저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
이학 박사학위 논문
다중 오믹스 데이터를 이용한 암 및
노화의 특성 분석
다중 오믹스 데이터를 이용한 암 및
노화의 특성 분석
지도교수 박 래 웅
이 논문을 의학 박사학위 논문으로 제출함.
2015년 8월
아 주 대 학 교 대 학 원
의학과/의학전공
감사의 글
대학원에 입학 한지도 어언 5 년이 지나갑니다. 이렇게 박사 학위 논문의 감사의 글을 쓰고 있자니 그간 일어난 많은 일들이 주마등처럼 스쳐지나 갑니다. 통계학과를 졸업하고 마냥 의학 통계학을 하고 싶단 생각으로 대학원에 진학하여, 석, 박사 통합과정으로 의료정보학과에 입학 하였습니다. 딱 맞아 떨어지는 전공은 아니었지만, 생물학 및 몇 가지 컴퓨터공학 학부 수업을 청강하며, 의료정보학의 기초지식을 쌓을 수 있었고, 생물정보학에 입문 하게 되면서 많은 것을 배우고 흥미를 느낄 수 있었습니다. 생물정보학의 생자도 모르던 제가 이렇게 졸업할 수 있기까지 많은 분들의 도움이 있었습니다. 먼저, 의료정보학과장님이시며 마지막엔 지도교수님으로서 저희가 어려운 상황에 처했을 때에 많은 도움을 주신 박래웅 교수님 감사합니다. 그리고 졸업하기 까지 많은 공동연구를 함께 진행하면서 제 지식과 견문을 넓혀주신 교수님들께 감사 드립니다. 연구실에 들어오면서 많은 조언을 해 주시고, 도움을 주신 혜진 언니, 만영 오빠, 우재 오빠 감사합니다. 힘들 때마다 항상 도와주시고, 많은 조언 아끼지 않는, 지금은 같은 방을 쓰면서 가장 오랜 시간 같이 지내는 은경 언니, 덕용 오빠 감사합니다. 그리고 연구실을 이동하게 되면서 많은 도움을 주고 더 가깝게 지내게 된 수연 언니, 다혜 고맙습니다. 처음 대학원 연구실을 접했을때, 잘 적응하도록 도와주고 함께 동고동락하고 힘든 시기 함께 이겨낸 효실 언니, 우진 오빠, 원표 오빠, 백효정 박사님을 비롯한 지금까지 거쳐간 연구원 선생님들께도 감사 드립니다. 그리고 힘든 시기 들어와서 아무 말 못하고 묵묵히 자기 자리 지켜준 정현이와 호준이, 많이 친해지지는 못했지만 여러 가지 많이 도와주신 임상섭 박사님, 고송 박사님 감사합니다. 시간이지나도 그때의 시절을 잊지 못할 것 같습니다. 그리고 연구실 선배로서, 친구로서 5 년동안 가족보다 더 많은 시간 함께 지내고 힘들때나 즐거울때나 항상 옆에서 투닥거린 경아.. 너가 있어서 대학원 생활이 덕분에 즐거울 수 있었고, 슬픔은 반이 될 수 있었던 것 같다. 내 생각해 주고 챙겨줘서 항상 고맙게 생각하고, 평생 지금처럼 잘 지내자. 그리고, 멀리 떨어져있지만 항상 응원해 주고 내 편이 되어주는 분들께 감사 합니다. 그리고.. 교수님, 이기영 교수님.. 교수님과의 첫 만남이 생각납니다. 삼촌 같은 인자한 웃음으로 뭐든 네가 원하는 연구를 하게 해주겠다고 호언장담 하시던 교수님.. 그 웃음에 넘어가 별 생각 없이 덜컥 대학원을 진학하게 되었습니다. 하지만 다시 생각해보면, 교수님 덕분에 제가 아주대학교 의료정보학과에 들어올 수 있었고, 교수님 덕분에 생물정보학에 흥미를 가질 수 있게 되었고, 교수님 덕분에 많이 부족한 제가 이렇게 졸업을 할 수 있게 되었습니다. 아직 교수님의 가르침을 더 받아야 되는데…,받고 싶은데.. 교수님의 부재가 이렇게 클 줄 몰랐습니다. 아직까지 교수님이 안 계신다는 게 믿기지가 않고, 곧 연구 안 하냐고 잔소리 하시며 나타나실 것 같은데.. 교수님과 더 많은 추억을 쌓지 못한 것이 아쉽습니다. 이렇게 빨리 우리 곁을 떠나신 교수님이 한편으로는 원망스럽기도 하지만 교수님 가르침, 말씀, 항상 마음에 새기고 잘 살아 나가겠습니다. 교수님은 정말 제 인생 최고의 스승이셨습니다. 제 능력을 과대평가하시고, 저를 믿어주셔서 감사했습니다. 언젠간 만나게 된다면 술도 마시고 배드민턴도 치고 등산도 하면 좋겠습니다. 마지막으로, 이기적인 딸래미 뒷바라지 하신다고 항상 고생하시는 엄마, 아빠.. 감사하고 사랑합니다. 많이 표현은 못하지만 부모님께 자랑스러운 딸이 되겠습니다. 그리고 하나 밖에 없는 내 동생, 힘든 시간
- 국문요약 -
다중 오믹스 데이터를 이용한 암 및 노화의 특성 분석
노화는 암을 포함한 많은 질병의 원인이 된다. 인간의 다양한 조직으로부터 추출한 DNA 메틸값을 이용해 실제 나이를 예측할 수 있다. 이는 DNA 메틸화가 노화의 지표가 될 수 있다는 의미이기도 하다. 하지만, 여러 조직의 정상과 종양 샘플의 많은 데이터 셋을 통합하여 노화 관련 DNA 메틸화 영역의 특징에 대해 밝힌 연구는 아직 많이 없는 실정이다. 본 연구에서는 16 개 독립적인 연구의 DNA 메틸화 및 유전자 발현 데이터를 통합하여 말초 혈액뿐만 아니라 유방, 자궁경부, 전립선, 뇌, 간, 대장 조직 등 다양한 조직의 대략 4,000 여개의 정상과 종양 샘플의 DNA 메틸화 영역에 대해 비교, 분석 하고 이들 영역의 유전자 발현과의 관계를 고찰 하였다. 이때, 선형과 비선형 통합 회귀 모형을 사용하여 조직 비 특이적으로 나이에 따라 변화하는 DNA 메틸화 영역을 노화 관련 DNA 메틸화 시그너처라고 명명하였다. 정상 조직에서 확인 한 127 개의 시그너처는 X 염색체에 많이 분포 되어 있었다. 특히, CpG islands (CGI) 영역에서는 구아닌(G) 과 시토신(C) 염기가 많이 분포된 나이에 따른 과 메틸화 영역이 많았고, CGI 를 제외한 non-CGI 영역은 아데닌(A) 과 티민(T) 염기가 많이 분포된 나이에 따른 저 메틸화 영역이 많았다. 반면에, 종양 샘플에서 26 개의 시그너처를 추출하였고, 정상 시그너처와의 겹치는 영역은 발견되지 않았다. 정상 시그너처에 존재하는 유전자의 생물학적과정에 대한 온톨로지를 살펴 보면, 대사 과정, 면역 체계 과정, 세포 증식 등으로 기존에 잘 알려진 노화와 관련된 기능을 하는 유전자가 많았다. 또한, 기존에 알려진 단백질 상호작용 정보를 이용하여 네트워크를 구축하고, 이 단백질 상호작용 네트워크 하에서, 정상 시그너처 영역에 존재 하는 단백질간의 상호작용은 거의 존재하지 않았지만 다른 단백질과는 평균보다 더 많이 상호작용 하는 것을 통계적으로 확인 할 수 있었다. 정상 시그너처의 DNA 염기 서열은 진화적 관점에서 잘 보존 되고 있고, 이들 시그너처를 이용하여 조직에 관계없이 나이를 예측 하는 회귀 모델을 구축하였다. 나이 예측 모델의 정확도는 상관계수(r)가 0.9 로 매우 높았다. 흥미롭게도 나이에 따른 DNA 메틸화의 증가 혹은 감소는 종양 샘플의 DNA 메틸값을 따라 가는 패턴을 확인하였다. 또한, 나이에 따른 DNA 메틸화와 유전자 발현과의 관계를 확인 하기 위해서 유전자 발현 프로파일을 이용해 정상 시그너처의 노화 관련 영역의 유전자의 발현 정도를 확인 하였다. 본 연구에서 제시한 정상과 종양 샘플에서 조직 비 특이적인 DNA 메틸화 시그너처의 특징은 발달 생물학이나 종양 연구를 하는데 있어 기저 연구로서 중요한 역할을 할 수 있을 것이다. 핵심어: DNA 메틸화, 유전자 발현, 후성 유전학, 노화 시그너처, 노화, 메타 분석, 시스템 생물학, 종양
차 례
국문요약 ··· ⅰ 차례 ··· ⅲ 그림차례 ··· ⅴ 표차례 ··· ⅶ 부록차례 ··· ⅷ Ⅰ. 서론 ··· 1 A. 연구 배경 ··· 1 1. 노화와 종양 ··· 1 2. DNA 메틸화 ··· 3 3. DNA 메틸화와 노화의 관계 ··· 5 B. 연구 목적 ··· 7 Ⅱ. 연구대상 및 방법 ··· 9 A. DNA 메틸화 프로파일 및 데이터 전처리 ··· 9 B. 노화 관련 시그너처 분석 ··· 11 C. 각 데이터 셋 별 순서 무작위 및 선택 분석 ··· 12 D. 나이 예측 모델 생성 ··· 13 E. 염색체 분포 분석 ··· 14 F. DNA 염기 서열 모티프 분석 ··· 15 G. 단백질 네트워크 및 DNA 염기서열 보존 분석 ··· 16Ⅲ. 결과 ··· 17 A. 노화와 관련된 DNA 메틸화 시그너처의 발견 ··· 17 B. 정상 조직에서의 CpG 영역과 나이의 관계 ··· 26 C. 정상 및 종양조직에서의 노화 관련 DNA 메틸화 시그너처 ··· 34 D. 조직 비 특이적인 노화 관련 DNA 메틸화 시그너처의 특징 ··· 53 E. 종양 조직에서의 노화 관련 DNA 메틸화 시그너처의 붕괴 ··· 66 F. DNA 메틸화와 mRNA 발현과의 관계 ··· 70 G. 노화 관련 DNA 메틸화 시그너처의 단백질 상호작용 네트워크와 DNA 염기서열 보존 분석 ··· 73 Ⅳ. 고찰 ··· 79 Ⅴ. 결론 ··· 83 참고문헌 ··· 84 ABSTRACT ··· 104
그림 차례
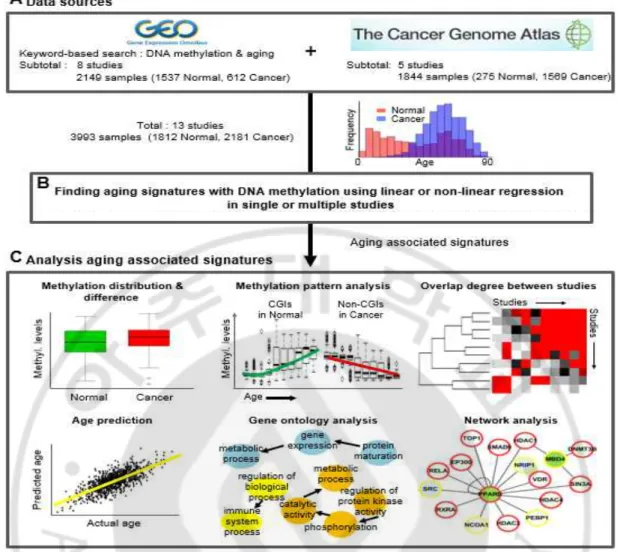
Fig. 1. Study overview ··· 19
Fig. 2. DNA methylation correlation between normal and cancer samples ··· 22
Fig. 3. DNA methylation patterns between normal and cancer samples according to genomic regions ··· 23
