Dissociation Curves of Transition Metal Compounds
고동혁, 송수환, 심은지*
화학과, 연세대학교, (50 Yonsei-ro Seodaemun-gu, Seoul 03722, KOREA)
E-mail: [email protected], [email protected] [email protected] Abstract We present a study of transition metal compounds using density functional theory (DFT), and density-corrected density functional theory(DC-DFT). By replacing the self-consistent density with that obtained from Hartree-Fock calculation, i.e., HF-DFT, the abnormality driven by self-interaction error is removed in several important cases. We discuss when and how HF-DFT works by examining 3d orbital dimers using approximate functionals and by comparing the results from self-consistent-DFT and HF-DFT with experimental values.
Introduction
Transition metals are of critical importance in many fields. Its application is broad including but not limited to material science[1] and catalysts in organic chemistry[2]. Therefore, it is important to understand the characteristics of transition metals fundamentally. However, getting accurate and meaningful values from transition metals is very challenging both experimentally and theoretically.
In experiments[3], it is extremely difficult to segregate the results of specific spin-states, and therefore observe principle characters of transition metals. Also, controlling thermodynamic conditions are challenging and generate unwanted errors.
Meanwhile in theoretical chemistry, it is known that transition metals are recommended to use ab-initio methods despite its computational cost[4], since density
functional theory(DFT)[5], a commonly used tool in theoretical chemistry often fails because of the degenerated state of 3d
orbitals and its large self interaction errors (SIE)[6] However, most ab-initio methods require high computational costs, and cannot calculate large molecules within applicable terms of time[7].
In this work, we propose a method that uses density corrected density functional
theory (DC-DFT) that has similar
computational costs with DFT, while improving results significantly in some important cases.
Theory
The electron density, 𝑛𝑛𝑛𝑛(r) in closed shell molecules can be written in terms of a sum of products of basis functions φ, which is given by
