이학석사학위논문
Loperamide enhances
bortezomib-mediated cell death
in colon cancer cells
아 주 대 학 교 대 학 원
의
2생
2명
2과
2학
2과
Loperamide enhances bortezomib-mediatedcell death
in colon cancer cells
by
MinJuneShim
A Dissertation Submitted to The Graduate School of
Ajou University in Partial Fulfillment of the Requirements for the
Degree of Master of Biomedical Sciences
Supervised by
Kyeong Sook Choi, Ph.D.
Major in Cancer Biology
Department of Biomedical Sciences
February, 2015
This certifies that the dissertation
ofMinJuneShim is approved.
SUPERVISORY COMMITTEE
심사위원장윤계순인
심사위원최경숙인
심사위원이종수인
The Graduate School, Ajou University
December, 19th, 2014
i
- ABSTRACT -
Loperamide enhances bortezomib-mediated cell death
in colon cancer cells
The destruction of proteins via the ubiquitin-proteasome system is a multi-step, complex process involving poly-ubiquitination of substrate proteins, followed by proteolytic degradation by the macromolecular 26S proteasome complex. Inhibitors of theproteasome promote the accumulation of proteins that are deleterious to cell survival. So, the proteasome is one of the most promising anti-cancer drug targets. Bortezomib, the first FDA approved 26S proteasome inhibitor, is now used in the treatment for multiple myeloma and mantle cell lymphoma. However, its efficacy in solid tumors is not satisfactory. In the present study, we show that combination of loperamide and bortezomib synergistically induces cell death in human colon cancer cells. Combined treatment with loperamide and bortezomib induced excessive dilation of the endoplasmic reticulum (ER) prior to induction of cell death. We found that co-treatment with loperamide further enhanced bortezomib-induced increase in the protein levels of CHOP and Noxa levels. Knockdown of CHOP or Noxa markedly attenuated the cell death induced by loperamide plus bortezomib, suggesting their critical involvement in this cell death. Combined treatment with loperamide and bortezomib increased mitochondrial ROS levels in DLD-1 cells. The general antioxidant, NAC, GSH, or the MnSOD mimetic, MnTBAP effectively blocked combined treatment-induced ER dilation and cell death. In addition, co-treatment with loperamide dispersed bortezomib-induced aggresome structures, possibly contributing to
ii
proteotoxicity in colon cancer cells. In conclusion, the combined regimen of loperamide and bortezomib may be useful to improve the effect of the proteasome inhibitor-based cancer therapy.
iii
TABLE OF CONTENTS
ABSTRACT ... i TABLE OF CONTENS ...iii LIST OF FIGURES ... vi I. INTRODUCTION ... 1 II. MATERIALS AND METHODS... 8 A. Chemicals and antibodies ... 8 B. Cell culture of various cancer cell lines and normal cells ... 8 C. Measurement of cell viability ... 9 D. Western blotting ... 9 E. Immunocytochemistry ... 10 F. Establishment of the stable cell lines in the fluorescence specifically endoplasmic reticulum ... 10 G. Measurement of mitochondrial superoxide anion ... 10 H. Measurement of cytosolic and mitochondrial Ca²⁺ levels ... 11 I. Small interfering RNAs ... 11 J. shRNA-mediated knockdown of proteins ... 12
iv
K. Aggresome staining ... 12 L. Flow cytometry for the analysis of DNA contents ... 13 M. Clonogenic cell survival assay ... 13 III. RESULTS ... 14 1. Synergistic cell death induced by loperamide plus bortezomib is neither apoptosis not autophagic cell death... 14 2. Intracellular Ca2+ influx is involved in the combined treatment-induced vacuolation . 27
3. Combined treatment with loperamide and bortezomib induces ER stress, and CHOP is critically involved in the cell death by loperamide plus bortezomib ... 33 4. ROS generation is important for the cell death induced by loperamide plus bortezomib. ... 42 5. Noxa is also critical factor for loperamide plus bortezomib-induced cell death ... 46 6. Disruption of aggresome formation may be involved in the cell death induced by loperamide and bortezomib ... 50 7. Combined treatment with loperamide and proteasome inhibitors does not induce cell death in normal colon cells ... 56 IV. DISCUSSION ... 60 V. REFERENCES ... 64
v
vi
LIST OF FIGURES
Figure 1. Combined treatment with loperamide and bortezomib induces the cell death in colon cancer cells. ... 17 Figure 2. Loperamide plus bortezomib synergistically induce cell death in colon cancer cells... ... 18 Figure 3. Effect of loperamide plus bortezomib on the long-term survival of DLD-1 cells.. . 19 Figure 4. Loperamide plus bortezomib induce cell death accompanied by vacuolation in colon cancer cells ... 20 Figure 5. z-VAD-fmk does not block the cell death by loperamide plus bortezomib in colon cancer cells ... 21 Figure 6. Combined treatment with loperamide and bortezomib induces caspase-independent cell death in DLD-1 and SW-480 cells ... 22 Figure 7. Combined treatment with loperamide and bortezomib does not induce apoptotic cell death in DLD-1 cells ... 23 Figure 8. Combined treatment with loperamide and bortezomib may not induce the release of mitochondrial cytochrome c in DLD-1 cells ... 24 Figure 9. Effect of autophagy inhibitors on combined treatment-induced cell death ... 25 Figure 10. Effect of autophagy inhibitors on combined treatment-induced vacuolation… .... 26 Figure 11. Effect of μ-opioid antagonist on combined treatment-induced cell death ... 29 Figure 12. The combination of loperamide and bortezomib significantly enhances calcium levels in DLD-1 cells ... 30
vii
Figure 13. Effect of various Ca2+-related inhibitors on the combination of loperamide and
bortezomib-induced cell death ... 31 Figure 14. BAPTA-AM delays the combination treatment-induced vacuolation, but not cell death ... 32 Figure 15. Vacuolation induced by loperamide plus bortezomib is originated from the ER in DLD-1 cells ... 36 Figure 16. Combination of loperamide and bortezomib inudces mitochondrial fragmentation… ... 37 Figure 17. The expression profiles of ER stress-related proteins in loperamide plus bortezomib-treated cells... 38 Figure 18. Loperamide plus bortezomib markedly increase CHOP levels in DLD-1 cells... 39 Figure 19. CHOP blocks vacuolation induced by loperamide plus bortezomib ... 40 Figure 20. CHOP is critically involved in cell death induced by loperamide plus bortezomib… ... 41 Figure 21. Loperamide plus bortezomib markedly increase mitochondrial ROS levels in DLD-1 cells ... 43 Figure 22. Effects of various antioxidants on the cell death by combination of loperamide and bortezomib ... 44 Figure 23. Various antioxidants delay the ER dilation induced by loperamide plus bortezomib in DLD-1 cells ... 45 Figure 24. The expression profile of Bcl-2-related proteins in loperamide plus bortezomib-treated cells... 47
viii
Figure 25. Noxa up-regulation critically contributes to the cell death by loperamide plus bortezomib ... 48 Figure 26. Effect of Noxa knockdown on the vacuolation induced by loperamide plus bortezomib ... 49 Figure 27. Co-localization of aggresome and p62 in DLD-1 cells treated with loperamide plus bortezomib ... 52 Figure 28. Co-treatment with loperamide does not increase bortezomib-induced accumulation of poly-ubiquitinated proteins in DLD-1 cells (immunocyto-chemistry)…. ... 53 Figure 29. Co-treatment with loperamide does not increase bortezomib-induced accumulation of poly-ubiquitinated proteins in DLD-1 cells (Western blotting) ... 54 Figure 30. CHOP induction may lie upstream of Noxa up-regulation in the cell death by loperamide plus bortezomib ... 55 Figure 31. Loperamide enhances the death of DLD-1 cells treated with other proteasome inhibitors ... 57 Figure 32. Loperamide and/or proteasome inhibitors do not induce cell death in normal colon cells ... 58 Figure 33. Loperamide and/or bortezomib do not induce CHOP and Noxa expression in normal colon cells ... 59
1
I. INTRODUCTION
Carcinogenesis is a multistep process that requires the accumulation of various genetic and epigenetic aberrations to drive the progressive malignant transformation of normal human cells. Ten major hallmarks of carcinogenesis that have been described are sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion & metastasis, degrading cellular energetics, avoiding immune destruction, genome instability & mutation and tumor-promoting inflammation (Hanahan and Weinberg, 2011). These properties have been targeted over the past decade in the development of therapeutic treatments for colorectal cancer (CRC), one of the most commonly diagnosed and lethal cancers worldwide. The treatment of solid tumor cancers such as CRC has been challenging due to the heterogeneity of the tumor itself and the chemoresistance of the malignant cells (Mathonnet et al., 2014).