Fig. 4. DNA methylation correlation and age-associated CpG loci across individual studies ··· 28
Fig. 5. Relation with the number of age-associated loci and sample information ··· 29
Fig. 6. Comparison of age-associated CpG loci across different studies with different tissue types ··· 32
Fig. 7. Example of age-associated DNA methylation signature independent of tissue type ··· 35
Fig. 8. Age-associated DNA methylation signature independent of tissue type ··· 37
Fig. 9. Manhattan plots of age-associated CpG signatures by chromosome ··· 39
Fig. 10. Manhattan plots of age-associated CpG loci in age-matched samples by chromosome ··· 43
Fig. 11. Fractions of age-associated CpG loci according to gender ··· 46
Fig. 12. Characteristics of age-associated DNA methylation signature ··· 49
Fig. 13. Tissue-type-specific age-associated CpG loci ··· 52
in age-associated signatures according to genomic regions ··· 56
Fig. 15. The hyper- or hypomethylated age-associated DNA loci patterns from
normal or cancer samples according to age groups in CGIs or non-CGIs ··· 57
Fig. 16. Nucleotide composition with surrounding sequences
of age-associated DNAm signatures ··· 59
Fig. 17. Nucleotide composition with surrounding sequences
of hypo- or hyper age-associated CpG loci ··· 61
Fig. 18. Disruption of age-associated DNA methylation signature in cancer ··· 68
Fig. 19. The correlation between expression value of age-associated genes and age ·· 72
Fig. 20. The human protein-protein interaction network of
age-associated DNA methylation signature in normal ··· 74
Fig. 21. Network characteristics of age-associated DNA methylation signature
in normal ··· 75
Fig. 22. DNA sequence conservation of
표 차례
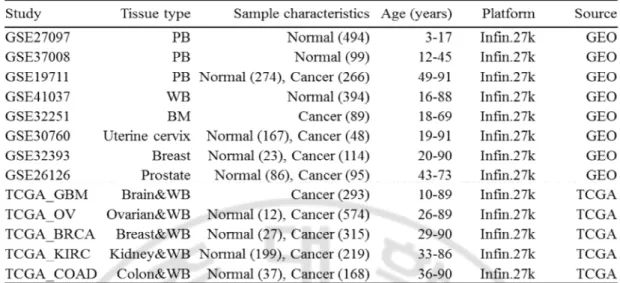
Table 1. DNA methylation data sets used in this study ··· 20
Table 2. Characteristics of other age-associated DNA methylation studies ··· 50
Table 3. Numbers of loci in the age-associated DNAm signatures
using the integrated data set ··· 54
Table 4. Top functional annotation clusters of significant differentially aging genes
in normal··· 63
Table 5. Top functional annotation clusters of significant differentially aging genes
in cancer ··· 65
Table 6. mRNA expression data sets used in this study ··· 70
Table 7. Top functional annotation clusters of significant differentially aging genes
부록 차례
I. 서 론
A. 연구 배경
1. 노화와 종양
노화는 나이가 들어가는 과정이다. 한 인간에서 시간의 흐름에 따른 변화의 축적이라고 할 수 있다(Bowen and Atwood, 2004). 특히 인간에서 노화는 육체적, 정신적, 사회적 변화 등 다양한 변화를 가져온다. 예를 들어, 육체적으로는 나이가 들수록 근육 축소로 인해 근력이 약화되고, 청력 손실이나 시각 손상을 동반한다. 또한, 노화는 종양, 신경계 관련 질환, 당뇨, 대사질환 등과 같은 인간 질병에 대한 위험 요소 중 가장 큰 이유 중의 하나 이다(Dillin et al., 2014). 크게 100,000 여명 이상이 노화와 관련된 질병에 의해 매일 사망하고 있다. 많은 연구자들이 노화 현상을 이해하고 풀기 위한 다양한 연구를 진행 하고 있고(Ahuja et al., 1998; Issa, 1999), 이러한 연구들을 통한 노화 시스템은 노화 현상을 정의하고 이해하는데 큰 도움을 주는 중요한 자료가 된다. 종양 발생에 대한 연구는 오랜 세월 진행 되어왔다. 여러 연구 결과, 종양은 젊은 사람에서 보다 나이든 사람에서 더 흔하게 나타나는 질병이라는 연구 결과들이 발표 되어 왔다(Svetlana V.ukrainteva, 2003). 종양 위험 요소와 나이의 관계에 대해 연구하는 대부분의 연구자들은 나이가 들어감에 따른 종양 발병률의 증가는 노화가 종양의 중요한 원인이라고 말한다(Issa, 1999). 종양 발생 기원의 원인이 무엇인가에 대한 의문에 많은 연구자들은 유전학적 영향이 우선인지 후성 유전학 영향이 우선인지에 대해 의견이 분분하다(Jones and Baylin,
2007). 특히, 후성 유전학 메커니즘으로서 DNA 메틸화 변화에 의한 유전자 제어는 노화뿐만 아니라 종양에서도 나타나는 특징이다(Holliday, 1987). 일찍이 시작된 종양 세포의 메틸화 연구와 함께 노화 세포 및 조직을 이용한 DNA 메틸화 변화 연구는 전 게놈 영역에서 특정 유전자의 저 메틸화(Hypo-methylation)가 일어난다고 밝혔다(Mays-Hoopes, 1989). 이러한 노화와 종양조직의 후성 유전학적 관계를 이용하여, 본 연구에서는 특히 DNA 메틸화 관점에서 노화와 종양에 관련된 메커니즘과 현상에 대해서 살펴 볼 것이다.
2. DNA 메틸화
후성 유전학이란, DNA 염기서열의 변화 없이 일어나는 유전자 발현을 제어하는 등의 기능 변화를 연구하는 학문이다(Portela and Esteller, 2010). 후성 유전의 변화는 크게 DNA 메틸화(DNA methylation), 히스톤 변형(Histone modification), 염색질 변형(Chromatin remodeling) 등 3 개의 카테고리로 나눌 수
있다. 이 중 DNA 메틸화는 시토신 염기가 DNA 메틸 전달효소에 의해 5 번 위치 탄소에 메틸기가 결합되어 유지되는 것을 말한다. 이러한 변형은 유전자 발현에 영향을 준다(Gal-Yam et al., 2008). 포유류 게놈에서 이 변화는 구아닌 앞에 있는 시토신(CpG dinucleotides)에서 거의 독점적으로 일어난다(Bird, 2002). 전체 게놈에서 CpG 영역은 거의 1-2% 정도를 차지한다(Zampieri et al., 2015). 그리고 CpG 영역은 일반적으로 대략 60-90%가 메틸화 되어있다(Johnson et al., 2012). 또한, CpG 영역이 매우 밀집된 게놈 영역이 있는데 이를 CpG islands
라고 한다. CpG islands 는 반대로 DNA 메틸화가 거의 일어나지 않는다(Bock and Lengauer, 2008). CpG islands 는 적어도 200 개의 염기 쌍 이상에서 GC 함량이
적어도 50% 이상 나타나는 영역이다(Portela and Esteller, 2010). CGI (CpG islands) 영역의 대략 60%는 유전자 프로모터 영역과 관련이 있고, 이 들 영역에는 거의 메틸화가 되어있지 않아 유전자 전사 기능을 가능케 한다(Suzuki and Bird, 2008; Jones, 2012). 반대로 유전자내의 CpG 영역은 거의 메틸화 된 상태로
존재하여 유전자 전사에 영향을 주지 않는다(Maunakea et al., 2010). 나머지 40%의 CGI 영역은 주로 유전자와 유전자 사이에 위치한다.