𝑛𝑛𝑛𝑛(r) = N � � 𝑃𝑃𝑃𝑃𝑢𝑢𝑢𝑢𝑢𝑢𝑢𝑢 𝑢𝑢𝑢𝑢
𝜑𝜑𝜑𝜑𝑢𝑢𝑢𝑢𝜑𝜑𝜑𝜑𝑢𝑢𝑢𝑢 𝑢𝑢𝑢𝑢
Where N is the number of electrons and P is the density matrix. The density is approximated self-consistently. Using this density, The Kohn-Sham(KS) total energy, of the dimer can be calculated by the following equation
𝑬𝑬𝑬𝑬[𝒏𝒏𝒏𝒏] = 𝑻𝑻𝑻𝑻𝒔𝒔𝒔𝒔[𝒏𝒏𝒏𝒏] + 𝑽𝑽𝑽𝑽𝒏𝒏𝒏𝒏𝒏𝒏𝒏𝒏[𝒏𝒏𝒏𝒏] + 𝑽𝑽𝑽𝑽𝒏𝒏𝒏𝒏𝒏𝒏𝒏𝒏[𝒏𝒏𝒏𝒏] + 𝑱𝑱𝑱𝑱[𝒏𝒏𝒏𝒏] + 𝑬𝑬𝑬𝑬𝒙𝒙𝒙𝒙𝒙𝒙𝒙𝒙[𝒏𝒏𝒏𝒏] (1)
where 𝑇𝑇𝑇𝑇𝑠𝑠𝑠𝑠[𝑛𝑛𝑛𝑛] is the kinetic energy of noninteracting elections, 𝑉𝑉𝑉𝑉𝑛𝑛𝑛𝑛𝑛𝑛𝑛𝑛[𝑛𝑛𝑛𝑛] is the nucleus election potential energy, 𝑉𝑉𝑉𝑉𝑛𝑛𝑛𝑛𝑛𝑛𝑛𝑛[𝑛𝑛𝑛𝑛] is the nuclear repulsion energy, 𝐽𝐽𝐽𝐽[𝑛𝑛𝑛𝑛] is the classical election-election repulsion energy, and 𝐸𝐸𝐸𝐸𝑥𝑥𝑥𝑥𝑥𝑥𝑥𝑥[𝑛𝑛𝑛𝑛] is the exchange-correlation energy. The functionals of KS-DFT varies by approximating the exchange correlation differently. Many researchers are focused on trying to find a better exchange-correlation that cancels the various errors that KS-DFT is known to have including the SIE, static correlation and dynamic errors by comparing approximations with experimental values[8]. However, many people seem to disregard the error that results from the approximated density. To solve this problem, we proposed to use a better density to correct the self-consistent DFT, the so called DC-DFT[9]. By using Hartree-Fock (HF)[10] wave functions which are inert of SIE to correct the approximated density (HF-DFT), we solved some important cases that had the SIE problem. HF-DFT proposes to use the following equation
𝑬𝑬𝑬𝑬[𝐧𝐧𝐧𝐧] = 𝑬𝑬𝑬𝑬𝒌𝒌𝒌𝒌{𝝓𝝓𝝓𝝓𝑯𝑯𝑯𝑯𝑯𝑯𝑯𝑯} + 𝑽𝑽𝑽𝑽𝒏𝒏𝒏𝒏𝒏𝒏𝒏𝒏{𝝓𝝓𝝓𝝓𝑯𝑯𝑯𝑯𝑯𝑯𝑯𝑯} + 𝑽𝑽𝑽𝑽𝒏𝒏𝒏𝒏𝒏𝒏𝒏𝒏{𝝓𝝓𝝓𝝓𝑯𝑯𝑯𝑯𝑯𝑯𝑯𝑯} + 𝑱𝑱𝑱𝑱(𝝓𝝓𝝓𝝓𝑯𝑯𝑯𝑯𝑯𝑯𝑯𝑯) + 𝑬𝑬𝑬𝑬𝑫𝑫𝑫𝑫𝑯𝑯𝑯𝑯𝑻𝑻𝑻𝑻𝒙𝒙𝒙𝒙𝒙𝒙𝒙𝒙[𝒏𝒏𝒏𝒏] (2)
where 𝐸𝐸𝐸𝐸𝑘𝑘𝑘𝑘{𝜙𝜙𝜙𝜙𝐻𝐻𝐻𝐻𝐻𝐻𝐻𝐻}, 𝑉𝑉𝑉𝑉𝑛𝑛𝑛𝑛𝑛𝑛𝑛𝑛{𝜙𝜙𝜙𝜙𝐻𝐻𝐻𝐻𝐻𝐻𝐻𝐻}, 𝑉𝑉𝑉𝑉𝑛𝑛𝑛𝑛𝑛𝑛𝑛𝑛{𝜙𝜙𝜙𝜙𝐻𝐻𝐻𝐻𝐻𝐻𝐻𝐻} and 𝐽𝐽𝐽𝐽(𝜙𝜙𝜙𝜙𝐻𝐻𝐻𝐻𝐻𝐻𝐻𝐻) are calculated using the HF wave function approximation’s density {𝜙𝜙𝜙𝜙𝐻𝐻𝐻𝐻𝐻𝐻𝐻𝐻} , and 𝐸𝐸𝐸𝐸
𝑥𝑥𝑥𝑥𝑥𝑥𝑥𝑥[𝑛𝑛𝑛𝑛]
energy is given from DFT. In normal cases, which means that the error driven by the density is smaller than the exchange correlation error, KS-DFT and HF-DFT do not differ much. However, in abnormal cases where the density driven error is significantly larger than the functional error, HF-DFT calculations give better values by using more accurate densities.
