에서 PWS과 AS으 로 진단을 받은 각각 90명, 30명의 환자들을 대상으 로 의무기록 검토를 통해 후향적인 연구를 시행하였 다. PWS과 AS에서 흔히 동반되는 것으로 알려진 임상양상을 분석하였고, 환자군에서 시행된 분자세 포유전학적 검사 결과(FISH, DNA methylation test)를 조사하였다.
결 과 : PWS의 경우 연령에 따라 발현하는 증상 에 차이를 보이는데 만 3세 미만의 환아들에서는 저 긴장증, 발달지연과 같은 비특이적인 증상을, 3세 이 후에는 식욕 증가에 따른 비만, 작은 손과 발, 저신 장, 특징적 얼굴 모양 등이 두드러졌다. FISH 검사 상 62명(74.6%)의 환자에서는 미세결실이 확인되 었고, 21명(25.3%)에서는 mUPD(15)가 그 원인이
었다. 미세결실형에서 저색소증과 눈의 이상이 더 흔히 동반되었으나, 이외의 임상양상은 차이가 없었 다. AS은 대부분의 환자에서 발달지연과 언어 장애 가 동반되었으며, PWS 에서보다 경련이 동반되는 경우가 많았다. FISH 검사상 미세결실이 확인된 환 아는 19명(95%)이었으며, 미세결실이 없었던 1명 의 환아에서 pUPD(15)가 확인되었다.
결 론 : PWS과 AS의 특징적인 임상양상을 숙지 함으로써 의심되는 환자에서 필요한 유전학적 검사 의 시행을 통해 조기 진단 및 치료적 접근이 가능할 것으로 기대된다.
References
1) Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet 2010;154C:365-76.
2) Prader A LA, Willi H. Ein Syndrome von Adipo- sitas, Kleinwuchs, Kryptorchismus und Oligoph- renie nach myatonieartigem Zustand im Neuge- borenenalter. Schweiz Med Wochenschr 1956;
86:1260-61.
3) Ramsden SC, Clayton-Smith J, Birch R, Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet 2010;11:70.
4) Cassidy SB, Driscoll DJ. Prader-Willi syndrome.
Eur J Hum Genet 2009;17:3-13.
5) Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and An- gelman syndromes. Annu Rev Genomics Hum Genet 2001;2:153-75.
6) Cassidy SB, Forsythe M, Heeger S, Nicholls RD, Schork N, Benn P, et al. Comparison of pheno- type between patients with Prader-Willi syn- drome due to deletion 15q and uniparental di- somy 15. Am J Med Genet 1997;68:433-40.
7) Mitchell J, Schinzel A, Langlois S, Gillessen- Kaesbach G, Schuffenhauer S, Michaelis R, et al. Comparison of phenotype in uniparental di- somy and deletion Prader-Willi syndrome: sex specific differences. Am J Med Genet 1996;65:
133-136.
8) Gillessen-Kaesbach G, Robinson W, Lohmann D, Kaya-Westerloh S, Passarge E, Horsthemke
B. Genotype-phenotype correlation in a series of 167 deletion and non-deletion patients with Prader-Willi syndrome. Hum Genet 1995;96:
638-43.
9) Torrado M, Araoz V, Baialardo E, Abraldes K, Mazza C, Krochik G, et al. Clinical-etiologic correlation in children with Prader-Willi syndrome (PWS): an interdisciplinary study. Am J Med Genet A 2007;143:460-8.
10) H. A. "Puppet" children : A report on three cases. Dev Med Child Neurol 1965;7:681-3.
11) Guerrini R, Carrozzo R, Rinaldi R, Bonanni P.
Angelman syndrome: etiology, clinical features, diagnosis, and management of symptoms. Pae- diatr Drugs 2003;5:647-61.
12) Williams CA. Neurological aspects of the An- gelman syndrome. Brain Dev 2005;27:88-94.
13) Fiumara A, Pittala A, Cocuzza M, Sorge G. Epi- lepsy in patients with Angelman syndrome. Ital J Pediatr 2010;36:31.
14) Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med 2010;12:385-95.
15) Hou JW, Wang PJ, Wang TR. Angelman syn- drome assessed by neurological and molecular cytogenetic investigations. Pediatr Neurol 1997;
16:17-22.
16) Gentile JK, Tan WH, Horowitz LT, Bacino CA, Skinner SA, Barbieri-Welge R, et al. A neuro- developmental survey of Angelman syndrome with genotype-phenotype correlations. J Dev Behav Pediatr 2010;31:592-601.
17) Lossie AC, Whitney MM, Amidon D, Dong HJ, CHen P, Theriaque D, et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet 2001;38:834-45.
18) Holm VA CS, Butler MG, Hanchett JM, Green- swag LR, Whitman BY. Prader-Willi syndrome:
consensus diagnostic criteria. Pediatrics 1993;
91:398-402.
19) Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, et al. An- gelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A 2006;
140:413-8.
20) Kim DS, Lim YL, Ko TS, Seo EJ, Yoo HW. A study of Prader-Willi Syndrome and Angelman Syndrome with the Deletion of Same Loci in 15 Chromosome. J Korean Child Neurology Soc 2002;10:226-34.
21) Lee JE, Moon KB, Hwang JH, Kwon EK, Kim SH, Kim JW, et al. Clinical characteristics and genetic analysis of Prader-Willi syndrome. J Korean Pedicatrics Soc 2002;45:1126-33.
22) Curfs LM FJ. Prader-Willi syndrome: a review with special attention to the cognitive and be- havioral profile. Birth Defects Orig Artic Ser 1992;28:99-104.
23) Carvalho DF, Cercato C, Almeida MQ, Mancini MC, Halpern A. Therapeutical approach of obe- sity in Prader-Willi Syndrome. Arq Bras Endo- crinol Metabol 2007;51:913-9.
24) Butler MG. Management of obesity in Prader- Willi syndrome. Nat Clin Pract Endocrinol Metab 2006;2:592-3.
25) Williams CA, Lossie A, Driscoll D. Angelman syndrome: mimicking conditions and phenotypes.
Am J Med Genet 2001;101:59-64.
26) Torrado M, Araoz V, Baialardo E, Abraldes K, Mazza C, Krochik G, et al. Clinical-Etiologic Correlation in Children with Prader-Willi Syn- drome. Am J Med Genet 2007;143:460-8.