종양과 같은 인간 질병에 있어서 이러한 DNA 메틸화는 단일 유전자 뿐 만 아니라 전 게놈영역에 걸쳐서 중요한 역할을 한다(Heyn and Esteller, 2012). 예를 들어, 정상 세포와 비교해서 종양 세포는 전반적으로 DNA 메틸화가 손실된다(Portela and Esteller, 2010). 동시에 특정 유전자 프로모터의 CGI 영역은 DNA 과 메틸화(Hyper-methylation) 양상을 보인다.
또한, 유전자 발현 정도와 DNA 메틸화는 유전적 변이와 관계 없이 높은 관련이 있다. 일반적으로, CGI 영역의 DNA 메틸화는 유전자 발현 억제에 기여하거나 원인이 된다고 알려져 있다(Portela and Esteller, 2010). DNA 메틸화는 다양한 메커니즘에 의해서 유전자 발현을 막을 수 있다. 메틸화 된 DNA 는 메틸화 된 CpG 영역에 특이적으로 붙는 단백질(methyl-CpG-binding domain protein)을 불러온다. 이 단백질은 메틸화 된 영역에 히스톤 변형(Histone
modification) 이나 염색질 재구조화(Chromatin remodeling) 에 영향을 미쳐
3. DNA 메틸화와 노화의 관계
노화는 인간에 따라 획일적이지 않고, 주로 환경이나 생활습관에 따라 다르게 나타난다(Zampieri et al., 2015). 인간의 노화에 영향을 주는 유전적 변화는 약 20-30%에 불과하고, 주로 비 유전적인 요소에 의해 변화한다(Herskind et al., 1996; Hirner and Rettenmeier, 2010). 이는 나이와 관련된 질병이 후성 유전학
변화에 의한 것일 수도 있다는 것을 의미한다(Huidobro et al., 2013). 종양 세포에서 CGI 영역의 DNA 메틸화의 변화가 많이 연구 되어 지고 있고(Hinshelwood and Clark, 2008), 대장암 세포에서 특정 유전자의 DNA 메틸화를 연구하는 과정에서 대장의 정상 세포에서도 이상 DNA 메틸화를 발견하였다(Issa et al., 1994). 이와 같이, 후성 유전학적인 변화는 직접적으로 노화와 관계가 있고,
나이가 들어감에 따라 변화한다(Vlaming and van Leeuwen, 2012; Huidobro et al., 2013). 후성 유전학적 변화 중, 정상 세포의 DNA 메틸화 변화는 노화에 따라
많은 영역에서 일어난다. 최근 연구들에 따르면, DNA 메틸화는 효모에서부터 인간에 이르기까지 많은 개체에서 노화와 관련이 있다고 알려져 왔다(Wilson et al., 1987; Longo and Kennedy, 2006; Johnson et al., 2012). 예를 들어, Horvath et al 의
논문에 따르면 DNA 메틸화가 게놈 전체적으로는 감소하지만, 특이적으로 CpG islands (CGI)에서 노화에 따라 과 메틸화 되는 양상을 보였다(Horvath et al., 2012).
이러한 양상은 종양 조직에서도 비슷한 패턴을 보이는 것을 알 수 있다. 몇 가지 다른 연구에서는 노화와 관련된 DNA 메틸화가 성별, 비만지수, 특정 조직이나 세포 등에 따라 달라진다고 알려져 있다(Bocklandt et al., 2011; Alisch et al., 2012; Numata et al., 2012; Hannum et al., 2013; Johansson et al., 2013). 위의 연구들은 노화와
DNA 메틸화의 관계를 설명하기 위해서 다양한 선형회귀분석 방법(Alisch et al.,
2012; Johansson et al., 2013), 가중상관네트워크 방법(Bocklandt et al., 2011; Hannum et
al., 2013), 다중차원크기비례분석 방법(Numata et al., 2012) 등을 이용하였다. 그러나,
대부분의 연구들이 특정 조직에 국한 되어있고, 제한된 샘플 수 혹은 샘플의 나이 범위가 제한 되어있다. 최근에, 몇 가지 연구에서 공개된 데이터 셋으로부터 다양한 조직에 대한 많은 샘플에서 노화와 관련된 더 신뢰할만한 CpG 영역을 찾았다(Christensen et al., 2009; Teschendorff et al., 2010; Day et al., 2013;
Horvath, 2013). 이 연구들은 주로 선형 기반 회귀 분석 방법을 이용했다. 기존에 알려진 연구에서 노화와 관련된 DNA 메틸화 변화양상이 선형뿐만 아니라 비 선형 패턴으로도 변화한다고 보고되었지만(Alisch et al., 2012), 실제로 비 선형 방법을 이용하여 노화에 따라 변화하는 DNA 메틸화 양상을 확인한 연구는 소수에 불과하다. 또한, 노화 관련 DNA 메틸화 시그너처(Signature)의 특징을 여러 조직의 정상 및 종양샘플에 적용한 연구가 많이 없는 실정이다.
B. 연구 목적
앞서 말한 바와 같이 기존의 노화와 DNA 메틸화와 관련된 연구가 많이 진행 되고 있고, 여러 조직이나 세포에 따른 노화 관련 DNA 메틸화 영역을 발견 하였다. 하지만 전 연령을 아우르는 노화와 DNA 메틸화 관계에 대한 연구는 많이 부족하고, 또한 조직에 관계 없이 일반적으로 정상 조직에서 변화하는 노화 관련 DNA 메틸화 영역에 대해서는 많은 연구가 진행되지 않았다. 정상 조직뿐만 아니라 종양 조직에서 노화에 따라 변화하는 DNA 메틸화 영역에 대한 보고 및 정상 조직의 영역과 비교한 자료가 거의 없는 실정이다. 따라서 본 연구의 목적은 정상과 종양 샘플에서 조직 비 특이적인 노화와 관련된 DNA 메틸화 시그너처를 찾기 위해 선형과 비 선형 회귀 모델을 이용한다. 또한, 신뢰 할 수 있는 노화 관련 CpG 영역을 찾기 위해 공개된 데이터베이스에서 정상뿐만 아니라 종양 샘플의 여러 가지 조직에 대한 다양한 연구 데이터 셋을 수집 하였다. 수집한 데이터 셋으로부터 전체 샘플에 대해서 정규화 한 DNA 메틸값을 추출하고, 이 값으로부터 노화 관련 DNA 메틸화 시그너처를 발굴한다. 발굴한 시그너처에 대해 기존에 알려진 결과와 비교함은 물론, 정상과 종양상태에서의 노화에 따른 DNA 메틸화 변화 양상, 유전자 온톨로지 분석, 단백질 네트워크 및 DNA 염기서열 보존 분석 등 다양한 분석 방법을 통해 노화 관련 DNA 메틸화 시그너처의 특징 및 생물학적 의미에 대해 고찰한다. 다음으로, 정상 조직에서의 특정 게놈 영역의 노화 관련 DNA 메틸화 시그너처의 DNA 메틸화 정도를 이용하여 나이 예측 변수로서의 역할을 할 수있는지 확인 하기 위해서 나이 예측 분석을 수행하여 다양한 조직에 비 특이적인 나이 예측 모델을 제안한다.
DNA 메틸화는 특정 영역에서 유전자 발현에 관여하여 유전자를 억제
하거나 발현 시킨다(Portela and Esteller, 2010). 이와 같은 현상을 확인 하기 위해, 본 연구에서는 공개된 데이터베이스에서 유전자 발현 프로파일 또한 수집 하고, 수집한 데이터 셋을 이용해 유전자 발현 정도가 노화에 따라 변화하는지 확인하고, 앞서 추출한 DNA 메틸화 시그너처의 영역과 그 영역에 놓인 유전자의 발현 정도를 비교 하고, 의미 있는 유전자들의 기능에 대한 추가 분석을 수행한다.
Ⅱ. 연구대상 및 방법
A. DNA 메틸화 프로파일 및 데이터 전처리
본 연구에서는 공개된 데이터베이스인 GEO (http://www.ncbi.nlm.nih.gov/geo/)와 TCGA (http://cancergenome.nih.gov/)에서 인간 정상 및 종양 조직에 대한 DNA 메틸화 프로파일과 mRNA 발현 프로파일 데이터를 수집 하였다. DNA 메틸화 분석 시 플랫폼 간의 기술적인 편차를 줄이기 위해서 Infinium HumanMethylation27 assay 로 제한하고, 각 데이터 셋의 샘플수가 10 개 미만인 데이터 셋이거나 종양이 아닌 다른 질병을 가진 샘플의 경우는 제외하였다. 결과적으로 13 개의 데이터 셋으로부터 1812 개의 정상 조직 샘플과 2181 개의 종양 조직 샘플을 수집 하였다(표 1). 데이터 셋 간에 DNA 메틸화 프로파일을 통합하고 비교하기 위해서 각 데이터 별 실험 어레이 결과의 메틸화(M) 신호 강도와 비 메틸화(U) 신호 강도를 추출하고, 각 데이터 셋 별로 정규화를 진행 하였다. 정규화 된 DNA 메틸값은 메틸화 신호 강도를 메틸화 신호 강도와 비 메틸화 신호강도의 합을 나눈 값으로[M/(M+U)] 0 에서 1 까지의 값을 가지게 되고, 0 에 가까울수록 메틸화가 일어나지 않는 것을 의미하고 1 에 가까울수록 완전히 메틸화 되었다는 것을 의미한다. 유전자 레벨의 메틸화 분석의 경우, 하나의 유전자 위에 놓인 CpG 영역들의 평균 DNA 메틸값으로 계산하였다. mRNA 발현 프로파일 데이터의 경우, 나이 정보를 가지고 있는 정상 조직의 데이터를 얻기 어려운 점이 있어 GEO 데이터베이스에서 각각 다른
조직의 3 개의 데이터 셋 만을 수집하여 분석을 수행 하였다. 각 데이터 셋 별로 발현량에 대한 정규화(Quntile normalization)를 수행 한 후, 유전자 레벨로 맵핑 하였다.