Abnormal cases may be identified by the following equation. Linear response theory can be applied in KS systems[11] δn(r) = � 𝑑𝑑𝑑𝑑3𝑟𝑟𝑟𝑟′𝜒𝜒𝜒𝜒 𝑠𝑠𝑠𝑠(𝑟𝑟𝑟𝑟, 𝑟𝑟𝑟𝑟′)𝛿𝛿𝛿𝛿𝑉𝑉𝑉𝑉𝑠𝑠𝑠𝑠(𝑟𝑟𝑟𝑟′) (3) and 𝜒𝜒𝜒𝜒𝑠𝑠𝑠𝑠(𝑟𝑟𝑟𝑟, 𝑟𝑟𝑟𝑟′) = � 𝑓𝑓𝑓𝑓𝑖𝑖𝑖𝑖− 𝑓𝑓𝑓𝑓𝑗𝑗𝑗𝑗𝜙𝜙𝜙𝜙𝑖𝑖𝑖𝑖 ∗(𝑟𝑟𝑟𝑟)𝜙𝜙𝜙𝜙 𝑗𝑗𝑗𝑗∗(𝑟𝑟𝑟𝑟)𝜙𝜙𝜙𝜙𝑖𝑖𝑖𝑖(𝑟𝑟𝑟𝑟′)𝜙𝜙𝜙𝜙𝑗𝑗𝑗𝑗(𝑟𝑟𝑟𝑟′) 𝜖𝜖𝜖𝜖𝑖𝑖𝑖𝑖− 𝜀𝜀𝜀𝜀𝑗𝑗𝑗𝑗+ 𝑖𝑖𝑖𝑖0 𝑖𝑖𝑖𝑖,𝑗𝑗𝑗𝑗 (4)
is the static density-density KS response function[12] where δn(r) is the change in density induced by the change of potential 𝑉𝑉𝑉𝑉𝑠𝑠𝑠𝑠(𝑟𝑟𝑟𝑟′), 𝑓𝑓𝑓𝑓 is the KS orbital occupation factor, 𝜙𝜙𝜙𝜙 is molecular orbitals, and 𝜖𝜖𝜖𝜖 is molecular orbital energy. Since the HOMO-LUMO gap[13] has the smallest value, it could indicate when an error is induced by the approximated density. In normal cases, HOMO-LUMO gaps are reasonably large, reducing difference in density despite a large potential difference. However, in abnormal cases, HOMO-LUMO gaps are small, resulting in a larger difference in density and therefore a larger error.
𝑛𝑛𝑛𝑛(r) = N � � 𝑃𝑃𝑃𝑃𝑢𝑢𝑢𝑢𝑢𝑢𝑢𝑢 𝑢𝑢𝑢𝑢
𝜑𝜑𝜑𝜑𝑢𝑢𝑢𝑢𝜑𝜑𝜑𝜑𝑢𝑢𝑢𝑢 𝑢𝑢𝑢𝑢
Where N is the number of electrons and P is the density matrix. The density is approximated self-consistently. Using this density, The Kohn-Sham(KS) total energy, of the dimer can be calculated by the following equation
𝑬𝑬𝑬𝑬[𝒏𝒏𝒏𝒏] = 𝑻𝑻𝑻𝑻𝒔𝒔𝒔𝒔[𝒏𝒏𝒏𝒏] + 𝑽𝑽𝑽𝑽𝒏𝒏𝒏𝒏𝒏𝒏𝒏𝒏[𝒏𝒏𝒏𝒏] + 𝑽𝑽𝑽𝑽𝒏𝒏𝒏𝒏𝒏𝒏𝒏𝒏[𝒏𝒏𝒏𝒏] + 𝑱𝑱𝑱𝑱[𝒏𝒏𝒏𝒏] + 𝑬𝑬𝑬𝑬𝒙𝒙𝒙𝒙𝒙𝒙𝒙𝒙[𝒏𝒏𝒏𝒏] (1)