Protein synthesis and degradation is a tightly regulated process that is essential for normal cellular homeostasis. Most intracellular proteins are degraded by the proteasome, which is the 26S macromolecular proteasome complex (Gallastegui et al., 2010) containing one catalytic core, the 20S proteasome, and two 19S regulatory complexes. The 19S regulatory particle can be further subdivided into lid and base components. Following recruitment to the proteasome, poly-ubiquitinated proteins undergo deubiquitination and unfolding. The removal of ubiquitin is accomplished by a family of deubiquitinase enzymes, some of which are associated with the 19S lid. Ubiquitin polypeptides that are removed from substrate proteins can be directly recycled by the cell. The 19S base component plays a key role in unfolding of the substrate
2
protein and delivery of the unfolded, deubiquitinated substrate into the 20S catalytic core particle. The 20S catalytic core particle consists of four layers of ring-like structures (Groll et al., 1997). The outer ring layers are composed of seven “alpha” subunits, α1-α7, while the inner “beta” rings are composed of seven beta subunits, β1-β7. The β1 subunits exhibit caspase-like (C-L) proteolytic activity, the β2 subunits exhibit trypsin-like (T-L) activity, and the β5 subunits exhibit chymotrypsin-like (CT-L) activity. Collectively, these subunits act to degrade substrate proteins into short oligopeptides.
The closely regulated ubiquitin-proteasome-system (UPS) clears the cell-plasma from damaged, misfolded and aged proteins. More than 80% of intracellular proteins are processed by the UPS (Crawford et al., 2013), the remaining proteins are handled by the lysosome system. UPS is also involved in the inactivation of regulatory proteins by initiating the posttranslational addition of multiple ubiquitin motifs which sorts intracellular proteins for degradation.
Degradation of proteins by the ubiquitin-proteasome system is accomplished in two major steps: 1) ubiquitination of the protein, and 2) proteolytic degradation of the poly-ubiquitinated protein by the macromolecular proteasome complex (Ciechanover, 2005; Orlowski et al., 2000; Shen et al., 2013). Each of these steps involves a complex series of protein interactions and biochemical events.
Poly-ubiquitination of substrate proteins first involves activation of the 76-amino acid ubiquitin polypeptide by the activating enzyme E1 (Shen et al., 2013). Ubiquitin activation is ATP dependent and is achieved by one of the two known E1 human enzymes. Activation involves covalent linkage between the carboxyl-terminus of ubiquitin and a cysteine residue present on E1, forming a thioester bond. The activated ubiquitin is then transferred to an E2
3
ubiquitin-conjugating enzyme forming again a thioester covalent linkage. At least 50 distinct E2 enzymes have been identified in humans. E2 enzymes define the position of ubiquitination (e.g. K48 vs. K63) and consequently determine the further destiny of the protein substrate (Shen et al., 2013). In a third step, an E3 ligase enzyme transfers the ubiquitin from E2 to the substrate protein. Since E3 proteins act to recognize and bind substrate proteins, it is not surprising that over 500 E3 enzymes appear to be encoded by the human genome (Liet al., 2008). The majority of E3 ligases are classified as RING finger E3s, and act by bringing substrates and E2 enzymes into close proximity. The RING finger E3s then directly transfer ubiquitin from E2 to the substrate, without forming an intermediate covalent bond. A minority of E3 ligases (roughly 30) are classified as HECT domain E3s, and act by forming an intermediate thioester linkage with ubiquitin before transfer to the substrate (Lauet al., 2012). The E3 enzymes ligate ubiquitin to lysine residues present on the substrate protein. Following mono-ubiquitination of the substrate, the process must be repeated to form an elongated chain of ubiquitin residues. Proper recognition of ubiquitinated substrates by the proteasome complex is thought to require a minimum of four ubiquitin residues in the poly-ubiquitin chain. Ubiquitin has seven lysine positions (K6, K11, K27, K29, K33, K48 and K63) with K48 and K63 being the most common positions where poly-ubiquitination occurs. The position of poly-ubiquitination determines whether a protein will be degraded (K48-linked) or will be activated (K63-linked) (Nathan et al., 2013). Little is known about ubiquitination at the other lysine positions.
Bortezomib (PS-341, Velcade), a cell-permeable dipeptidyl boronic acid, is a first-in-class reversible inhibitor of the proteasome that has achieved considerable success in the treatment of certain hematologic malignancies. Notably, the United States Food and Drug Administration
4
(US FDA) has approved the use of bortezomib for multiple myeloma and mantle cell lymphoma (Bross et al., 2004; Fisher et al., 2006; Kaneet al., 2003; Kaneet al., 2007; Richardson et al., 2003; Richardson et al., 2005).
Several independent investigators have found that bortezomib inhibits activation of the transcription factor nuclear factor κB (NF-κB) by preventing proteasomal degradation of IκBα (Hideshima et al., 2001; Russo et al., 2001; Sunwoo et al., 2001; Hideshima et al., 2002; Tan et al., 2002; Ma et al., 2003; Cusack et al., 2001). Bortezomib treatment leads to activation of JNK in multiple myeloma (Hideshima et al., 2003; Chauhan et al., 2004) and nonsmall cell lung cancer cells (Yang et al., 2004). Proteasome inhibition has also been shown to stabilize the cyclin-dependent kinase inhibitors p21 and p27, the tumor suppressor p53 (Hideshima et al., 2001; Shah et al., 2001; Williams et al., 2003; Breitschopf et al., 2000; Li et al., 2000). A recently published study found that bortezomib prevented activation of caveolin-1 in multiple myeloma cells (Podar et al., 2004). Activation of caveolin-1, a protein that functions in cell motility or migration in a number of tissues, requires phosphorylation. Inhibition of the proteasome leads to up-regulation of pro-apoptotic members of the Bcl-2 protein family, including Noxa, Bax, and Bik (Fribley et al., 2006; Li et al., 2008; Perez-Galan et al., 2007; Qin et al., 2005; Voortman et al., 2007; Zhu et al., 2005; Zhu et al., 2005). bortezomib can trigger the accumulation of anti-apoptotic factors responsible for a protective antitumor effect. This includes Mcl-1 anti-apoptotic protein. Bortezomib-induced apoptosis was associated with Mcl-1 cleavage regardless of Mcl-1L accumulation (Gomez-Bougie et al., 2007). Treatment of cells with proteasome inhibitors has been shown to up-regulate expression of DR4 and DR5, enhancing sensitivity to TRAIL (Liu et al., 2007; Nikrad et al., 2005; Seki et al., 2010; Shanker
5
et al., 2010; Voortman et al., 2007; Yoshiba et al., 2011). Bortezomib was also reported to elicit the unfolded protein response, which is activated when the physiologic environment of the endoplasmic reticulum is altered. The induction of ER stress induces reactive oxygen species (ROS) (Fribley et al., 2004; Obeng et al., 2006; Weniger et al., 2011).
Diarrhea is commonly seen with the use of bortezomib, a proteasome inhibitor used in the treatment of multiple myeloma and mantle cell lymphoma. In the pivotal studies with this agent, diarrhea occurred in 51% of patients, with 8% of the events being severe (Berenson et al., 2005). Bortezomib binds in reversible fashion to the β5 subunit, inhibiting the CT-L activity of the 20S catalytic core particle (Chen et al., 2011). However, bortezomib is not entirely specific, with modest inhibitory activity against the β1 subunit, as well as inhibitory activities against a variety of serine proteases, including cathepsins A and G, chymase, dipeptidyl peptidase II, and HtrA2/Omi (Arastu-Kapur et al., 2011). It has been proposed that these nonspecific activities contribute to the high rate of peripheral neuropathy that has been observed in bortezomib-treated patients (Arastu-Kapur et al., 2011) Bortezomib-induced Peripheral neuropathy, a representative side effect of bortezomib, occurred in 37–44% of clinical trial patients, requiring dose modification and potential changes in the treatment plan when it occurs (Cavaletti et al., 2010; Argyriou et al., 2010). Due to the reversible nature of bortezomib, prolonged inhibition of the proteasome in vivo may require relatively frequent administration, although this is somewhat mitigated by the slow rate of bortezomib dissociation from the β5 subunit. And also, bortezomib displays marked pro-apoptotic effects in hematological malignant cells, it possesses limited effects on solid malignant tumor cells. Therefore, there is a growing need for the development of optimal combinations of these novel agents to inhibit tumor progression.
