저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Master
’s Thesis in the Department of
Biomedical Sciences
Ischemic Vulnerability in the Brain
of an Alzheimer
’s Mouse Model
Ajou University Graduate School
Major in Neuroscience
Ischemic Vulnerability in the Brain
of an Alzheimer
’s Mouse Model
Supervised by
Jin Soo Lee, Ph.D.
I submit this thesis as the Master’s thesis in the Department of Biomedical Sciences.
February, 2019
Ajou University Graduate School
Major in Neuroscience
The Master’s thesis of Keoung Sun Son in Department of Biomedical Sciences in hereby
approved.
Thesis Defense Committee President Member Jin Soo Lee seal Member Ji Man Hong seal Member Chan Bae Park seal
Ajou University Graduate School
December 24, 2018
i
-Abstract-
Ischemic Vulnerability in the Brain of an Alzheimer’s Mouse Model
Alzheimer's disease (AD) is a condition that mostly affects elderly individuals who are also at an increased risk of cerebrovascular disease. It is not well known if AD pathology results in poorer outcomes for cerebral ischemia. In the current study, we hypothesized that a brain affected by AD is vulnerable to cerebral ischemia, resulting in greater necrosis and neuronal cell death by inducing cerebral amyloid beta (Aβ) and tau protein accumulation, as well as degenerative changes. In this study, the effect of transient cerebral ischemia induced by bilateral carotid artery occlusion, which is often used as a vascular dementia model, was assessed in a transgenic (TG) mouse model of AD (3xTG-AD). After 15 minutes of bilateral carotid artery occlusion, followed by reperfusion, we evaluated cognitive changes, extent of neuronal death, and dementia-specific neuropathological changes compared with wild-type (WT) mice. Results of behavioral studies demonstrated that TG mice exposed to cerebral ischemia showed greater deficits in spatial learning and memory than similarly treated WT mice. Cognitive impairment in TG mice might be attributed to greater neuronal cell death due to brain ischemia compared with WT mice. However, similar Aβ accumulation was observed in both groups. Expression of caspase-3, Iba-1, and GFAP was increased in ischemic TG mice. In conclusion, our results suggest that Alzheimer’s neuropathology can
ii
trigger changes that make AD brains more vulnerable to cerebral ischemia/reperfusion.
Key word: Alzheimer’s disease, cerebral ischemia, 3xTG-AD mice, amyloid beta, tau protein, neuronal change
iii
TABLE OF CONTENTS
ABSTRACT ... ⅰ TABLE OF CONTENTS... ⅲ LIST OF FIGURES ... v I. INTRODUCTION ... 1II. MATERIALS AND METHODS ... 4
A. Animal care and study time frame ... 4
B. Surgery ... 4
C. Behavior tests ... 5
1. Morris water maze test ... 5
2. Y maze test ... 6
D. Tissue processing ... 7
E. Cresyl violet staining ... 7
F. Immunohistochemistry ... 7
G. Immunofluorescence staining... 8
H. Thioflavin S ... 9
I. Statistical analyses ... 9
III. RESULTS ... 10
iv
B. Neuronal cell death ... 13
C. Changes in Alzheimer’s pathology ... 16
1. Amyloid plaques ... 16
2. Amyloid precursor protein ... 18
3. Tau protein ... 21
D. Glial changes ... 23
IV. DISCUSSION ... 27
V. REFERENCES ... 31
v
LIST OF FIGURES
Figure 1. Effect of tBCCAO on memory as measured by the Morris
water maze test and spontaneous alternation test in the Y maze ... 11
Figure 2. Effect of tBCCAO on neuronal cell death ... 14
Figure 3. Amyloid plaques’ Thioflavin S expression in TG ... 17
Figure 4. Amyloid precursor Y188 expression ... 19
Figure 5. Tau protein AT8 expression ... 22
1
I. Introduction
With an aging society, the incidence of Alzheimer’s disease (AD) has been increasing, along with a characteristic declining cognitive function in elderly individuals. In parallel, the incidence of vascular dementia, a vascular disease, is also known to be increasing (Gorelick, Scuteri et al. 2011). AD and ischemic stroke both involve pathological brain changes. Both pathological changes may cause synergistic effects on neuronal damage, resulting in further memory loss and learning disability (Swaab, Hofman et al. 1994). In addition, recent evidence has shown that ischemia/reperfusion (I/R) exacerbates amyloid pathology, which is associated with AD, and that vascular stress in animal models contributes significantly to cognitive deficiency (Song, Ao et al. 2013).