B. 노화 관련 시그너처 분석
본 연구에서는 노화에 따른 DNA 메틸화 변화에 대한 분석을 수행하기 위해서 회귀 모델을 이용하였다. 선형 회귀 분석을 이용하여, 각 데이터 셋에서 각각의 CpG 영역에 대한 나이에 따른 DNA 메틸화 변화에 대해 분석 하였다. 또한, 조직 비 특이적인 노화 관련 시그너처를 추출하기 위하여 2 차와 3 차의 다중 비 선형 회귀 모델을 이용 하였다. 모든 회귀 모델은 R 프로그램의 “lm” 기능을 이용하여 분석 수행 하였다. 또한 다중 비교를 위해서 R 프로그램의 “p.adjust” 기능을 이용하여 FDR(False discovery rate)값을 계산 하였다. 통합 된샘플에서 각각의 다른 샘플들과의 상관계수의 중간값이 0.5 미만일 경우는 샘플의 이상치 라고 간주하고 제외하였다. 이러한 방법은 유전자 레벨의 DNA 메틸화 분석에서도 똑같이 적용 하였다. 노화 관련 시그너처의 임계치는 FDR 과 상관계수, 회귀모형의 잔차 에러를 반영 하였다.
C. 각 데이터 셋 별 순서 무작위 및 선택 분석
모든 순서 무작위 및 선택 분석은 R 프로그램을 이용하였다. 본 분석은 실제로 찾은 노화 관련 영역들의 개수들이 무작위 추출에 비해서 의미 있는지를 확인하기 위하여 수행 하였다. 각 데이터 셋 별 CpG 영역에 대해 순서 무작위 유의 확률(p-value) 에 대한 계산은 각 데이터 셋 별로 나이를 순서 무작위로 치환하여 총 100 회의 회귀 분석을 수행 하였다. 순서 무작위 회귀 분석 결과, 통계적으로 의미 있는 CpG 영역의 개수에 대한 분포 하에서 실제 데이터 셋의 노화 관련 CpG 영역의 개수에 대해 Z – test 의 p-value 를 평가 한 값이다(그림 4C). 각 데이터 셋의 노화 관련 CpG 영역들의 겹치는 정도를 확인하기 위해서,본 연구에서 사용한 Infinium HumanMethylation27 assay 플랫폼의 모든 CpG 영역인 27,551 개 CpG 영역 중에서 각 데이터 셋에서 추출한 노화 관련 CpG 영역의 개수를 무작위로 선택 하였다. 이때, 겹치는 정도(Overlap degree)의 p-value 는 무작위로 선택 한 데이터 쌍 간의 겹치는 개수의 분포 하에서 실제로
D. 나이 예측 모델 생성
Genetic 알고리즘(Scrucca, 2013)은 사용하고자 하는 통계적 모델을 최적화 하기 위한 변수를 추출하는 알고리즘이다. 본 알고리즘은 생물 정보학, 진화 생물학, 컴퓨터 공학, 경제, 화학, 수학 등 많은 분야에서 사용하고 있다. 본 연구에서 발굴한 정상 시그너처 127 개 영역의 DNA 메틸값을 이용하여 실제 나이를 예측하는 모델을 생성하고자 genetic 알고리즘을 적용하였다. Genetic 알고리즘은 R 패키지의 “genalg”을 이용 하였고, genetic 알고리즘 결과로 추출 된 CpG 영역의 DNA 메틸값을 이용하여 다중 선형 회귀 모형을 갖는 나이 예측 모델을 생성 하였다. 그 결과로서 정상 노화 관련 127 개 CpG 시그너처 중 20 개의 CpG 영역을 추출 하였고, 이 최적화 셋을 이용하여 전통적인 선형 회귀 모형을 모델링 하였다. 이 때, 모델의 정확도는 실제 나이와 모델에 적용하여 추출 된 나이간의 상관계수로 평가 하였다. 또한, 뽑은 20 개의 CpG 영역의 개수가 무작위에 비해 의미 있는지를 확인하기 위해서 전체 27,551 CpG 영역 중에서 무작위로 20 개의 CpG 영역을 선택하여 선형 회귀 분석 시뮬레이션을 수행 하였다.E. 염색체 분포 분석
추출한 정상 및 종양 시그너처의 게놈상의 위치를 확인하기 위해서 염색체 분포에 따른 분석을 수행하였다. 이를 위해 NCBI Human Genome (build 36.1)과 HumanMethylation 27 플랫폼에서 제공하는 Illumina annotation 을 이용해서
각 CpG 영역의 염색체와 해당 위치를 확인 하였다. 염색체 분포의 시각화는 R 프로그램을 이용 하여 맨하튼 그림으로 나타내었고, 각 염색체마다 몇 개의 노화 관련 CpG 영역이 있고, 그 개수가 의미 있는 결과인지를 확인하기 위하여 통계 분석을 수행 하였다. 각 염색체 별로 초기하 분석(Hypergeometiric test)을 수행하여 나이에 따른 과 메틸화 혹은 저 메틸화 되는 영역의 개수에 대해 p-value 를 이용하여 통계적으로 의미 있게 분포되는 염색체 및 나이에 따른 패턴을 확인 하였다.
F. DNA 염기 서열 모티프 분석
노화와 관련 된 CpG 영역 주위의 DNA 염기 서열 모티프를 확인 하기 위해서, DNA 메틸화 실험 플랫폼에 존재하는 27,551 CpG 영역의 CpG 주위의 20 개 DNA 염기 서열에 대해서 확인 하였다. 이들의 염기 서열을 각각 하나의 포인트 염기 마다 어떤 염기가 많이 분포 되는지, 비율이 어느 정도 인지를 계산하기 위해서 전체 27,551 개 CpG 영역 주위의 20 개의 염기 서열을 무작위로 10,000 번 뽑았을 때의 염기 서열을 분포를 추출 하였다. 무작위로 뽑은 염기 서열의 분포 하에서 127 개의 정상 시그너처 주위의 DNA 염기 서열과 비교 분석 하였다. 염기 서열의 시각화는 R 패키지의 “seqLogo”를 이용하였다(그림 16, 17)(Schneider and Stephens, 1990).G. 단백질 네트워크 및 DNA 염기서열 보존 분석
인간 단백질-단백질 상호작용 네트워크를 생성하기 위해서, HPRD (Keshava Prasad et al., 2009), BioGRID (Chatr-Aryamontri et al., 2013), IntAct (Kerrien et
al., 2012), MINT (Licata et al., 2012), Reactome (Chelliah et al., 2013), iRefWeb(Turner et
al., 2010)등의 잘 알려진 공개된 데이터베이스에서 단백질 상호작용 정보를 추출 하여 통합 하였다. 통합 된 네트워크는 인간 단백질 14,216 개가 136,489 개의 상호작용을 한다. 정상 노화 관련 시그너처의 단백질 네트워크를 알기 위해, 127 개의 노화 관련 CpG 영역에 해당하는 단백질뿐만 아니라, 이들과 제일 가깝게 상호작용하는 단백질도 모두 추출 하여, 최종적으로 1,163 개의 단백질과 12,620 의 상호작용을 하는 확장 네트워크를 구성 하였다. 네트워크 시각화는
Cytoscape 프로그램을 이용 하였다(Lopes et al., 2010).
다음으로, 진화의 관점에서 정상 노화 시그너처의 DNA 염기서열의 보존 정도를 확인하기 위해서 UCSC Genome Browser(Meyer et al., 2013)의 진화 점수인 phyloP 점수를 이용 하였다. 전체 게놈 영역의 phyloP 점수의 분포를 정규 분포라 가정하고, 이 분포의 Z 값의 p-value 가 0.05 보다 낮은 값을 가지는 영역을 추출 하였다. 또한 잘 보존 되어 있다고 여겨지는 영역의 개수에 대한 통계적 의미를 확인하기 위하여, 전체 게놈 영역의 phyloP 점수 분포 하에서 무작위로 127 개의 점수를 뽑았을 때, 얼마나 많은 영역이 phyloP 점수가 높은지를 확인 하였다.
Ⅲ. 결 과
A. 노화와 관련된 DNA 메틸화 시그너처의 발견
노화와 관련된 DNA 메틸화 시그너처를 찾기 위해, 공개된 데이터 베이스인 Gene Expression Omnibus (GEO) 데이터베이스 (http://www.ncbi.nlm.nih.gov/geo/; 그림 1)에서 여러 가지 DNA 메틸화 프로파일 데이터를 검색하고, 다운로드 하였다. 다운 받은 데이터 셋 중에서 나이에 대한 정보가 없거나, 샘플수가 10 개 미만인 데이터 셋은 제외 하였고, 종양 데이터 셋을 제외한 다른 질병에 대한 데이터 셋 또한 제외 하였다. 이전의 연구들에서 다른 마이크로어레이 플랫폼 간의 기술적인 편차가 존재한다는 것이 알려져 있다(Bock, 2012). 그렇기 때문에 다운로드 한 프로파일 데이터 셋 중에 가장 많이 사용되어지는 Illumina Infinium HumanMethylation27 Bead Chip 어레이를 사용하였다. 결과적으로, GEO 데이터베이스의 8 개의 데이터 셋으로부터 정상조직이 1537 개, 종양조직 612 개인 총 2149 개의 샘플을 수집 하였고, 추가적으로, The Cancer Genome Atlas (TCGA) 컨소시움 데이터베이스로부터 이용 가능한 5 개의 종양 조직(유방암, 난소암, 뇌종양, 신장암, 대장암)에 대한 총 1844 개의 DNA 메틸화 프로파일을 수집 하였고, 이 중 정상조직의 개수는 275 개, 종양조직의 개수는 1569 개이다(Noushmehr et al., 2010; Cancer Genome Atlas Research, 2011; Cancer
Genome Atlas, 2012b, a; Cancer Genome Atlas Research, 2013). 최종적으로 1812 개의
사용하였다. 위 샘플들은 10 여개의 다양한 조직을 포함하고 나이의 범위는 0 - 91 세로 넓은 분포를 가진다(표 1).