6
Loperamide, an FDA-approved antidiarrhea drug, acts on the -opioid receptors in the mesenteric plexus of large intestines (Loetchutinat et al., 2003). Loperamide is widely used in the clinic to control diarrhea induced by digestive disorders, chemotherapy, and radiotherapy. Loperamide at submicromolar concentrations blocks L-type calcium channels and inhibits binding of verapamil to such channels. The antisecretory properties of loperamide may be due in part to blockade of calcium channels (Burleigh et al., 1988). Other sites of action for loperamide have been documented. Loperamide at high micromolar concentrations blocks high voltage dependent calcium channels and N-methyl-D-aspartate evoked responses in rat and mouse hippocampal pyramidal neurons (Church et al., 1994). However, when administered intrathecally, loperamide has been reported to produce significant antinociceptive effect in the formalin test. Loperamide showed a direct inhibitory effect on calcium-influx, the analgesic effect of intrathecally injected loperamide might be due to its blockade of the voltage-dependent calcium channels at the terminals of the primary afferent fibers (Kumar et al., 2012; Hagiwara et al., 2003).
In humans, many key organs including brain, liver, and heart express permeability glycoprotein (P-gp), and P-gp serves to minimize retention of toxic substances in normal cells. Recent studies have shown that loperamide acts as a P-gp substrate in the blood–brain barrier (BBB) (Ludwig et al., 2006; Nobili et al., 2006). Loperamide molecules normally do not cross the BBB to any significant extent, but those that cross the BBB are quickly exported from the brain by the P-gp in the BBB.
Some solid malignant tumor cells have resistant or become refractory to ongoing bortezomib treatment. To improve the efficacy of proteasome inhibitor–based treatments and to overcome
7
resistance, drugs augmenting the antitumor properties of bortezomib are required. We identified loperamide, clinically used for the treatment of diarrhea, as an effective sensitizer of bortezomib to improve its anti-cancer effect. Our results show that ER stress, possibly due to ER dilation, increased mitochondrial ROS and intracellular Ca2+ levels may be critically involved in the cell
death by combination of loperamide and bortezomib in colon cancer cells. In addition, induction of CHOP and Noxa may play an important role in this cell death by loperamide plus bortezomib. Taken together, our findings on the underlying mechanisms involved in the cell death by the combined treatment with loperamide and bortezomib may improve the proteasome inhibitor-based cancer therapeutic strategies.
8
II. MATERIALS AND METHODS
A. Chemicals and antibodies
N-acetylcysteine (NAC), cycloheximide (CHX), reduced glutathione (GSH), 1,2-bis(o-aminophenoxy)ethane-N,N,N’N’-tetraacetic acid acetoxymethyl ester (BAPTA-AM), and ruthenium red were purchased from Sigma (St. Louis, MO). MitoTracker-Red, Fluo-3 AM, Rhod-2-AM, MitoSOX-Red, 4’6-diamidino-2-phenylindole (DAPI), calcein acetosymethyl ester (calcein-AM) and ethidium homodimer (EthD-1) were purchased from Molecular Probes (Carlsbad, CA). Caspases inhibitors benzyloxy-carbonyl-Val-Ala-Asp-(OMe) fluoromethyl ketone (z-VAD-fmk) was from R&D systems (Minneapolis, MN). Loperamide (Lop) was obtained from Tocris Bioscience(United Kingdom, CA). Bortezomib was purchased from Selleckchem. The following antibodies were used: anti-Noxa (Calbiochem); anti-ubiquitin, ATF4, and Mcl-1 (Santa Cruz Biotechnologies, Santa Cruz, CA); anti-CHOP (GADD153), phospho-ERK1/2, total ERK1/2, phosphor-eIF2α, and total eIF2α (Cell Signaling, Beverly, MA); anti-α-tubulin (Abcam); anti-p62 (Sigma, St. Louis, MO).
B. Cell culture of various cancer cell lines and normal cells
The DLD-1,SW-480, HCT116WT and HCT116 p53(-/-) human colon cancer cell lines, and CCD-112, and CCD-841 human normal colon cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). These various cancer cell lines and normal colon
9
cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics (GIBCO-BRL, Grand Island, NY). Cells were maintained in a humidified atmosphere containing 5% CO2at 37℃. Cell culture passage number less than five was used in the present
study. Loperamide (>98% purity, Sigma) and bortezomib (Selleckchem) were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 50 mM and 2.6 mM, and stored at -20℃. These stock solutions were diluted to the required concentration when need.
C. Measurement of cell viability
Cell viability was assessed by double labeling of cells with 2 μM calcein-AM and 4 μM EthD-1. The calcein-positive live cells and EthD-1-positive dead cells were visualized using a fluorescence microscope (Axiovert 200M using Axiovision Release 4.4 and Axiocam HRM digital camera; Zeiss, Oberkohen, Germany) and counted.
D. Western blotting
Cells were washed in PBS and lysed in boiling sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (6.25 mM Tris [pH 6.8], 1% SDS, 10% glycerol, and 5% β-mercaptoethanol). The lysates were boiled for 5 min, separated by SDS-PAGE, and transferred to an Immobilion membrane (Millipore, Bredford, MA, USA). After blocking nonspecific binding sites for 1 h using 5% skim milk, membrane were incubated for 2 h with specific Antibodies. Membranes were then washed three times with TBST and incubated further
10
for 1 h with horseradish peroxidase-conjugated anti-rabbit, -mouse antibody. Visualization of protein bands was accomplished using ECL (Advansta).
E. Immunocytochemistry
DLD-1 colon cancer cells were plated on chamber slides before drug exposure. Cells were fixed with 4% paraformaldehyde, permeabilized using 0.5% Triton X-100, and incubated overnight with indicated primary antibodies. Fluorescent secondary antibodies were used to visualize protein localization. Images were obtained using a fluorescence microscope (Axiovert 200M, Carl Zeiss).
F. Establishment of the stable cell lines in the fluorescence specifically endoplasmic
reticulum
To establish the stable cell lines expressing the fluorescence specifically in the ER, DLD-1 cells were transfected with the pEYFP-ER vector (Clontech Laboratories, Mountain View, CA). Stable cell lines over-expressing pEYFP-ER (YFP-Mito of YFP-ER) were selected with the complete media containing 500 μg/mL G418 (Calbiochem, San Diego, CA). Images of ER were obtained from the fluorescence microscopy using a Zeiss filter set #10.
11
Generation of superoxide was determined using a previously established fluorescence microscopy or flow cytometry technique. 20 μM loperamide plus 40 nM bortezomib-treated cells were stained with 2.5 μM MitoSOX red for 20 min and samples were observed under a fluorescence microscope equipped with Zeiss filter set #20 (excitation; band pass (BP) 546 nm, emission; BP 575-640 nm).
H. Measurement of cytosolic and mitochondrial Ca²
⁺ levels
To measure cytosolic Ca²⁺ levels,treated cells were incubated with 2.5μM Fluo-3-AM at 37℃ for 20 min, washed with HBSS, and analyzed immediately by flow cytometry. To measure mitochondrial Ca²⁺ levels treated cells were incubated with 2.5μM Rhod-2-AM at 4℃ for 30 min, washed with HBSS, further incubated with HBSS at 37℃ for 20 min, and then analyzed by flow cytometry.
I. Small interfering RNAs
The small interfering RNA (siRNA) duplexes used in this study were purchased from Invitrogen and have the following sequences: Noxa (Santa cruze Cat. No. sc-37305). Negative Universal ControlTM (Invitrogen) was used as the control. After annealing of the pairs of siRNA oligos,
cells were transfected with siRNA oligonucleotides. To confirm successful siRNA-mediated knockdown, we performed Western blotting of the proteins of interest.
12
J. shRNA-mediated knockdown of proteins
Knockdown of the CHOP protein in colon cancer cells was achieved by lentiviral infections of viral vectors that express different shRNA directed to CHOP mRNA and were purchased from Sigma-Aldrich (ShRNA MISSION). Viruses were generated by transfection of 293T cells with MISSION shRNA vectors and DNRF vector encoding for gag- pol, and CMV-VSVG encoding for envelop glycoprotein of vesicular stomatitis virus. The medium of transfected 293T cells containing lentiviruses was used to infect myoblasts that were further selected with puromycin (3 mg/ml). Knockdown efficiency was analyzed by western blotting. Viral particles that caused maximal repression of CHOP expression relative to control particles were chosen for the knockdown experiments.