where 𝑇𝑇𝑇𝑇𝑠𝑠𝑠𝑠[𝑛𝑛𝑛𝑛] is the kinetic energy of noninteracting elections, 𝑉𝑉𝑉𝑉𝑛𝑛𝑛𝑛𝑛𝑛𝑛𝑛[𝑛𝑛𝑛𝑛] is the nucleus election potential energy, 𝑉𝑉𝑉𝑉𝑛𝑛𝑛𝑛𝑛𝑛𝑛𝑛[𝑛𝑛𝑛𝑛] is the nuclear repulsion energy, 𝐽𝐽𝐽𝐽[𝑛𝑛𝑛𝑛] is the classical election-election repulsion energy, and 𝐸𝐸𝐸𝐸𝑥𝑥𝑥𝑥𝑥𝑥𝑥𝑥[𝑛𝑛𝑛𝑛] is the exchange-correlation energy. The functionals of KS-DFT varies by approximating the exchange correlation differently. Many researchers are focused on trying to find a better exchange-correlation that cancels the various errors that KS-DFT is known to have including the SIE, static correlation and dynamic errors by comparing approximations with experimental values[8]. However, many people seem to disregard the error that results from the approximated density. To solve this problem, we proposed to use a better density to correct the self-consistent DFT, the so called DC-DFT[9]. By using Hartree-Fock (HF)[10] wave functions which are inert of SIE to correct the approximated density (HF-DFT), we solved some important cases that had the SIE problem. HF-DFT proposes to use the following equation
𝑬𝑬𝑬𝑬[𝐧𝐧𝐧𝐧] = 𝑬𝑬𝑬𝑬𝒌𝒌𝒌𝒌{𝝓𝝓𝝓𝝓𝑯𝑯𝑯𝑯𝑯𝑯𝑯𝑯} + 𝑽𝑽𝑽𝑽𝒏𝒏𝒏𝒏𝒏𝒏𝒏𝒏{𝝓𝝓𝝓𝝓𝑯𝑯𝑯𝑯𝑯𝑯𝑯𝑯} + 𝑽𝑽𝑽𝑽𝒏𝒏𝒏𝒏𝒏𝒏𝒏𝒏{𝝓𝝓𝝓𝝓𝑯𝑯𝑯𝑯𝑯𝑯𝑯𝑯} + 𝑱𝑱𝑱𝑱(𝝓𝝓𝝓𝝓𝑯𝑯𝑯𝑯𝑯𝑯𝑯𝑯) + 𝑬𝑬𝑬𝑬𝑫𝑫𝑫𝑫𝑯𝑯𝑯𝑯𝑻𝑻𝑻𝑻𝒙𝒙𝒙𝒙𝒙𝒙𝒙𝒙[𝒏𝒏𝒏𝒏] (2)
where 𝐸𝐸𝐸𝐸𝑘𝑘𝑘𝑘{𝜙𝜙𝜙𝜙𝐻𝐻𝐻𝐻𝐻𝐻𝐻𝐻}, 𝑉𝑉𝑉𝑉𝑛𝑛𝑛𝑛𝑛𝑛𝑛𝑛{𝜙𝜙𝜙𝜙𝐻𝐻𝐻𝐻𝐻𝐻𝐻𝐻}, 𝑉𝑉𝑉𝑉𝑛𝑛𝑛𝑛𝑛𝑛𝑛𝑛{𝜙𝜙𝜙𝜙𝐻𝐻𝐻𝐻𝐻𝐻𝐻𝐻} and 𝐽𝐽𝐽𝐽(𝜙𝜙𝜙𝜙𝐻𝐻𝐻𝐻𝐻𝐻𝐻𝐻) are calculated using the HF wave function approximation’s density {𝜙𝜙𝜙𝜙𝐻𝐻𝐻𝐻𝐻𝐻𝐻𝐻} , and 𝐸𝐸𝐸𝐸
𝑥𝑥𝑥𝑥𝑥𝑥𝑥𝑥[𝑛𝑛𝑛𝑛]
energy is given from DFT. In normal cases, which means that the error driven by the density is smaller than the exchange correlation error, KS-DFT and HF-DFT do not differ much. However, in abnormal cases where the density driven error is significantly larger than the functional error, HF-DFT calculations give better values by using more accurate densities.