AD caused by mutations in the APP and PS1 genes increases amyloid beta production and accumulation in the brain. Amyloid beta 42 oligomerization and deposition as diffuse plaques are activated by microglia and astrocytes and affects synapses. As a result, synaptic and neuritic injury causes oxidative injury, kinase/phosphatase is activated, and neurofibrillary tangles accumulate. This process leads to cell death and tissue loss in the brain due to the accumulation of amyloid beta plaques and neurofibrillary tangles (Hardy and Selkoe 2002). This
2
reduces the brain size and affects the brain function (Moolman, Vitolo et al. 2004). In addition, changes in cerebral blood flow (CBF) contribute to the pathogenesis of AD (Montagne, Nation et al. 2016, Kisler, Nelson et al. 2017), resulting in neurovascular deficits such as disruption of neuronal function and accumulation of amyloid in the brain. Thus, both cognitive impairment and AD are associated with neurovascular dysfunction.
Vascular dementia (VaD) is the second most common type of dementia after AD. Cerebral hypoperfusion contributes to the development and progression of dementia through inflammation and oxidative stress pathways (Cechetti, Pagnussat et al. 2012, Fan, Wan et al. 2014, De Silva and Miller 2016). Despite the involvement of many other mechanisms in VaD, there is increasing evidence that cerebral hypoperfusion plays an important role in the onset and progression of VaD by eliciting inflammatory responses (Xu, Zhang et al. 2016, Kisler, Nelson et al. 2017) which involve activation and accumulation of inflammatory cells, such as microglia and astrocytes, in the hippocampus (Jo, Law et al. 2014, Xu, Zhang et al. 2016).
Tau protein is present in a part of the microtubule. Hyperphosphorylation of tau protein by AD causes an accumulation of abnormal filaments (Delacourte, David et al. 1999). This leads to microtubule structure collapse and
3
neurofibrillary tangles. Activation of tau protein activates microglia (Leyns and Holtzman 2017). This causes a deterioration in microgliosis and astrocyte co-activation (Ferrer, Lopez-Gonzalez et al. 2014). Thus, pro-inflammatory cytokine secretion increases toxicity and impairs function. Gliosis and inflammatory signaling can affect tau phosphorylation and improve aggregation. The chronic neuroinflammation associated with tau pathology is worse the neurodegeneration (Lull and Block 2010, Ransohoff 2016).
We previously found that cognitive decline had an additive effect in the AD model when mild chronic cerebral ischemia was induced (Lee, Im et al. 2011). However, it is not well known whether transient stronger cerebral ischemia would induce cognitive decline or changes in AD pathologies. Here, we evaluated the cognitive function and neurological deficits associated with I/R in 3x transgenic (TG) AD mice.
4
II. MATERIALS AND METHODS
A. Animal care and study time frame
3x TG-AD mice (n=8 ischemia/5 non-ischemia/1 positive control) and wild-type (WT) (C57BL/6J) mice (n=7 ischemia/5 non-ischemia) were used in this study. Thirteen 14-month-old female mice carrying the APPSwe, tauP301L, and PS1M148V transgenes (3x TG-AD) were used. Twelve WT littermates served as controls. Mice had ad libitum access to food and water, were kept in cages maintained at humidity level of 55±10% and a constant temperature of 22±2°C, and maintained under 12-h/12-h light/dark cycles (lights on from 7:00 to 19:00). All animal protocols were approved by the Institutional Animal Management and Use Committee of Ajou University School of Medicine.