K. Aggresome staining
After combined treatment with loperamide and bortezomib, cells were fixed in 4% paraformaldehyde for 30 minutes at room temperature (RT) and permeabilized with 0.5% Triton X-100 in 16 assay buffer for 5 minutes at RT. Cells were washed with 16assay buffer for two times and stained with ProteoStat Aggresome dye (Enzo Life Sciences, PA) for 30 minutes at RT and washed with 16 assay buffer. The stained cells were examined with a fluorescence microscope (Axiovert 200M, Carl Zeiss) and images were captured at 663 objective with red filter.
13
L. Flow cytometry for the analysis of DNA contents
Trypsinized and floating cells were pooled, washed with PBS, and fixed in 70% (v/v) ethanol. DNA contents were assessed by staining cells with propidium iodide after treatment with 50 μg/ml RNase A and monitoring by FACScan (Becton Dickinson, Mountain View, CA, USA). DNA content was assessed with a ModFit LT program (Verify Software House, Inc., Topsham, MA, USA).
M. Clonogenic cell survival assay
DLD-1 cells were plated in 12 well plates at a density of 1 X 103 cells per well in triplicate and
treated with loperamide and/or bortezomib for 4 hours. And then cells were treated with fresh drug-free medium and incubated for an additional 8 days. After 8 days, cells were fixed in cold-methanol, and stained with 0.5% crystal violet.
14
III. RESULTS
1.Synergistic cell death induced by loperamide plus bortezomib is neither
apoptosis not autophagic cell death
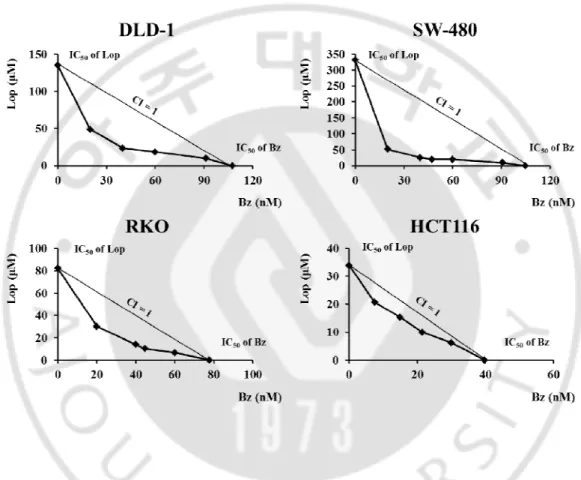
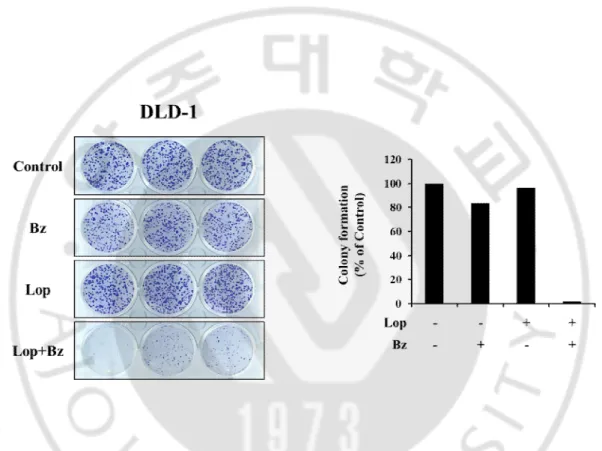
Although bortezomib, the first FDA approved proteasome inhibitor (PI), is effective in the treatment of multiple myeloma and mantle cell lymphoma, its anti-cancer effects against solid tumors including colon cancer are not satisfactory. Therefore, identification of the effective sensitizers of the proteasome inhibitor may improve the efficacy of PI-based cancer therapy. In this study, we first investigated whether loperamide could overcome the resistance of colon cancer cells to bortezomib. DLD-1, SW-480, HCT116, and RKOcolon cancer cells were treated with various doses of loperamide and/or bortezomib for 24 h and the cell viability assay using calcein-AM and EthD-1, to detect live and dead cells, respectively, was performed. While treatment withbortezomib or loperamide alone did not induce cell death, combined treatment with loperamide and bortezomib significantly and dose-dependently enhanced cell death (Fig. 1).Isobologram analysis showed that loperamide and bortezomib synergistically induced cell death in these colon cancer cells (Fig. 2). We next compared the effect of loperamide plus bortezomib on the long-term survival of DLD-1 cells. Combined treatment with 20 μM loperamide and 40 nM bortezomib for 4 h was sufficient to reduce the clonogenicity of these cells (Fig. 3). When we observed cellular morphologies following treatment with loperamide and/or bortezomib, treatment of four different colon cancer cells with loperamide alone for 24 h
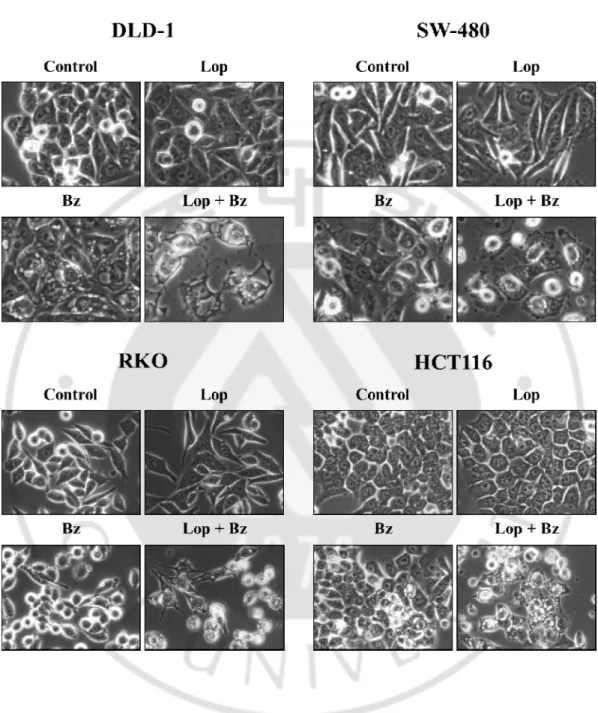
15
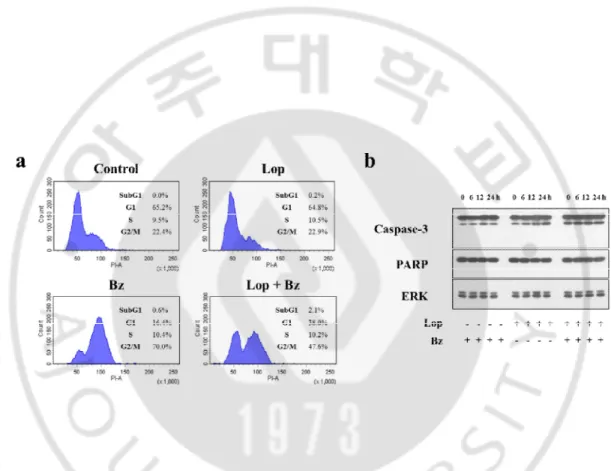
did not notably alter the cellular morphologies (Fig. 4). Interestingly, while treatment with borteomib alone for 24 h induced vacuolation in (20-30%) treated colon cancer cells, combined treatment with loperamide and bortezomib induced a dramatic vacuolation prior to cell death in all the treated cells. Because bortezomib was reported to induce apoptosis in many cancer cells (Patricia et al., 2007), we examined whether the combined treatment with loperamide and bortezomib kills the tested colon cancer cells via caspase-mediated apoptosis. However, cell deathinduced by the combined treatmentwas not significantly rescued by pretreatment with the pan-caspase inhibitor, z-VAD (Fig. 5, 6). Flow cytometry to measure the DNA content also showed that treatment of DLD-1 cells with 20 μM loperamide and 40 nM bortezomib for 24 h did not increase the sub-G1 DNA content (Fig. 7a). Furthermore, loperamide and bortezomib did not induce the proteolytic processing of caspase-3 and PARP, a substrate of caspase-3 (Fig. 7b). Immunocytochemical analysis of cytochrome c and COX IV, a mitochondrial protein, further revealed that the combined treatmentdid not induce the release of mitochondrial cytochrome c (Fig.8). Taken together, these results indicate that apoptosis may not critically contribute to the cytotoxicity of loperamide and bortezomib toward the tested cancer cells.We next examined whether vacuolation and subsequent cell death induced by the combined treatment might be associated with autophagy. First, we tested the effects of various autophagy inhibitors on the cell death induced by loperamide and bortezomib. However, the cell death induced by the combined treatment was not affected by autophagy inhibitors, including bafilomycin A1, 3-MA, chloroquine or wortmannin (Fig. 9). In addition, pretreatment with these autophagy inhibitors did not affect the vacuolation induced by loperamide and bortzomib (Fig. 10).Taken together, autophagy may not be associated with the vacuolation and subsequent cell
16
17
Figure 1. Combined treatment with loperamide and bortezomib induces the cell death in colon cancer cells. DLD-1, SW-480, RKO, and HCT-116 colon cancer cells were treated with
the indicated concentrations of combined treatment with loperamide and/or bortezomib for 24 h and then viability was assessed using calcein-AM and EthD-1. * p<0.05 vs. Control.