Abnormal cases may be identified by the following equation. Linear response theory can be applied in KS systems[11] δn(r) = � 𝑑𝑑𝑑𝑑3𝑟𝑟𝑟𝑟′𝜒𝜒𝜒𝜒 𝑠𝑠𝑠𝑠(𝑟𝑟𝑟𝑟, 𝑟𝑟𝑟𝑟′)𝛿𝛿𝛿𝛿𝑉𝑉𝑉𝑉𝑠𝑠𝑠𝑠(𝑟𝑟𝑟𝑟′) (3) and 𝜒𝜒𝜒𝜒𝑠𝑠𝑠𝑠(𝑟𝑟𝑟𝑟, 𝑟𝑟𝑟𝑟′) = � 𝑓𝑓𝑓𝑓𝑖𝑖𝑖𝑖− 𝑓𝑓𝑓𝑓𝑗𝑗𝑗𝑗𝜙𝜙𝜙𝜙𝑖𝑖𝑖𝑖 ∗(𝑟𝑟𝑟𝑟)𝜙𝜙𝜙𝜙 𝑗𝑗𝑗𝑗∗(𝑟𝑟𝑟𝑟)𝜙𝜙𝜙𝜙𝑖𝑖𝑖𝑖(𝑟𝑟𝑟𝑟′)𝜙𝜙𝜙𝜙𝑗𝑗𝑗𝑗(𝑟𝑟𝑟𝑟′) 𝜖𝜖𝜖𝜖𝑖𝑖𝑖𝑖− 𝜀𝜀𝜀𝜀𝑗𝑗𝑗𝑗+ 𝑖𝑖𝑖𝑖0 𝑖𝑖𝑖𝑖,𝑗𝑗𝑗𝑗 (4)
is the static density-density KS response function[12] where δn(r) is the change in density induced by the change of potential 𝑉𝑉𝑉𝑉𝑠𝑠𝑠𝑠(𝑟𝑟𝑟𝑟′), 𝑓𝑓𝑓𝑓 is the KS orbital occupation factor, 𝜙𝜙𝜙𝜙 is molecular orbitals, and 𝜖𝜖𝜖𝜖 is molecular orbital energy. Since the HOMO-LUMO gap[13] has the smallest value, it could indicate when an error is induced by the approximated density. In normal cases, HOMO-LUMO gaps are reasonably large, reducing difference in density despite a large potential difference. However, in abnormal cases, HOMO-LUMO gaps are small, resulting in a larger difference in density and therefore a larger error.
Calculation
In this paper, we studied dissociation potential energy surfaces of transition metal compounds with DFT and HF-DFT. We also calculated CCSD(T)[14] which is called the “golden standard” in ab-initio calculations as a reference. Dissociation curves calculated with DFT are well known to be largely affected by SIE. Also, transition metal compounds are very challenging systems for the DFT due to their degenerated 3d orbitals. For practical comparisons between functionals and densities, molecular vibrational frequency is omitted. Various dissociation curves are graphed by calculating the binding energy, 𝐸𝐸𝐸𝐸𝑏𝑏𝑏𝑏 which is given by
𝐸𝐸𝐸𝐸𝑏𝑏𝑏𝑏(R) = 𝐸𝐸𝐸𝐸𝐴𝐴𝐴𝐴𝐴𝐴𝐴𝐴𝑡𝑡𝑡𝑡𝑡𝑡𝑡𝑡𝑡𝑡𝑡𝑡(𝑅𝑅𝑅𝑅) − (𝐸𝐸𝐸𝐸𝐴𝐴𝐴𝐴+ 𝐸𝐸𝐸𝐸𝐴𝐴𝐴𝐴) (5)
where 𝐸𝐸𝐸𝐸𝐴𝐴𝐴𝐴𝐴𝐴𝐴𝐴𝑡𝑡𝑡𝑡𝑡𝑡𝑡𝑡𝑡𝑡𝑡𝑡(𝑅𝑅𝑅𝑅) is the energy of the dimer at distance R, and 𝐸𝐸𝐸𝐸𝐴𝐴𝐴𝐴 and 𝐸𝐸𝐸𝐸𝐴𝐴𝐴𝐴 are the energy of each atom.
First, we calculated the K2 dimer which has no electrons in the 3d orbital to test the HF density. While evaluating the results, we chose three DFT functionals to examine ScH and ScH+ DC-DFT and HF-DFT calculations - PBE and BLYP as generalized gradient approximates(GGA)[15], and B3LYP as a hybrid functional[16].