B. Surgery
Female 3xTG and C57BL/6J mice were anesthetized with 2% isoflurane and maintained at 1.5% isoflurane via a face mask. Rectal temperature was measured during surgery and maintained at 37°C on a heating pad. Short-term cerebral ischemia was induced by transient bilateral common carotid artery occlusion (tBCCAO). Briefly, the BCCAO was isolated and occluded with a microvascular clip.
5
Fifteen minutes following ischemia induction, the clips were removed from both arteries to allow blood recirculation. C. Behavioral tests
1. Morris water maze test
The Morris water maze (MWM) was used to assess changes in cognition and behavior among groups. Three days after tBCCAO, the mice underwent the MWM test for a period of 5 days. The MWM pool was 2 m in diameter and filled with white water at a temperature of 25 ± 1°C and a depth of 25 cm. During capture training, a translucent acrylic platform (diameter 10 cm) was placed in the center of the northeast quadrant at a depth of 1.5 cm below the water surface. The pool had spatial clues and was located in the center of the experiment room, which was equipped with a video camera attached to the ceiling that was used to record the swimming path.
The mice were tested 3 times a day for 5 consecutive days. In each of the 3 tests, the mouse was gently released into the water, facing the tank wall, at four different starting positions that were equally spaced around the pool wall. Mice were allowed 60 seconds to find the hidden platform. Upon reaching the platform, the mouse was left for 10 seconds. If the mouse was unable to find the platform within 60 seconds, the training ended and the maximum score of 60 seconds was assigned. In such cases, the mouse was
6
guided by hand to the hidden platform, where it was allowed to remain for 15 seconds. The latency to escape to the hidden platform was recorded as a metric of spatial learning performance. In order to assess spatial memory, a probe test was performed 24 hours after the last training test. In this test, we removed the platform from the pool, and mice swam freely for 60 seconds before they were removed from the water.
2. Y maze test
The Y maze test was performed as described previously. Spatial cognition was investigated using spontaneous alternation in a Y-type maze device, a horizontal maze consisting of three arms (40 cm in length, 3 cm in width, 12 cm in height), with the arms at an angle of 120° to each other. Animals were initially placed in the center of the maze, and the number of arm entries made in a period of 8 minutes were manually recorded for each animal. Spontaneous alternation was defined as the consecutive entry into all 3 arms. The maze was thoroughly washed with water to remove any residual odor after the completion of a test before the next mouse was placed in the maze. The percentage of alternations was defined as % = (number of alternations)/ (all entries in entry) × 100. The total number of arm entries was also used as an indicator of walking activity.
7
D. Tissue processing
After surgery, mice were anesthetized and transcardially perfused with 30 ml of 1X phosphate-buffered saline (PBS) followed by 30 ml of 4% paraformaldehyde (PFA). Perfused brains were extracted, cut along the midline, post-fixed in 4% PFA for 24 hours, and dehydrated with 30% sucrose. The brain was followed by OCT processing, and 20-μm coronal sections were cut using a Leica cryostat. The brain tissue was stored in a stock solution at 4°C. E. Cresyl violet staining
Neuronal damage to the hippocampus was visualized by cresyl violet staining. Brain sections were mounted on coated slidesthat were allowed to dry at room temperature overnight. Brain sections were stained with 0.1% cresyl violet solution for 5 minutes. The stained brain slices were washed in distilled water. The brain slices were then dehydrated with ethanol, immersed in xylene for 5 minutes, and mounted on slides in a Vecta mounting solution.