18
Figure 2. Loperamide plus bortezomib synergistically induce cell death in colon cancer cells.Classic isobologram at IC50. Cells were treated with loperamide and bortezomib for 24 h.
Isoboles for the combination of loperamide with bortezomib that were isoeffective (IC50) for
inhibition of cell viability are shown. The dashed line indicates the zero interaction of the isobole. IC50, the concentration of the drug required for 50% inhibition of cell viability; CI,
19
Figure 3.Effect of loperamide plus bortezomib on the long-term survival of DLD-1 cells.DLD-1 cells were treated with 20 μM loperamide plus 40 nM bortezomib for 4 h and
clonogenic assay was done as described in Matrials and Methods. Representative dishes after clonogenic assay are shown and colony-forming units were enumerated and expressed as the percentages of control cells.
20
Figure 4. Loperamide plus bortezomib induce cell death accompanied by vacuolation in colon cancer cells. DLD-1, SW-480cellswere treated with 20 μM loperamide plus 40 nM
bortezomib and RKO, HCT-116 cells were treated with 20 μM loperamide plus 10 nM bortezomib for 24 h and observed.
21
Figure 5.z-VAD-fmk does not block the cell death by loperamide plus bortezomib in colon cancer cells. DLD-1, SW-480, RKO, and HCT-116 colon cancer cells were untreated or
pretreated with 10 μM z-VAD-fmk for 30 min and further treated with 20 μM loperamide plus 40 nM bortezomib for 24 h. Cellular viability was assessed using calcein-AM and EthD-1.* p<0.05 vs. Control.
22
Figure 6. Combined treatment with loperamide and bortezomib induces caspase-independent cell death in DLD-1 and SW-480 cells. DLD-1, SW-480, RKO, and HCT-116
cells were untreated or pretreated with 10 μM z-VAD-fmk for 30 min and further treated with 20 μM loperamide plus 40 nM bortezomib (DLD-1, SW-480)/10 nM bortezomib (RKO, HCT-116) for 24 h and observed.
23
Figure 7. Combined treatment with loperamide and bortezomib does not induce apoptotic cell death in DLD-1 cells.(a) DLD-1 cells were treated with 20 μM loperamide plus 40 nM
bortezomib for 24 h. DNA content was analyzed by flow cytometry. (b) DLD-1 cells were treated with 20 μM loperamide plus 40 nM bortezomib for the indicated at time points. Whole cell extracts were prepared and subjected to western blotting using caspase-3 and anti-PARP antibodies. Anti-ERK antibody was used as a loading control.
24
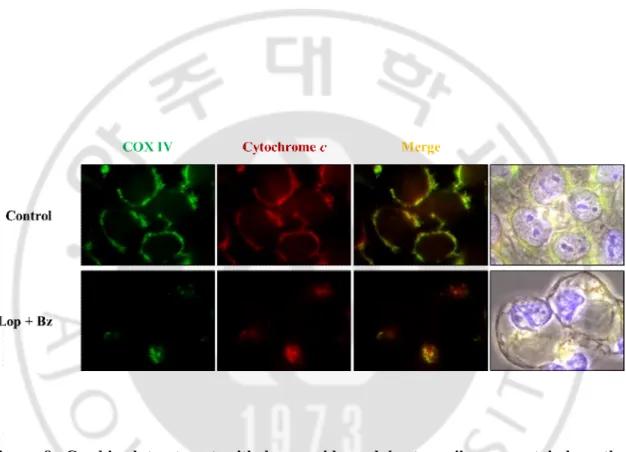
Figure 8. Combined treatment with loperamide and bortezomib may not induce the release of mitochondrial cytochrome c in DLD-1 cells. Immunocytochemistry using
anti-cytochrome c and anti-COX IV antibodies was performed in DLD-1 cells treated with combination of 20 μM loperamide and 40 nM bortezomib for 24 h. Representative fluorescence microscopic images of cells are shown.
25
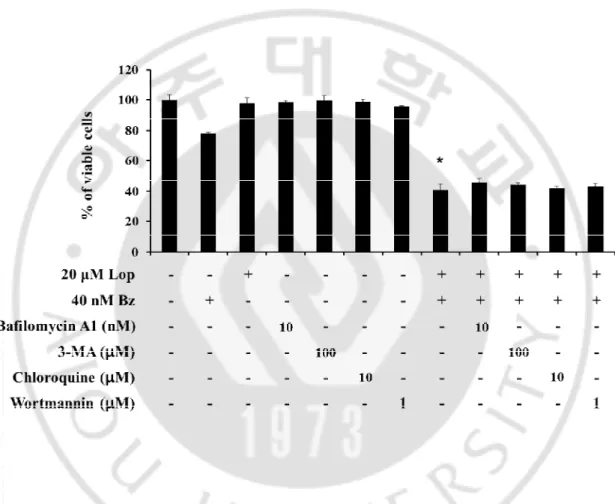
Figure 9.Effect of autophagy inhibitors on combined treatment-induced cell death.DLD-1
cells were untreated or pretreated with bafilomycin A1, 3-MA, chloroquine and wortmannin at the indicated concentrations for 30 min and further treated with 20 μM loperamide and 40 nM bortezomib for 24 h.* p<0.05 vs. Control.
26
Figure 10.Effect of autophagy inhibitors on combined treatment-induced vacuolation.DLD-1 cells were untreated or pretreated with 10 nM bafilomycin A1, 100 μM
3-MA, 10 μM chloroquine and 1 μM wortmannin at the indicated concentrations for 30 min and further treated with 20 μM loperamide and 40 nM bortezomib for 24 h and observed.
27
2. Intracellular Ca
2+influx is involved in the combined treatment-induced
vacuolation
Loperamide is an antidiarrheal drug with a minimal side effects (Awouters et al., 1983). The antidiarrheal effectofloperamide isassociated with the action on the μ-opioid receptors in the mesenteric plexus of large intestines (Loetchutinat et al., 2003). Also, loperamide at submicromolar concentrations blocks L-type calcium channels and augments intracellular calcium levels, when SOC channels are activated (Burleigh,1988; Harper et al., 1997). We investigated whether the sensitizing effect of loperamide on bortezomib-mediated cell death was derived from its action on the μ-opioid receptors. When we pretreated DLD-1 cells with naloxone, μ-opioid antagonist, vacuolation and cell death induced by loperamide plus bortezomib was not affected by it, suggesting that loperamide may enhance bortezomib-mediated cell death via independently of opoid receptor (Fig. 11a, b). Since loperamide elevates intracellular Ca2+ levels via activated SOC channels, we checked
whether the combined treatment affected perturbation of intracellular Ca2+ homeostasis in
DLD-1 cells. Flow cytometry using Fluo-3, a cell permeable Ca2+-indicator dye, showed
that treatment with 20 μM loperamide and 40 nM bortezomib for the indicated time points dramatically increased intracellular Ca2+ levels ([Ca2+]
i) in DLD-1 cells. Flow cytometry
using Rhod-2, an indicator dye for mitochondrial Ca2+, revealed that combined treatment
transiently increased mitochondrial Ca2+ levels ([Ca2+]
m) with a peak at 18 h (Fig. 12). We
28
inhibited combination-induced vacuolation and cell death.While treatment with each antagonist alone did not affect the viability of DLD-1 cells, treatment with 20 μM loperamide and 40 nM bortezomib for 24 h reduced their viability up to about 40%. Pretreatment with either BAPTA (a chelator of extracellular Ca2+), BAPTA-AM (a
chelator of free cytosolic Ca2+), ruthenium red (RR, mitochondrial Ca2+ uniporter
inhibitor)(Yoon et al., 2012), and Ru360 (dinuclear analog of RR) (Matlib et al., 1998) did not appear to affect combination treatment-induced cell death in these cells (Fig 13),suggesting that the simple scavenging of extracellular or intracellular Ca2+ does not
alter combined treatment-induced cell death. As shown in Fig. 14, chelating calcium withBAPTA-AM significantly delayed loperamide plus bortezomib-induced cellularvacuole formation. Interestingly, BAPTA, RR, and Ru360 did not affect cellular vacuolation induced by combined treatment (Fig. 14). These data suggestthat the increase in intracellular free calcium is related to the combined treatment with loperamide and bortezomib-induced vacuolation.