Since the spin state of the dimer and dissociated atoms could be different, multiplicity is considered. The logical spin multiplicities for each distance is calculated and the lower energy value is used. All
calculations are done with the Turbomole program.[17]
Results and Discussion
A. K2
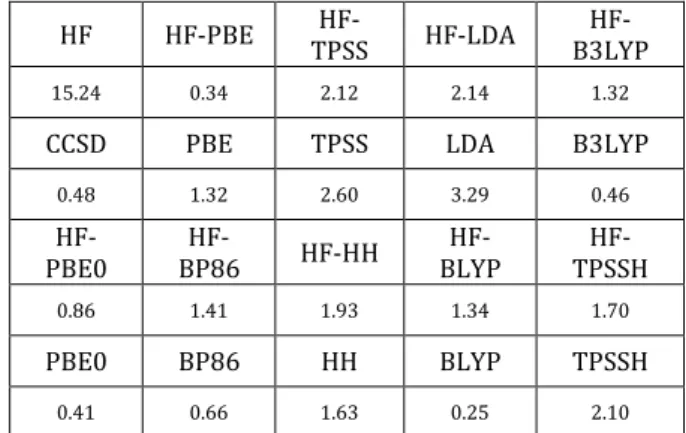
As shown in Table 1, BLYP and HF-PBE meet chemical convergence. B3LYP is also chosen since it is a popular functional used in various fields.
HF HF-PBE TPSS HF- HF-LDA B3LYP
HF-15.24 0.34 2.12 2.14 1.32
CCSD PBE TPSS LDA B3LYP
0.48 1.32 2.60 3.29 0.46 HF-PBE0 BP86 HF- HF-HH BLYP HF- TPSSH HF-0.86 1.41 1.93 1.34 1.70 PBE0 BP86 HH BLYP TPSSH 0.41 0.66 1.63 0.25 2.10
Table 1. Ground state binding energy error(kcal/mol)
compared with experimental value (3.9 Å, -12kcal/mol)[18]
The HOMO-LUMO gap of K2 shown in Figure 1 implies that K2 is a normal case, where functional driven error is larger than density driven error.
Figure 1. HOMO-LUMO gap of K2
0 1 2 3 4 5 3 4 5 E( Ev ) R(Å) HF pbe b-lyp b3-lyp
B. ScH
In Figure 2, HF-DFT shows better results, indicating ScH is an abnormal case. This is supported by the small HOMO-LUMO gap of DFT as shown in Figure 3 The discontinuous point in CCSD(T) most likely occurred because of the HF approximations used for CCSD(T) calculations, are done with single reference slater determinents.
Figure 2. Dissociation curve of ScH
Figure 3. HOMO-LUMO gap of ScH C. ScH+
In Figure 3, the PBE functional seems to follow the reference values, while HF-DFT values under-bind the compound, indicating a normal case. However, the HOMO-LUMO gap shown in Figure 4 suggests that the PBE density
approximation could also have a large error. This should be investigated in further research
Figure 4. Dissociation curve of ScH+
Figure 5. HOMO-LUMO gap of ScH+
Conclusion
The dissociation curve of ScH, a 3d orbital transition metal shows that errors were diminished by using better densities, rather than better functionals. However, DFT using the PBE functional were better at approximating dissociation curves than HF-DFT when the gap showed a possible density error. This needs further investigation, while developing other methods to identify abnormal cases.
A
CKNOWLEDGMENTSThis research was supported by the EDISON -80 -60 -40 -20 1 2 3 Eb (K ca l/ m ol ) R(Å) HF pbe Hfpbe b3-lyp HFb3-lyp b-lyp HFb-lyp CCSD(T) 0 3 6 9 1 2 3 Eb (K ca l/m ol ) R(Å) HF pbe b-lyp b3-lyp -80 -30 1 2 3 Eb (K ca l/m ol ) R(Å) HF pbe Hfpbe b3-lyp HFb3-lyp b-lyp HFb-lyp CCSD(T) 0 5 10 1 2 3 Eb (K ca l/m ol ) R(Å) HF pbe b-lyp b3-lyp
B. ScH
In Figure 2, HF-DFT shows better results, indicating ScH is an abnormal case. This is supported by the small HOMO-LUMO gap of DFT as shown in Figure 3 The discontinuous point in CCSD(T) most likely occurred because of the HF approximations used for CCSD(T) calculations, are done with single reference slater determinents.