Fig. 1. Study overview. (A) Sources of DNA methylation data with sample information.
Eight studies from GEO and five from open TCGA data were included. (B) Identifying an
age-associated DNA methylation signature. Linear and nonlinear regression models using
single or combined studies were applied. (C) Age prediction and characterization of
identified age-associated signatures. Various analyses using DNA methylation patterns and
distributions, gene ontology, and protein networks in normal and cancer tissues were
Table 1. DNA methylation data sets used in this study.
Infin.27k : Illumina HumanMethylation27 BeadChip (HumanMethylation27_270596_v.1.2) WB : Whole blood, PB : Peripheral whole blood, BM : Bone marrow
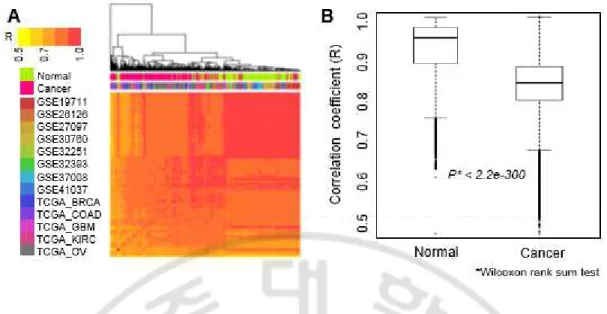
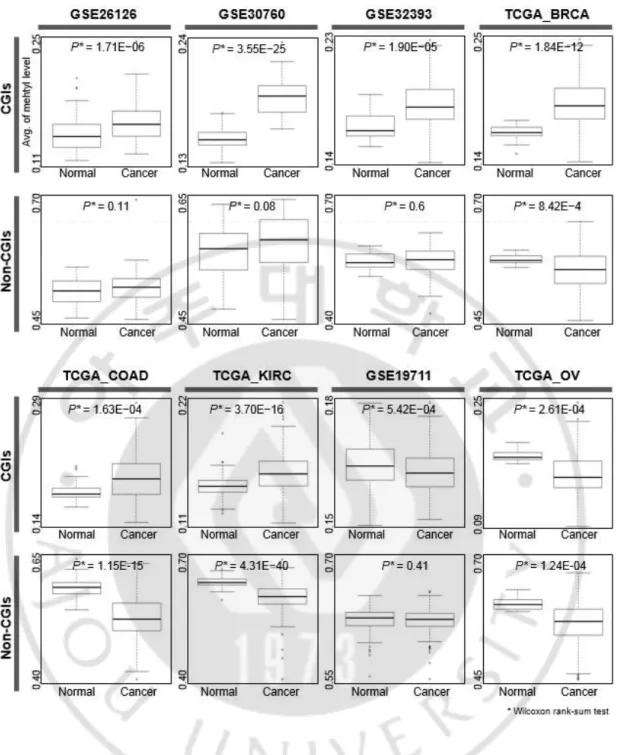
다음으로, DNA 메틸화 프로파일 데이터의 메틸값을 정규화 하기 위하여 메틸화 신호 강도를 메틸화 신호 강도와 비 메틸화 신호강도의 합을 나눈 베타값([M/(M+U)])으로 변환 하였고, 정규화 된 DNA 메틸값이 샘플 별로 얼마나 차이가 나는지 확인하기 위하여 샘플간 상관관계를 비교 하였다. 그 결과 상관계수(r)가 주로 0.6 이상으로 정상 샘플과 종양 샘플간에도 비교적 높은 상관관계를 가지고 있었으나, 오직 정상 샘플간의 상관계수가 주로 0.8 이상으로 상관관계가 더 높은 것을 확인 하였다(그림 2). 또한, 정상과 종양 조직 모두에서 각 샘플의 CGI 영역의 DNA 메틸값이 비 CGI 영역보다 더 낮은 것을 확인 하였다(그림 3). 게다가, 정상과 종양조직의 DNA 메틸값은 게놈 영역에 따라 다른 양상을 보이는 것을 확인 하였다. 예를 들어, GSE26126 데이터의 CGI 영역에서 평균 DNA 메틸값은 정상 조직에서 보다 종양 조직에서 보다 높았다. 이러한 패턴은 TCGA 데이터베이스의 난소암 샘플을 제외하고, 모든 데이터 셋에서 나타나는 현상인 것을 확인 하였다(그림 3).
Fig. 2. DNA methylation correlation between normal and cancer samples. (A) Heat map
representing the Pearson’s correlation coefficients between all samples of normal and cancer
tissues used in this study. A hierarchical clustering was used. (B) Box plot representing the
correlations between normal samples or between cancer samples. P-value was calculated
Fig. 3. DNA methylation patterns between normal and cancer samples according to
(Cancer) samples according to genomic regions (CGIs and non-CGIs) for individual studies.
몇 가지 선행 연구에서 DNA 메틸값은 나이에 따라서 비선형적으로 변할 수 있다는 보고가 있다(Alisch et al., 2012). 예를 들어, DNA 메틸화의 변화는 어린 나이에서 급격하게 변화하고 성인이 될수록 완만하게 변화한다. 이러한 현상을 반영하기 위해서, 본 연구에서는 선형 회귀 모델뿐만 아니라 비 선형 회귀 모델도 적용하여 정규화 된 DNA 메틸값과 관계 있는 노화 관련 시그너처를 추출 하였다. 13 개 각각의 데이터 셋과 통합한 데이터를 이용하여, 통계적으로 의미 있는 노화 관련 DNA 메틸화 영역을 찾은 후, 이 영역들에서의 나이와 DNA 메틸화 패턴의 관계, 각 영역에 위치하는 유전자 온톨로지 분석, DNA 염기서열 보존 분석, 단백질 네트워크 분석 등을 통해 시그너처의 특징과 생물학적 의미를 확인 하였다. 또한, 노화 관련 DNA 메틸화 영역에 대해 각각의 데이터 셋, 조직 타입간에도 비교, 분석 하였다. 조직 비 특이적인 노화 관련 DNA 메틸화 시그너처를 찾기 위해서, 이상치인 샘플을 제외하고, 각각 정상 조직과 종양 조직, 전체 샘플을 통합 하였다. 마지막으로, 정상 조직을 이용해 조직 타입에 관계 없는 노화 관련 DNA 메틸화 영역을 찾은 후, 메틸값을 이용하여 의미 있는 나이 예측 모델을 개발 하였다.
B. 정상 조직에서의 CpG 영역과 나이의 관계
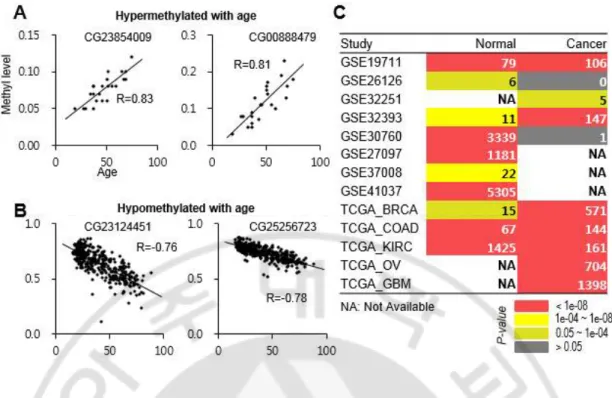
특정 게놈 영역에 따라 DNA 메틸값의 분포는 정상 조직과 종양 조직 간에 의미 있는 차이를 보인다. 먼저 각각의 정상 샘플과 종양 샘플을 가지고 선형 회귀 모델을 이용하여 노화 관련 DNA 메틸화 영역이 있는지 확인 하였다. 예를 들어, GEO 데이터베이스 아이디인 GSE32393 데이터 셋(Zhuang et al., 2012)의 정상 샘플의 경우, 19 번 염색체의 62802940 번째에 위치한 CG23854009
영역과 20 번 염색체의 19141824 번째에 위치한 CG00888479 영역은 나이와 DNA 메틸값의 관계인 상관계수(r; correlation coefficient)값이 각각 0.83 과 0.81 로 나이에 따라 선형적으로 과 메틸화(Hypermethylated) 되는 양상을 보였다(그림 4A). 반대로, GSE41037 데이터 셋(Horvath et al., 2012)의 정상 샘플의 경우, 1 번
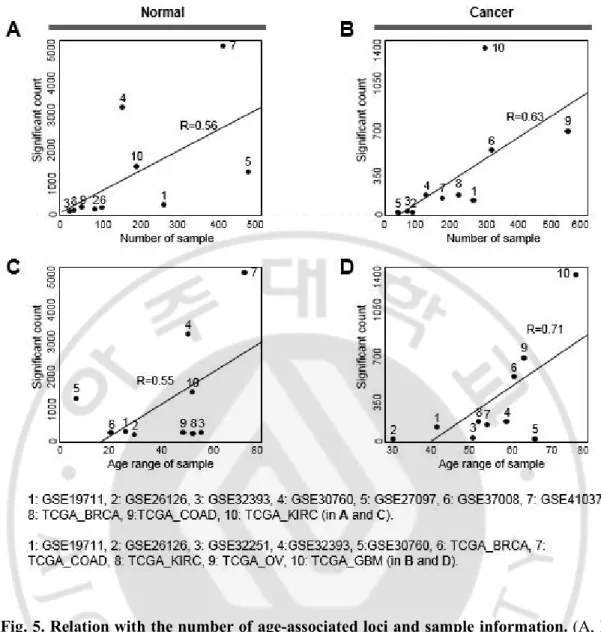
염색체의 167822568 번째에 위치한 CG167822568 영역과 20 번 염색체의 37878077 번째에 위치한 CG23124451 영역의 각각 r 값이 -0.78 과 -0.76 으로 나이에 따라 선형적으로 저 메틸화(Hypomethylated) 되는 양상을 확인 하였다(그림 4B). 다음으로, 각 데이터 셋 별로 통계적으로 유의한 노화 관련된 CpG 영역의 개수를 추출 하였다(선형 회귀 분석 결과, p-value < 0.0001). 유의한 CpG 영역의 개수는 데이터 셋 마다 약간씩 차이가 있었는데(그림 4C), 이는 주로 각 데이터 셋마다 샘플 수에 차이가 있거나 나이의 범위가 상이하기 때문이다(그림 5). 예를 들어, GSE41037 및 GSE30760 데이터 셋은 넓은 나이 분포와 많은 샘플
되었다. 추출한 노화 관련된 CpG 영역의 개수의 유의성을 확인하기 위하여 각 데이터 셋 별로 나이에 대한 순서 무작위 분석(Random permutation test)을 100 번씩 수행 하였다. 그 결과 정상 조직의 각 데이터 셋 마다 유의한 영역의
개수가 다양했지만, 사용한 모든 데이터 셋에서 무작위로 기대 했던 결과보다는 유의하게 더 많은 수의 노화관련 CpG 영역개수가 나온 것을 확인 하였다(Z test, p-value < 0.05; 그림 4C). 하지만, 2 개 데이터 셋의(GSE26126, GSE30760) 종양
조직 샘플은 노화 관련 유의한 CpG 영역이 각각 0 개와 1 개로 개수가 아주 작았고, 그렇기 때문에 무작위 분석에서도 통계적으로 의미 있지 않은 결과를 도출 하였다.