29
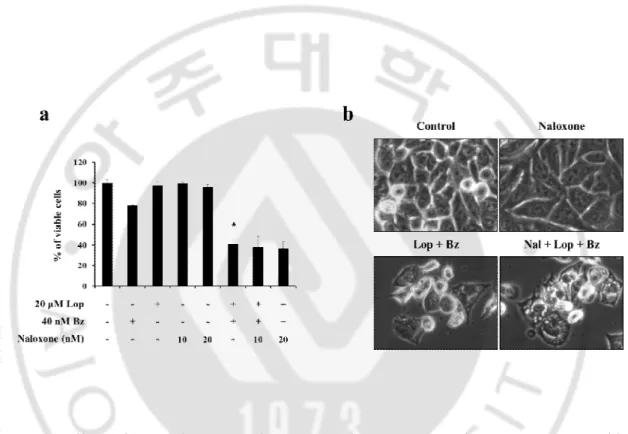
Figure 11.Effect of μ-opioid antagonist on combined treatment-induced cell death.(a)
DLD-1 cells were untreated or pretreated with naloxone at the indicated concentrations for 30 min and further treated with 20 μM loperamide and 40 nM bortezomib for 24 h and (b) observed.* p<0.05 vs. Control.
30
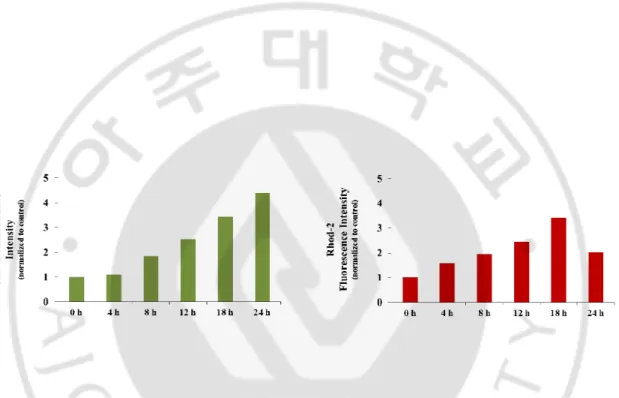
Figure 12. The combination of loperamide and bortezomib significantly enhances calcium levels in DLD-1 cells. DLD-1 cells treated with 20 μM loperamide and 40 nM bortezomib for
the indicated time points were stained with 2.5 μM Fluo-3 or 2.5 μM Rhod-2, and processed for FACS analysis. Intensity of Fluo-3 or Rhod-2 fluorescence were assessed and denoted in the graph.
31
Figure 13.Effect of various Ca2+-related inhibitors on the combination of loperamide and
bortezomib-induced cell death.DLD-1 cells were pretreated with the indicated concentrations
of various Ca2+ modulators and further treated with or without 20 μM loperamide and 40 nM
bortezomib for 24 h. Cellular viability was assessed using calcein-AM and EthD-1.* p<0.05 vs. Control.
32
Figure 14. BAPTA-AM delays the combination treatment-induced vacuolation, but not cell death. DLD-1 cells were pretreated with the indicated Ca2+ modulators (10 μM BAPTA, 10
μM BAPTA-AM, 5 μM RR, 5 μM Ru360, 1 mM EGTA) for 30 min, further treated with 20 μM loperamide plus 40 nM bortezomib for 10 h, and observed.
33
3. Combined treatment with loperamide and bortezomib induces ER stress, and
CHOP is critically involved in the cell death by loperamide plus bortezomib
Since scavenging of intracellular Ca2+ attenuated the vacuolation induced by loperamide
plus bortezomib, as shown in Fig. 14, we investigated whether these vacuoles are derived from mitochondria or the ER, which are known as major reservoirs for intracellular Ca2+
(Pivovarova et al., 2002). For this purpose, we established the DLD-1 sublines transfected with the ER-YFP plasmid for ER labeling. Fluorescent microscopy in YFP-ER cells showed that the ER exhibited as reticulate structure in untreated YFP-ER cells (Fig. 15). While treatment with 20 M loperamide alone did not affect the ER structures, treatment with 40 nM bortezomib alone started to induce small ER-derived vacuoles in about 20 % cells from 16 h of post-treatment, although it did not noticeably alter the ER structures. In contrast, combined treatment markedly induced ER-derived vacuoles in all the treated cells from 6 h. Afterwards, the sizes of these vacuoles were increased (Fig. 15), whereas their numbers were decreased, possibly suggesting that fusion among the swollen ER was progressed. At 18 h of the combined treatment, cells were fully occupied by several dramatically expanded the ER-derived vacuoles. At time points beyond 24 h, cells with reduced sizes were floated, suggesting that swollen ER structures might be collapsed in the progression of death. We further examined the changes in mitochondrial structures using the DLD-1 sublines stably transfected with YFP-Mito plasmid. While mitochondria in untreated YFP-Mito cells exhibited an elongated morphology, loperamide and/or
34
bortezomib similarly induced mitochondrial fragmentation (Fig. 16). Collectively, these results suggest that co-treatment with loperamide mainly accelerates the dilation and fusion of the ER induced by bortezomib, possibly leading to irreversible cell death.
It has been documented that proteasome inhibitors induce extensive cellular vacuolation likely due to ER stress (Wen-Xing et al., 2007). Therefore, we next investigated whether loperamide plus bortezomib also activates the signals associated with ER stress. We found that treatment of DLD-1 cells with 40 nM bortezomib or 20 M loperamide alone alone increased eIF2 phosphorylation, but combined treatment did not dramatically increased it. Bortezomib markedly increased ATF protein levels at 6 h, compared to those at 12 h, but combined treatment did not further increased them (Fig. 17). Although treatment with bortezomib alone induced up-regulation of CHOP from 6 h, its combination with loperamide more strongly up-regulated CHOP protein levels than bortezomib alone. Fluorescent microscopy using anti-CHOP antibody also showed that CHOP levels were dramatically increased in DLD-1 cells treated with loperamide plus bortezomib for 12h, compared to those treated with bortezomib alone (Fig. 18). Since among the tested proteins related to ER stress, only CHOP demonstrated time-dependent increase by the combined treatment, we next examined its functional significance in this cell death by employing the lentiviral system for CHOP knockdown. When we first examined the effect of shRNA-mediated CHOP knockdown, the dilation of the ER induced by treatment with loperamide plus bortezomib for 10 h was markedly attenuated by it (Fig. 19). Furthermore, suppression of CHOP expression significantly inhibited cell death by loperamide plus bortezomib (Fig. 20). Collectively, our results demonstrate that the induction of CHOP
35
36
Figure 15. Vacuolation induced by loperamide plus bortezomib is originated from the ER in DLD-1 cells. YFP-ER/DLD-1 cells were treated with combination of 20 μM loperamide and
37
Figure 16.Combination of loperamide and bortezomib inudces mitochondrial fragmentation.YFP-Mito cells were treated with 20 μM loperamide and 40 nM bortezomib for
38
Figure 17. The expression profiles of ER stress-related proteins in loperamide plus bortezomib-treated cells. DLD-1 cells treated with 20 μM loperamide and 40 nM bortezomib
for the indicated time points were subjected to Western blotting of the indicated proteins. ERK was examined to verify equal loading.
39
Figure 18. Loperamide plus bortezomib markedly increase CHOP levels in DLD-1 cells.
DLD-1 cells were treated with 20 μM loperamide and 40 nM bortezomib at time points. Immunocytochemistry using anti-CHOP antibodies were performed and the representative images of cells are shown.
40
Figure 19. CHOP blocks vacuolation induced by loperamide plus bortezomib. DLD-1 cells
stably expressing shCHOP were incubated with 20 μM loperamide and 40 nM bortezomib for 10 h and observed.
41
Figure 20. CHOP is critically involved in cell deathinduced by loperamide plus bortezomib.
DLD-1 cells stably expressing short-hairpin (sh) RNA complementary to CHOP (shCHOP) were incubated with 20 μM loperamide and 40 nM bortezomib for 10 h. Suppression of CHOP expression by its shRNA transduction was confirmed by Western blotting.* p<0.05 vs. Control, # p<0.05 vs. Lop + Bz.