Figure 2. Dissociation curve of ScH
Figure 3. HOMO-LUMO gap of ScH C. ScH+
In Figure 3, the PBE functional seems to follow the reference values, while HF-DFT values under-bind the compound, indicating a normal case. However, the HOMO-LUMO gap shown in Figure 4 suggests that the PBE density
approximation could also have a large error. This should be investigated in further research
Figure 4. Dissociation curve of ScH+
Figure 5. HOMO-LUMO gap of ScH+
Conclusion
The dissociation curve of ScH, a 3d orbital transition metal shows that errors were diminished by using better densities, rather than better functionals. However, DFT using the PBE functional were better at approximating dissociation curves than HF-DFT when the gap showed a possible density error. This needs further investigation, while developing other methods to identify abnormal cases.
A
CKNOWLEDGMENTSThis research was supported by the EDISON -80 -60 -40 -20 1 2 3 Eb (K ca l/ m ol ) R(Å) HF pbe Hfpbe b3-lyp HFb3-lyp b-lyp HFb-lyp CCSD(T) 0 3 6 9 1 2 3 Eb (K ca l/m ol ) R(Å) HF pbe b-lyp b3-lyp -80 -30 1 2 3 Eb (K ca l/m ol ) R(Å) HF pbe Hfpbe b3-lyp HFb3-lyp b-lyp HFb-lyp CCSD(T) 0 5 10 1 2 3 Eb (K ca l/m ol ) R(Å) HF pbe b-lyp b3-lyp
Program through the National Research Foundation of Korea(NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2011-0020576)[19]
R
EFERENCE[1] Tegus, O., Brück, E., Buschow, K. H. J., & De Boer, F. R. Nature, 150-152. (2002)
[2] Aiken, John D., and Richard G. Finke. Journal of Molecular Catalysis A: Chemical 145.1 (1999) [3] Decurtins, S., Gütlich, P., Köhler, C. P., Spiering, H., & Hauser, A. Chemical physics letters, 105(1), (1984) [4] Kresse, G., & Furthmüller, J. Computational Materials Science, 6(1), 15-50 (1996).
[5] Kohn, Walter, and Lu Jeu Sham. Physical review 140.4A (1965)
[6] Cohen, Aron J., Paula Mori-Sánchez, and Weitao Yang. Chemical Reviews 112.1 (2011)
[7] Carter, Emily A. Science 321.5890 (2008)
[8] Becke, Axel D. The Journal of chemical
physics 98.2 (1993)
[9] Kim, Min-Cheol, Eunji Sim, and Kieron Burke. The Journal of chemical physics 140.18 (2014) [10] Boys, S. Francis. Proceedings of the Royal Society of London A: Mathematical, Physical and Engineering Sciences. Vol. 200. No. 1063. The Royal Society, (1950)
[11] Kim, M. C., Park, H., Son, S., Sim, E., & Burke, K.
The journal of physical chemistry letters, 6(19) (2015) [12] Kim, Min-Cheol, Eunji Sim, and Kieron Burke. Physical review letters 111.7 (2013).
[13] Kim, Min-Cheol, Eunji Sim, and Kieron Burke.
The Journal of Chemical Physics (2011)
[14] Stanton, John F., and Rodney J. Bartlett. The Journal of chemical physics 98.9 (1993)
[15] Perdew, John P., Kieron Burke, and Matthias Ernzerhof Physical review letters 77.18 (1996)
[16] Becke, Axel DThe Journal of chemical
physics 98.7 (1993)
[17] TURBOMOLE V7.1 2016, a development of
University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH,
since 2007; available
from http://www.turbomole.com.
[18] Furche, Filipp, and John P. Perdew The Journal of chemical physics 124.4 (2006)