F. Immunohistochemistry
Brain sections were mounted on coated slides and washed with 1X phosphate buffered saline for 5 minutes. Endogenous peroxidase activity was inhibited by adding 0.3 % hydrogen peroxide for 5 minutes followed by 0.25%
8
Triton x-100 for 10 minutes. After washing the brain sections, they were blocked with 10% normal horse serum for 1 hour and allowed to react with primary antibodies (anti-Iba-1, 1:1000 dilution; Wako Chemicals GmbH, Neuss, Germany; anti-GFAP, 1:500 dilution; DAKO, Glostrup, Denmark; anti-active cleaved caspase-3, 1:20 dilution, Millipore, Darmstadt, Germany; anti-AT8, 1:100 dilution Thermo Fisher Scientific, Malvern, PA, USA; anti-APP-Y188, 1:750 dilution, Abcam, Princeton, NJ, USA) overnight at 4℃. Subsequently, brain sections were incubated with biotinylated secondary antibody (1:200 dilution, Vector Laboratories, Inc., Burlingame, CA, USA) for 2 hours and reacted with an avidin–biotin-complex reagent (ABC, Vector Laboratories, Inc., Burlingame, CA, USA) for 1 hour. Nickel-enhanced 3,3′-diaminobenzidine (DAB, Vector Laboratories, Inc., Burlingame, CA, USA) was used for visualization of immunoreactivity.
G. Immunofluorescence staining
Brain sections were mounted on coated slides and washed with 1X phosphate buffered saline for 5 minutes. Endogenous peroxidase activity was inhibited by adding 0.3 % hydrogen peroxide for 5 minutes followed by 0.25% Triton x-100 for 10 minutes. After washing the brain sections, they were blocked with 10% normal horse serum for 1 hour and incubated with Anti-APP-Y188 (1:750
9
dilution, Abcam, Princeton, NJ, USA) in combination with anti-NeuN (1:500 dilution, Millipore, Darmstadt, Germany) antibodies overnight for 4°C. Sections were then incubated with Alexa Fluor 488 (1:200 dilution, Invitrogen, Eugene, OR, USA) or Alexa 555 (1:500 dilution, Invitrogen) secondary antibody. Slides were embedded with a Vecta shield mounting solution (Vector Laboratories, Inc.) containing 4′,6-diamidino-2-phenylindole (DAPI).
H. Thioflavin S
Brain sections were mounted on coated slides and washed with 1X phosphate buffered saline. Brain sections were stained with 1% thioflavin-S (T1892, Sigma, St. Louis, MO) solution for 5 minutes. Brain sections were then differentiated in 70% ethanol for 5 minutes and rinsed twice in dH2O. Slides were embedded with a Vecta shield mounting solution (Vector Laboratories, Inc.) containing 4′,6-diamidino-2-phenylindole (DAPI).
I. Statistical analyses
The data are presented as means ± standard error of the mean (S.E.M.) and statistically analyzed using a Student’s t-test in GraphPad’s Prism program. Differences were considered to be significant for P values <0.05.
10
III.
RESULTS
A. Behavioral testing after tBCCAO
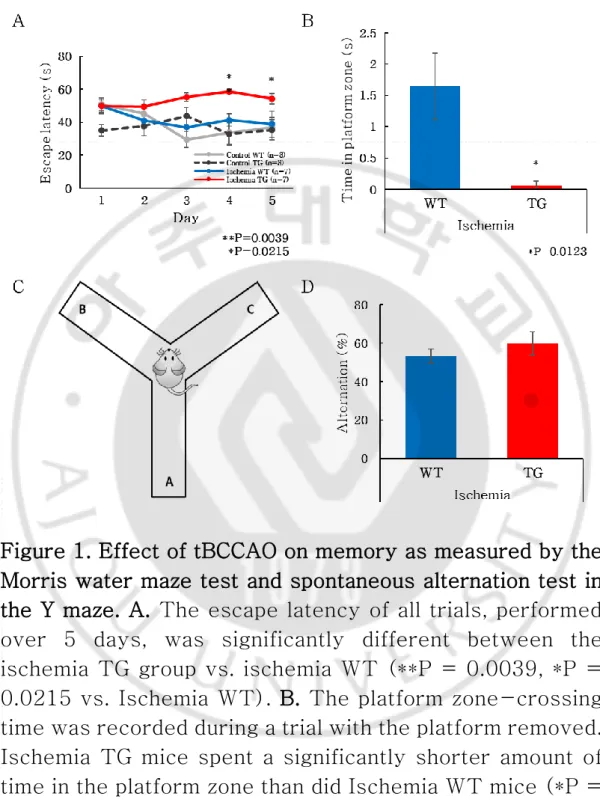
The Morris water maze (MWM) test was used to assess the effect of tBCCAO on learning memory in the AD mice. The TG mice showed no improvement in latency in the MWM test over time after exposure to ischemia, whereas the WT mice showed decreased latency as the trial proceeded, irrespective of exposure to ischemia (Fig. 1A). In addition, in the ischemia group, TG mice differed significantly from WT mice in terms of the time spent in the platform zone after removal of the platform (P = 0.0123; Fig. 1B). These results suggest that ischemia had a greater effect on learning memory in TG than in WT mice. To measure the willingness to explore the new environment, the behavior spontaneous alternation behaviors were measured using Y maze (Fig. 1C). However, there was no statistical difference in Y maze alteration tests (Fig. 1D).