42
4. ROS generation is important for the cell death induced by loperamide plus
bortezomib
Bortezomib was shown to induce ROS accumulation in tumor cells (Andrew et al., 2004; Takae et al., 2005; Pérez-Galán et al., 2006; Kawabata et al., 2012). Mitochondrial oxidative stress causes an imbalance in mitochondrial fission–fusion, resulting in mitochondrial fragmentation (Shengnan et al., 2011). Therefore, we attempted to investigate whether ROS played a critical role in colon cell death induced by loperamide plus bortezomib. Fluorescent microscopy using MitoSOX-Red, a fluorescent probe targeted to the mitochondria and highly selective to oxidation by superoxide (Lieven et al., 2006) showed that mitochondrial superoxide levels were markedly increased in DLD-1 cells treated with loperamide and bortezomib, but not in cells treated with loperamide or bortezomib alone (Fig. 21a). Flow cytometry using MitoSOX revealed that the combined treatment progressively increased mitochondrial superoxide levels (Fig. 21b). To examine the functional significance of increased mitochondrial superoxide levels in this cell death, we examined the effect of MnSOD mimetic, MnTBAP. MnTBAP pretreatment dose-dependently inhibited the cell death by loperamide plus bortezomib. In addition, pretreatment with the general antioxidants, NAC or GSH, also significantly inhibited this cell death (Fig. 22). Furthermore, pretreatment with NAC, GSH, and MnTBAP effectively blocked the vacuolationinduced by combined treatment (Fig. 23). These results suggest that ROS generation critically contributes to the cell death induced by loperamide plus bortezomib.
43
Figure 21. Loperamide plus bortezomib markedly increase mitochondrial ROS levels in DLD-1 cells. (a) Fluorescent microscopic observation of the combination of loperamide and
bortezomib-induced changes in the mitochondrial superoxide levels. DLD-1 cells were treated with 20 μM loperamide and 40 nM bortezomib for 24 h, and loaded with 2.5 μM MitoSOX-Red for 20 min. Fluorescence images of MitoSOX Red were obtained under the fluorescence microscope (b) DLD-1 cells were treated with 20 μM loperamide and 40 nM bortezomib for the indicated time points, exposed to 2.5 μM MitoSOX-Red for 20 min and analyzed by flow cytometry.
44
Figure 22.Effects of various antioxidants on the cell death by combination of loperamide and bortezomib.DLD-1 cells were pretreated with various antioxidants(NAC, GSH and
MnTBAP) at the indicated concentrations for 30 min and further treated with 20 μM loperamide and 40 nM bortezomib for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. * p< 0.05 vs. Control, # p< 0.05 vs. Lop + Bz.
45
Figure 23. Various antioxidants delay the ER dilation induced by loperamide plus bortezomib in DLD-1 cells. DLD-1 cells were pretreated with the indicated antioxidants (2
mM NAC, 1 mM GSH, 200 uM MnTBAP) for 30 min, further treated with 20 μM loperamide plus 40 nM bortezomib for 10 h, and observed.
46
5. Noxa is also critical factor for loperamide plus bortezomib-induced cell death
Several members of the Bcl-2 family are known targets ofbortezomib; thus, we studied the effect of 24 h of bortezomibtreatment on anti-apoptotic (Mcl-1)and pro-apoptotic (Bim and Noxa)protein expression. Previous reports showedthat bortezomib exposure leads to Mcl-1L accumulation (Qin et al., 2005; Perez-Galan et al., 2006) and Noxa up-regulation (Sachiko et al., 2011; Patricia et al., 2007).While Bcl-xL protein levels were not altered by loperamide and/or bortezomib, Mcl-1 protein levels were up-regulated similarly by bortezomib alone or loperamide plus bortezomib (Fig. 24). We found that bortezomib treatment up-regulated Noxa protein levels and co-treatment with loperamide further increased it. To investigate the possible role of Noxa up-regulation in the cell death induced by loperamide and bortezomib, we employed Noxa siRNA. We found that siRNA-mediated Noxa knockdown significantly rescued DLD-1 cells from the cell death by loperamide plus bortezomib (Fig. 25). However, vacuolation induced by the combined treatment was not notably affected by Noxa knockdown (Fig. 26). Taken together, our results demonstrate that accumulation of Noxa may contribute to the cytotoxicity of combined treatment with loperamide and bortezomib, but not ER dilation.
47
Figure 24.The expression profile of Bcl-2-related proteins in loperamide plus bortezomib-treated cells.DLD-1 cells bortezomib-treated with 20 μM loperamide and 40 nM bortezomib for the
indicated time points were subjected to Western blotting of the indicated proteins. ERK was examined to verify equal loading.
48
Figure 25. Noxa up-regulation critically contributes to the cell death by loperamide plus bortezomib. DLD-1 cells were treated with 40 nM control RNA, Noxa siRNA, and then treated
with 20 μM loperamide and 40 nM bortezomib for 10 h. Suppression of Noxa expression by its siRNA transfection was confirmed by Western blotting.* p<0.05 vs. Control, # p< 0.05 vs. Lop + Bz.
49
Figure 26.Effect of Noxa knockdown on the vacuolation induced by loperamide plus bortezomib.DLD-1 cells stably expressing siNoxa were incubated with 20 μM loperamide and
50
6. Disruptionof aggresome formation may be involved in the cell death induced by
loperamide and bortezomib
Aggresome formation by proteasome inhibition has been proposed as one of the resistance mechanism of bortezomib in pancreatic cancer cells (Nawrocki et al., 2006). Next, we investigated whether loperamide overcomes the resistance of colon cancer cells to bortezomib via modulation of aggresome formation. Staining using ProteoStat Aggresome dye showed that treatment of DLD-1 cells with bortezomib alone using showed the evident aggresome structures with circular shape, whereas loperamide treatment did not induce any change. In contrast, combined treatment induced the staining patterns with scattered and irregular shape (Fig. 27). Immunocytochemistry of ubiquitin and p62 also revealed that while treatment with bortezomib alone induced marked co-localization patterns with circular shape at perinuclear regions,ubiquitin and p62 was not colocalized by the combined treatment(Fig. 28). These results suggest that bortezomib-induced aggresome formation may be interfered by co-treatment with loperamide, possibly contributing to proteotoxicity due to failure in the aggresome-mediated sequestration of potentially toxic unfolded proteins.When we examined the protein levels of poly-ubiquitinated proteins, treatment with bortezomib resulted in the time-dependent accumulation of poly-ubiquitinated proteins in DLD-1 cells as assessed by western blotting,whereas accumulation of poly-ubiquitinated proteins was similarly induced at 12 h of the combined treatment but it was reduced at 24 h of post-treatment, compared with that by
51
bortezomib treatment alone (Fig.29). There results suggest that loperamide sensitizesDLD-1 cells to bortezomib not via further accumulation of poly-ubiquitinated proteins.
Since CHOP and Noxa have critical roles in thecell death by loperamide plus bortezomib, we checked whether CHOP or Noxa hasany effect on the accumulation of ubiquitinated proteins in DLD-1 cells treated with loperamide plus bortezomib. However, neither knockdown of CHOP nor Noxa did affect the accumulation of poly-ubiquitinated proteins(Fig. 30). These results suggest that accumulation of poly-ubiquitinated proteins may localize upstream of induction of CHOP or Noxa. Or we cannot exclude the possibility that accumulation of poly-ubiquitinated proteins may be controlled independent of induction of CHOP or Noxa. Interestingly, we found that CHOP knockdown markedly reduced the up-regulation of Noxa by loperamide plus bortezomib, suggesting that CHOP induction may play a role in Noxa up-regulation during the cell death by loperamide plus bortezomib. Taken together, these results indicate that loperamide disrupts the bortezomib-induced formation of aggresomes, contributing to overcoming the resistance of colon cancer cells to bortezomib.
52
Figure 27. Co-localization of aggresome and p62 in DLD-1 cells treated with loperamide plus bortezomib. DLD-1 cells were treated with 20 μM loperamide and/or 40 nM bortezomib
for 24 h. Immunocytochemistry using anti-p62 and ProteoStat® aggresome assay were performed and the representative images of cells are shown.
53
Figure 28. Co-treatment with loperamide does not increase bortezomib-induced accumulation of poly-ubiquitinated proteins in DLD-1 cells (immunocyto-chemistry).
DLD-1 cells were treated with 20 μM loperamide and/or 40 nM bortezomib for 24 h. Immunocytochemistry using anti-Ub and anti-p62 were performed and the representative images of cells are shown.
54
Figure 29.Co-treatment with loperamide does not increase bortezomib-induced accumulation of poly-ubiquitinated proteins in DLD-1 cells (Western blotting). DLD-1
cells treated with 20 μM loperamide and 40 nM bortezomib for the indicated time points were subjected to Western blotting of the anti-Ub antibodies. ERK was examined to verify equal loading.