Fig. 4. DNA methylation correlation and age-associated CpG loci across individual
studies. (A) Examples of CpG loci (i.e., CG23854009 and CG0888479) hypermethylated
with age in the GSE32393 study. (B) Examples of CpG loci (i.e., CG23124451 and
CG25256723) hypomethylated with age in the GSE41037 study. R: correlation coefficient.
(C) The number of significant age-associated CpG loci in normal or cancer tissues from
individual studies. The number and color in each cell corresponds to the number of
significant age-associated CpG loci and its significance in terms of a P-value, respectively.
P-value: Z-test result using the random distribution of the 100 age-permutation tests. NA: not
Fig. 5. Relation with the number of age-associated loci and sample information. (A, B)
Each point is a study corresponding to the count of significant loci and the sample number in
normal (A) or cancer (B). (C, D) Significant counts and age ranges of samples in individual
studies in normal (C) or cancer (D). Many age-associated loci were observed in the
GSE41037 study because of the wide age range (16–88 years) and in the GSE30760 study
relatively small number of loci were identified as age-associated because of the narrow age
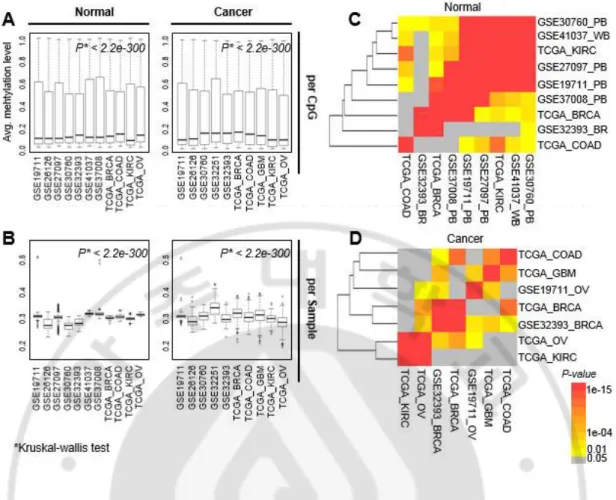
본 연구에서 모은 각각의 데이터 셋은 다른 나이 분포를 가진 다양한 조직 타입의 샘플들로 이루어져 있다(표 1). 그렇기 때문에, 각 데이터 셋마다 정상 샘플뿐만 아니라 종양 샘플에 대해 CpG 영역에 대한 평균 DNA 메틸값의 분포가 매우 차이 난다는 것을 확인 하였다. Kruskal-Wallis test 결과, 정상 샘플의 경우 p-value 가 <2.2e-200, 종양 샘플의 경우도 p-value 가 <2.2e-200 인 것을 확인 하였다(그림 6A). 비슷한 결과로 샘플 당 평균 DNA 메틸값 분포도 매우 차이가 난다는 것을 확인 하였다(그림 6B). DNA 메틸값은 조직에 따라 차이가 있다(Johansson et al., 2013). 그러므로 각 데이터의 조직 타입에 따라 추출한 노화 관련 CpG 영역이 얼마나 겹치는지 확인하기 위해서, 각각 두 개의 데이터 쌍 마다 겹치는 CpG 영역의 개수를 확인 하였다. 그리고 겹치는 정도(overlap degree)의 유의성을 확인하기 위하여 같은 개수의 무작위 선택 분석(Random
selection tests with same counts)을 수행 하였다. 그 결과, 정상 조직 샘플에서
대부분의 데이터 쌍 간에서 무작위보다 더 유의하게 노화 관련 CpG 영역이 잘 겹치는 것을 확인 하였다(그림 6C). 또한 겹치는 개수를 p-value 를 이용하여 계층 군집 분석(Hierarchical clustering analysis)을 수행한 결과, 조직에 관계없이 대부분의 데이터 셋 간에 잘 겹치는 것을 확인 하였다. 종양 샘플의 경우에는 노화 관련 CpG 영역의 겹치는 정도가 유의한 결과를 보였지만, 정상 샘플에 비해서는 겹치는 정도가 떨어지는 것을 보아, 종양 샘플의 DNA 메틸화의 차이가 더 큰 것을 알 수 있다(그림 6D).
Fig. 6. Comparison of age-associated CpG loci across different studies with different
tissue types. (A) Box plots of average methylation values (y-axis) per CpG unit in normal
and cancer tissues across individual studies (x-axis). (B) Box plots of average methylation
values per sample unit in normal or cancer tissue across individual studies. P-values were
calculated by Kruskal–Wallis tests. (C, D) We checked the degree of overlap of
age-associated CpG loci between studies by calculating the number of common CpG loci. We
normal (C) and cancer (D). P-value: a Z-test result using the distribution of 10,000 random
C. 정상 및 종양 조직의 노화 관련 DNA 메틸화 시그너처
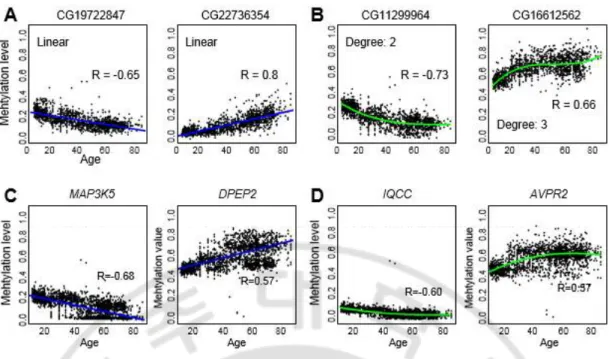
조직 비 특이적인 노화 관련 CpG 영역을 찾기 위해서, DNA 메틸값의 이상치를 가지는 샘플을 제거하고, 전체 데이터 셋에서 정상 조직 샘플의 정규화 된 DNA 메틸값을 통합 하였다. 통합한 데이터 셋을 이용하여 먼저 나이에 따라 DNA 메틸값이 선형적으로 변화하는 CpG 영역을 추출 하였다. 조직 타입에 관계없이, 예를 들어, 12 번 염색체 위의 30740381 번째에 위치한 CG19722847 영역은 상관 계수 r 값 (correlation coefficient)이 -0.65 로 나이에 따라 선형적으로 저 메틸화 되고, 6 번 염색체 위의 18230698 번째에 위치한 CG22736354 영역은 r 값이 0.8 로 선형적으로 과 메틸화 되는 것을 확인 하였다(그림 7A). 그러나 몇몇의 CpG 영역의 DNA 메틸값은 나이에 따라 비 선형적인 양상으로 변화 하였다(그림 7B). 비 선형 분석을 위해서 degree2, degree3 의 다중 회귀 모형을 이용 하였다. 예를 들어, 정상 샘플에서 9 번 염색체 위의 127509604 번째에 위치한 CG11299964 영역은 r 값이 -0.73 으로 나이에 따라 비 선형적으로 저 메틸화 되고, 22 번 염색체의 28042045 번째에 위치한 CG16612562 영역은 r 값이 0.66 으로 나이에 따라 비 선형적으로 과 메틸화 되는 양상을 확인 하였다(그림 7B). 비 선형 관계를 가지는 경우에, 이전 연구 결과와 동일하게 나이가 어릴수록 DNA 메틸값이 더 빠르게 변화하는 것을 확인 하였다 (Alisch et al., 2012; Numata et al., 2012). 또한, CpG 영역을 유전자 레벨로 맵핑한Fig. 7. Example of age-associated DNA methylation signature independent of tissue
type. (A, B) Examples of age-associated CpG loci with linear (A) or nonlinear (B)
relationships identified in integrated normal samples. (C, D) Examples of age-associated
DNA methylation genes with a linear (C) or nonlinear (D) relationship. Gene-level
따라서 본 연구에서는 선형뿐만 아니라 추가로 비선형 다중(degree 2 and 3) 회귀 모델을 이용해서 조직 비 특이적인 노화 관련 DNA 메틸화 시그너처를
동정 하였다. 이 때, 조직 타입에 따른 특이성을 줄이기 위해서 False discovery rate (FDR), 상관계수(correlation coefficient), 회귀 모형의 잔차 에러(residual error)등
3 개의 임계치를 이용해서 노화 관련 DNA 메틸화 CpG 영역을 찾았다. 각각의 임계치는 다음과 같다. FDR < 0.01, r >= 0.55, residual error < 0.15. 그 결과, 1812 개의 통합된 정상 조직 샘플에서 127 개의 노화 관련 DNA 메틸화 CpG 영역을 동정 하였고, 아래부터 이 영역을 노화 관련 DNA 메틸화 시그너처라고 부른다. 이 시그너처 중에서 80 개의 CpG 영역은 나이에 따라 선형 관계를 가지고, 나머지 47 개의 CpG 영역은 비 선형 관계를 가진다(부록 1). 또한, 77 개의 영역은 나이에 따라 저 메틸화 패턴을 가지고, 50 개의 영역은 과 메틸화 패턴을 가졌다. 통합한 2181 개의 종양 샘플에 대해서도 같은 방법으로 노화 관련 DNA 메틸화 시그너처에 대해 확인 하였다. 정상 샘플에 반해서, 종양 샘플은 26 개의 노화 관련 CpG 영역을 찾았다(그림 8A). 흥미롭게도, 정상 샘플과 종양 샘플의 노화 시그너처 간에 겹치는 CpG 영역은 없었다(그림 8B). 이와 같은 현상은 유전자 레벨로 맵핑한 경우에도 동일한 결과를 얻을 수 있었다(그림 8C, D). 정상 샘플과 종양 샘플을 모두 합친 데이터 셋의 경우(combined)에는 18 개의 노화 관련 시그너처를 찾을 수 있었다.
Fig. 8. Age-associated DNA methylation signature independent of tissue type. (A) Venn
diagrams showing the numbers of age-associated CpG loci with three regression models
(linear and second- and third-degree nonlinear) in integrated normal, cancer, or all samples.
(D) Venn diagram showing the number of age-associated CpG loci among integrated normal,
cancer, and all samples. (C) Venn diagrams showing the numbers of age-associated genes in
three regression models (linear and second- and third-degree nonlinear) in integrated normal,
cancer, or all samples. (D) Venn diagram showing the number of age-associated genes in
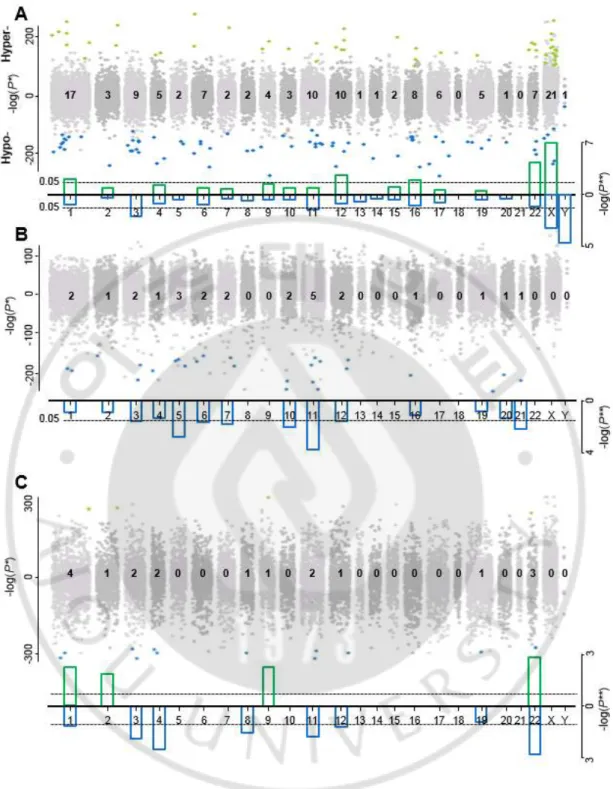
다음으로, 각각 정상 샘플(그림 9A)과 종양 샘플(그림 9B), 모두 합친 샘플(그림 9C)에 대한 노화 관련 시그너처의 염색체 위치에 대한 분포를 저 메틸화 영역과 과 메틸화 영역을 나누어 확인 하였다. 정상 샘플의 노화 관련 127 개 CpG 영역은 18 번, 21 번 염색체를 제외하고 비교적 인간 게놈 전체에 걸쳐 분포 되었다. 남아 소아 샘플을 이용하여 분석한 이전의 연구 결과(Alisch et al., 2012)와 반대로 노화 관련 CpG 영역이 X 염색체에 많이 나타났다. 이 결과는 성별, 나이 범위, 혹은 조직타입에 대한 차이에 의해 일어난 결과라고 추측한다. 또한, 초기하 분석을 통하여 각 염색체 별 분포된 CpG 영역의 개수가 유의미한지 확인 하였다. 그 결과, X 염색체는 p-value 가 8.1e-08, 22 번 염색체는 1.3e-03, 12 번 염색체는 1.7e-02, 1 번 염색체는 4.9e-02 로 나이에 따라 선호적으로
과 메틸화되는 영역이 많이 모여 있는 것을 알 수 있었다. 반면에, Y 염색체의 경우 p-value 가 3.4e-05, X 염색체는 9.4e-04, 3 번 염색체는 9.5e-03, 11 번 염색체는 4.3e-02 로 나이에 따라 저 메틸화 영역이 많이 분포 되어 있었다. 특히 정상 샘플의 경우, 노화 관련 시그너처가 X 염색체에서 많이 나타났다(그림 9A). 종양 샘플의 염색체 별 분포에 대한 결과를 보면, 3 번 염색체에서 p-value 가 0.03, 5 번 염색체에서 1.7e-03, 6 번 염색체에서 0.03, 7 번 염색체에서 0.02, 10 번 염색체에서 0.01, 11 번 염색체에서 1.8e-04, 21 번 염색체에서 0.01 로 나이에 따라 저 메틸화 되는 영역이 많이 분포된 것을 확인 할 수 있었다(그림 9B). 흥미롭게도, 종양 샘플에서는 X 염색체나 Y 염색체에 노화관련 시그너처는 존재 하지 않았다.
Fig. 9. Manhattan plots of age-associated CpG signatures by chromosome. (A)
Hypermethylated CpG loci with age are shown with a –log(P-value) and hypomethylated
loci are shown with a log(P-value). The most significant P-values among linear and
nonlinear models were chosen. Significant loci are marked as green (hypermethylated) or
blue (hypomethylated) dots. The numbers of significant age-associated CpG loci by
chromosome. Bar plots of P-values with hypergeometric tests for the degrees of significance
of the numbers of the loci. (B) Manhattan plot of age-associated CpG loci in integrated
cancer samples by chromosome. (C) Manhattan plot of age-associated CpG loci in combined
본 연구에서 사용된 정상과 종양 샘플의 나이 분포는 약간의 차이를 보인다. 정상의 경우, 3.6 세 – 90 세로 평균 39.88 세의 나이 분포를 가지고, 종양의 경우, 10 세 - 91 세로 평균 60.29 세의 나이 분포를 가진다. 정상 샘플에서 종양 샘플보다 더 많은 소아 샘플을 가진다. 그러므로 정상과 종양에서 노화 관련 DNA 메틸화 시그너처 특징의 차이가 나이 분포에 의함인지를 확인하기 위해서, 두 가지 방법을 통해서 나이 분포를 맞추어 같은 분석 방법으로 새롭게 노화 관련 영역을 추출하여 비교하였다. 첫 번째로 종양 샘플의 나이가 10 세에서 91 세이기 때문에 정상의 나이가 10 세에서 90 세를 가지는 샘플만을 추출하였다. 그 결과 29 개의 노화 관련 CpG 영역을 추출하였고, 기존과 동일하게 29 개의 CpG 영역들이 대부분의 염색체 위에 존재 했고, 특히 X 염색체 위에 많이 분포하는 것을 확인 하였다(그림 10A). 두 번째는 정상과 종양의 나이의 분포를 무작위 선택하여 완전히 일치 시켰다. 기존의 시그너처를 찾기 위한 임계치가 너무 엄격하여, 본 분석에서는 임계치를 약간 조정하여 회귀 분석 결과의 R 값이 > 0.32 이상인 영역만 추출 하였다. 그 결과 정상에서 444 개의 나이 관련 CpG 영역을 추출 하였고, 종양에서는 61 개의 영역을 추출 하였다. 정상의 444 개 CpG 영역은 기존의 정상 시그너처와 동일하게 전체 게놈의 염색체상에 존재 하는 것을 확인 하였다. 하지만, 이들 영역은 X 염색체 위에 많이 분포 하지 않는 것을 알 수 있었다(그림 10B). 이전에 연구된 결과 중에서 DNA 메틸화의 변화가 아동기에서 빠르게 변화한다는 것이 알려져 있다(Alisch et al., 2012). 본 연구 결과로 아동기에서의 변화가 X 염색체에서 많이 변화 할 수도 있을 것이라고 추측해 본다. 종양의 경우, 기존의 종양 시그너처와
같이 모두 나이에 따른 저 메틸화 영역만 존재했고, 정상간의 겹치는 영역은 존재 하지 않았다(그림 10C).