55
Figure 30.CHOP induction may lie upstream of Noxa up-regulation in the cell death by loperamide plus bortezomib.DLD-1 cells were expressed short-hairpin (sh) RNA
complementary to CHOP (shCHOP) or treated with 40 nM control RNA, Noxa siRNA, and then treated with 20 μM loperamide and 40 nM bortezomib for 24 h. Treated proteins were subjected to Western blotting of the indicated proteins.
56
7. Combined treatment with loperamide and proteasome inhibitors does not
induce cell death in normal colon cells
Next, we examined the effect of loperamide plus other proteasome inhibitors (Carfilzomib, MLN9708,Epoxomicin, MG-132) on DLD-1 cells. We found that co-treatment with loperamide enhanced cell death, when combined with other proteasome inhibitors, similar to bortezomib (Fig. 31). When we examined the effect of loperamide and/or any proteasome inhibitor on the viability of CCD-112 and CCD-841 normal colon cells, neither a single treatment nor any combined treatment did induce cell death (Fig. 32). These results suggest that the combined treatment with loperamide and any proteasome inhibitor may selectively kill colon cancer cells, sparing normal cells. When we further examined the expression of CHOP and Noxa in these normal colon cells, treatment with loperamide and/or bortezomib did not up-regulate CHOP and Noxa, differently from those in DLD-1 cells (Fig. 33). These results indicate that the combination treatment with loperamide and bortezomib may selectively kill colon cancer cells via up-regulation of CHOP and Noxa.
57
Figure 31.Loperamide enhances the death of DLD-1 cells treated with other proteasome inhibitors.DLD-1 cells were treated with the indicated concentrations of combined treatment
with 20 μM loperamide and other proteasome inhibitors (Carfilzomib, MLN9708, Epoxomicin, MG-132)for 24 h and then viability was assessed using calcein-AM and EthD-1.* p<0.05 vs. Control.
58
Figure 32.Loperamide and/or proteasome inhibitors do not induce cell death in normal colon cells.Normal colon cells were treated with the indicated concentrations of combined
treatment with 20 μM loperamide and other proteasome inhibitors (Carfilzomib, MLN9708, Epoxomicin, MG-132)for 24 h and then viability was assessed using calcein-AM and EthD-1.
59
Figure 33.Loperamide and/or bortezomib do not induce CHOP and Noxa expression in normal colon cells. Normal colon cells were treated with 20 μM loperamide and 40 nM
bortezomib for the indicated time points were subjected to Western blotting of the indicated proteins. α-tubulin was examined to verify equal loading.
60
IV. DISCUSSION
Bortezomib is a 26S proteasome inhibitor that was approved by the Food and Drug Administration for the treatment of relapsed/refractory multiple myeloma and mantle cell lymphoma (Richardson et al., 2003; Richardson et al., 2005). Previous studies proposed that apoptosis induced by bortezomib requires the up-regulation of pro-apoptotic members of the Bcl-2 protein family, including Noxa, Bax, and Bik (Fribley et al., 2006; Li et al., 2008; Perez-Galan et al., 2007; Qin et al., 2005; Voortman et al., 2007; Zhu et al., 2005). In contrast, accumulation of anti-apoptotic factors, including Mcl-1, was reported to confer cancer cells resistance against bortezomib. On the other hand, bortezomib-induced apoptosis was associated with Mcl-1 cleavage regardless of Mcl-1L accumulation (Gomez-Bougie et al., 2007). In our study, we demonstrated that combination of loperamide and bortezomib could induce the synergistic killing of various colon cancer cells. Loperamide, an FDA-approved antidiarrhea drug, acts on the μ-opioid receptors in the mesenteric plexus of large intestines (Loetchutinat et al., 2003). The cell death by the combination treatment was non-apoptotic and non-autophagic in DLD-1 cells (Fig. 1-9). Since we observed dramatic ER-derived vacuolation prior to cell death by loperamide plus bortezomib, we hypothesized that this cell death might be related to ER stress. Indeed, loperamide plus bortezomib induced a marked induction of CHOP/GADD153, a marker of the ER stress signaling. In addition, co-treatment with loperamide further enhanced bortezomib-induced Noxa up-regulation. Knockdown of CHOP or Noxa could effectively rescue from the cell death by loperamide plus bortezomib, suggesting their critical involvement. In addition, we found that Noxa was down-regulated by CHOP
61
knockdown, suggesting the possible mechanistic link between the CHOP and Noxa of DLD-1 cells. We also found that the synergistic cell death may be dependent on ROS generation. While ROS play an important role in physiological cellular functions by activating several enzymatic cascades and transcription factors (Droge, 2002), excessive ROS signals, however, are detrimental, causing Ca2+ overload, mitochondrial depolarization, lipid peroxidation,
transcription factor activation and DNA damage, and lead to apoptotic and/or non-apoptotic cell death (Yanet al., 2006). In our study, loperamide plus bortezomib induced the generation of mitochondrial superoxide (MitoSOX-red). Scavenging of ROS by antioxidants (NAC and GSH) and MnSOD mimetic (MnTBAP) attenuated not only the vacuolation but also cell death induced by combined treatment. Aggresome has cytoprotective response that is activated in response to proteasome inhibition perhaps by shuttling ubiquitinated proteins to lysosomes for degradation (Garcia-Mata et al., 2002). We found that co-treatment with loperamide could disrupt aggresome formation induced by bortezomib, possibly contributing to the cell death by the combination. Since active protein synthesis appears to be required for induction of vacuolation and aggresome formation (Wasik et al., 2011; Steffan et al., 2008), we tested the effect of the translation inhibitor cycloheximide on vacuolation and cell death by loperamide plus bortezomib. We found that pretreatment with cycloheximide almost completely inhibited not only the dilation of the ER but also cell death(data not shown). We further tested whether cyclohemixide pretreatment has any effect on the key signals involved in the cell death by loperamide plus bortezomib. Interestingly, co-treatment with cycloheximide remarkably reduced the accumulation of poly-ubiquitinated proteins as well as induction of CHOP and Noxa. Taken together, these results indicate that protein synthesis is required for ER-derived dilation and
62
subsequent cell death induced by the combined treatment. When we analyzed the cell cycle following treatment with loperamide and/or bortezomib, bortezomib-induced G2/M arrest was abrogated by co-treatment with loperamide without increase in subG1 cell population. Therefore, we speculate that loperamide-mediated cell cycle progression may interfere bortezomib-induced G2 phase, accelerating protein synthesis and aggravating proteotoxicity. Further study on the effect of loperamide and/or bortezomib on cell cycle is required to understand the underlying mechanism of proteotoxicity by the combined treatment. Furthermore, the combined treatment with loperamide and bortezomib could induce intracellular and mitochondrial Ca2+
accumulation. BAPTA-AM pretreatment alleviated the ER dilation but not cell death, suggesting that the increased intracellular Ca2+ levels may be related with ER dilation but not
cell death. However, it remains to be clarified how the combined treatment with loperamide and bortezomib induces ROS generation and induction of CHOP/Noxa during the progression of cell death.
Because both bortezomib and loperamide are used in the clinic, safety of the respective drug is already approved. It has been reported that diarrhea is commonly seen with the use of bortezomib in the treatment of multiple myeloma and mantle cell lymphoma patients. In the pivotal studies with this agent, diarrhea occurred in 51% of patients, with 8% of the events being severe (Berenson et al., 2005). In addition to diarrhea, peripheral neuropathy is also a significant side effect of bortezomib. Peripheral neuropathy induced by bortezomib is occurred in 37-44% of clinical trial patients, requiring dose modification and potential changes in the treatment plan when it occurs (Cavaletti et al., 2010; Argyriou et al., 2010). In addition to the antidiarrheal effect, loperamide has been shown to demonstrate the analgesic effect (Kumar et
63
al., 2012). Therefore, co-treatment with loperamide is expected to not only enhance the anti-cancer effect of bortezomibbut also reduce the required dose of bortezomib, contributing to minimize its side effects. Furthermore, since combination of loperamide and bortezomib effectively induces cell death in various colon cancer cells, but not in normal cells, this combination regimen may provide a safe and effective cancer therapeutics against colon cancer that is resistant to anti-cancer effect of the proteasome inhibitor.
In conclusion, our results show that combined treatment with loperamide and bortezomib effectively kills colon cancer cells via up-regulation of CHOP and Noxa, ROS generation, and disruption ofaggresomes.