Fig. 10. Manhattan plots of age-associated CpG loci in age-matched samples by
between normal and cancer samples. We only included normal samples in the age range of
cancer since the normal age range is wider than cancer. We identified 29 age-associated CpG
loci in normal. (B, C) In addition, we exactly matched the age distributions between normal
and cancer samples by random sampling with smaller numbers of samples between normal
and cancer according to age groups. Here, we used the threshold of R > 0.32 and identified
new 444 (B) and 61 (C) loci in normal and cancer as age-associated, respectively.
Hypermethylated CpG loci with age are shown with a –log(P-value) and hypomethylated
loci are shown with a log(P-value). The most significant P-values among linear and
nonlinear models were chosen. Significant loci are marked as green (hypermethylated) or
blue (hypomethylated) dots. The numbers of significant age-associated CpG loci by
또한, 이전의 몇몇 연구 중에서 성별에 따라 DNA 메틸값에 차이가 있다고 발표해왔다(Hannum et al., 2013). 그렇기 때문에, 추가적으로 성별에 따라 노화 관련 시그너처를 추출 하여 비교 분석 하였다(그림 11). 본 연구의 정상 샘플에서 여성은 859 명, 남성은 944 명이고, 종양 샘플에서 여성은 1594 명, 남성은 491 명이다. 분석 결과, 정상 샘플의 남성에서는 560 개의 노화 관련 CpG 영역을 추출했고, 이 중 87 개 영역이 노화에 따른 과 메틸화, 473 개 영역이 저 메틸화 영역 이었다. 여성의 경우, 152 개의 노화 관련 CpG 영역을 추출했고, 이 중 103 개 영역이 과 메틸화, 49 개 영역이 저 메틸화 영역이었다. 전체적으로 성별에 따라 노화 관련 CpG 영역의 개수에 대해서는 차이가 있었지만, 염색체 별 비율에 대한 분포를 보면 비슷하다는 것을 확인 하였다(그림 11A). 또한, X 염색체 위에 존재하는 노화 관련 저 메틸화 혹은 과 메틸화에 따른 CpG 영역의 개수가 성별에 따라 차이가 있는지를 확인 하기 위하여 Fisher’s test 를 수행 하였고, p-value 가 0.222 로 차이가 없었다(그림 11B).
Fig. 11. Fractions of age-associated CpG loci according to gender. (A) Bar plot for the
fractions of age-associated loci by chromosome in normal male (as dark blue) or female (as
pink) samples. (B) The numbers of hyper- or hypomethylated loci on X chromosome with
다음으로, 127 개의 조직 비 특이적 정상 노화 관련 시그너처를 이용하여 실현 가능한 나이 예측 모델을 고안 하였다. 이를 위해서 genetic 알고리즘을 이용하여 시그너처 중 모델에 적용 할 하위 그룹(CpG 영역)을 찾고, 이들을 이용하여 다중 선형 회귀 모형을 생성 하였다. 결과적으로, 전체 시그너처 중 20 개의 CpG 영역이 나이 예측 모델에 선택 되었다(부록 1). 고안 된 모델의 성능을 평가하기 위해 실제 나이와 예측된 나이의 상관관계를 보았을 때 r 값이 0.91 로 매우 잘 예측 하는 것을 확인 하였다(그림 12A). 또한 추출한 20 개의 CpG 영역의 유의성을 확인하기 위해, 전체 CpG 영역 중에서 무작위로 20 개의 영역을 선택하여 새롭게 생성한 예측 모델에 적용하여, 10,000 회 무작위 선택 분석(Random selection tests)을 수행 하였고, 본 연구에서 고안한 나이 예측 모델은 p-value 가 0.0002 로 매우 유의하게 실제 나이를 잘 예측 하는 결과를 얻었다. 이 모델은 조직 비 특이적으로 노화와 관련된 시그너처의 DNA 메틸값을 이용하여 나이를 예측할 수 있다는 것을 의미 한다. 추출한 127 개의 정상 시그너처가 이전 연구결과와 얼마나 일치하는 지를 확인 하기 위하여, 이전 연구 결과에서 추출한 노화 관련 시그너처를 비교, 분석 하였다(표 2). 대부분의 이전 연구들은 선형 모델을 이용하여 FDR 임계치를 통해 노화 관련 영역을 추출 하였기 때문에, 본 연구의 FDR <0.01 의 임계치를 이용한 선형 회귀 분석 결과로부터 추출한 430 개의 노화 관련 CpG 영역을 이용하여 비교 하였다. 예를 들어, 최근 나이가 14 세에서 94 세를 가지는 421 명의 건강한 사람의 혈액세포를 가지고 Illumina Human 450K 어레이를 이용해서 선형 회귀 모델을 통해 137,993 개의 노화 관련 CpG 영역을 찾은 연구
결과가 있었다(Johansson et al., 2013). 이들 137,993 개의 CpG 영역 중 본 연구의 Illumina 27K 어레이에 존재하는 영역이 6,696 개였고, 이 영역 중 430 개의 노화
관련 CpG 영역과 일치하는 영역은 313 개 영역으로 73%가 일치 하였다. 또 다른 Day et al.연구(Day et al., 2013)에서는 뇌 조직을 포함한 4 개의 조직 타입을 가지고
선형 회귀 분석 방법을 통해 4747 개의 CpG 영역을 찾았고, 본 연구결과 430 개의 CpG 영역과는 47%가 일치 하였다. 특히, 어떠한 질병을 포함한 샘플에 대해 연구한 결과보다 오직 정상 샘플만 이용한 연구와 더 잘 일치 하는 것을 확인 할 수 있었다(그림 12B, 표 2). 본 연구의 노화 관련 127 개 시그너처 중 16 개의 영역이 이전 연구에서 발견되지 않은 영역이었고, 흥미롭게도 이 새로 발견한 16 개의 영역 중 13 개영역이 X 염색체 위에 위치하는 것을 알 수 있었다(부록 1).
Fig. 12. Characteristics of age-associated DNA methylation signature. (A) Age
prediction using the age-associated normal DNAm signature. Age was predicted with the
normal signature using a multivariate linear regression after using a genetic algorithm to
identify a feasible set of loci. (B) Degrees of overlap with age-associated DNAm signatures
identified in previous studies. Overlap percentages were calculated by the common numbers
divided by the smaller number of total loci in either study. The studies with only normal
Table 2. Characteristics of other age-associated DNA methylation studies.
Paper Platform Tissue type # of
Sample Sample characteristics Method
# of age-associated
CpGs
Overlap (%)
Teschendorff et al. Illumina 27K WB 261 Healthy wonmen (148) A robust linear regression model
(FDR < 0.05) 589 25.1
Ovarin cancer (113)
Alish et al. Illumina 27K PB 398 Health pediatric males A linear fixed-effects regression
model (FDR < 0.01) 2078 54.2
Horvath et al. Illumina 27K Brain &
WB 1688
Healthy (365)
Weighted linear network model
1000 (age-hyper methylation) 10 Schizophrenia (293) Type1 diabetics (190) Healthy older women (348)
Brain (492)
Johansson et al. Illumina 450K White
blood cells 421 Healthy (421)
A linear regression model (Bonferroni < 0.05)
137993
(27K : 6696) 73
Hannum et al. Illumina 450K WB 482 Healthy (482) A multivariate linear regression
model (FDR < 0.05) 71 (27K : 7) 1.2
5
0
-본 연구에서 취합한 정상 조직의 DNA 메틸화 프로파일은 다양한 조직 타입을 포함한다. 추가적으로, 조직 특이적인 노화 관련 시그너처를 추출 하였다(그림 13). 정상 샘플에서 노화 관련 CpG 영역의 수는 1 개(전립선 조직의 경우)에서 2713 개(혈액 조직의 경우)까지 다양하게 존재 했지만(그림 13A), 각 조직 쌍 별로 뽑은 시그너처들이 얼마나 많이 겹치는지에 대해서 분석 한 결과 전립선 조직을 제외하고는 거의 대부분의 조직들이 무작위에 비해서 잘 겹치는 것을 확인 하였다(그림 13B). 종양 조직의 경우, 본 연구에서 사용한 샘플과 임계치로 조직 특이적인 노화 관련 시그너처를 찾을 수 없었다.
Fig. 13. Tissue-specific associated CpG loci. (A) The numbers of significant
age-associated CpG loci according to distinct types of normal tissue. Numbers in parentheses
indicate the counts of samples. (B) Hierarchical clustering results using the overlap degrees
of age-associated CpG loci between different tissue types in normal. P-value: a Z-test result
D. 조직 비 특이적인 노화 관련 DNA 메틸화 시그너처의 특징
본 연구에서 추출한 노화 관련 DNA 메틸화 시그너처의 특징들을 살펴 보기 위해서 먼저, 정상 샘플 혹은 종양 샘플의 시그너처들의 게놈상의 위치에 대해서 확인 하였다. 정상 샘플의 127 개 시그너처 중에서, 78 개 CpG 영역이 CGI 영역에 위치 했고, 나머지 49 개 CpG 영역은 non-CGI 영역에 위치 하였다 (표 3). 반면에 종양 샘플의 경우, 22 개 영역이 CGI, 4 개영역이 non-CGI 영역에위치 하였다. Fisher 분석결과 p-value 가 0.02 로 정상 및 종양 샘플 모두 non-CGI 영역보다 non-CGI 영역에 차이 나게 많이 위치하는 것을 확인 할 수 있었다.
Table 3. Numbers of loci in the age-associated DNAm signatures using the integrated
data set.
(A) Numbers of CpG-level signatures
(B) Numbers of gene-level signatures