11
Figure 1. Effect of tBCCAO on memory as measured by the Morris water maze test and spontaneous alternation test in the Y maze. A. The escape latency of all trials, performed over 5 days, was significantly different between the ischemia TG group vs. ischemia WT (**P = 0.0039, *P = 0.0215 vs. Ischemia WT). B. The platform zone-crossing time was recorded during a trial with the platform removed. Ischemia TG mice spent a significantly shorter amount of time in the platform zone than did Ischemia WT mice (*P =
12
0.0123 vs. Ischemia WT). C. Y Maze. D. There were no significant changes between WT groups and TG groups.
13
B. Neuronal cell death
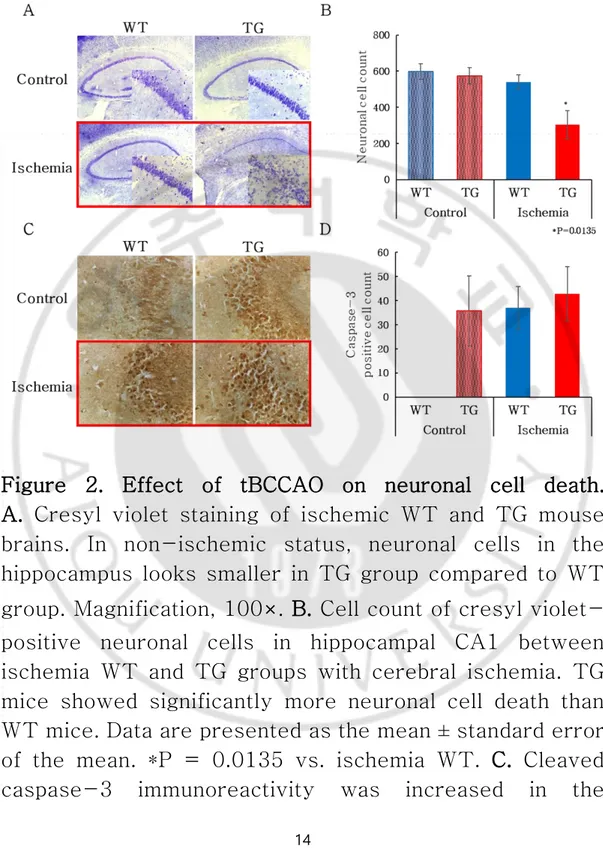
Staining of cresyl violet and caspase-3 was performed to confirm neuronal cell death due to ischemia. Neuronal cell death shown by cresyl violet staining was greater in the ischemic TG group compared to ischemic WT group (P = 0.0135) (Fig. 2A-B). Apoptosis stained by the caspase-3 did not show a significant difference between ischemic TG and ischemic WT groups (Fig. 2C). In addition, apoptosis was greater in the ischemia TG group than in the ischemia WT group, although the results of the caspase-3-positive cell count confirmed only an increasing tendency in the ischemia TG group (Fig. 2D). This finding reveals that a greater extent of necrosis and apoptosis was induced in ischemia TG mice and that these mice are more vulnerable to ischemia.
14
Figure 2. Effect of tBCCAO on neuronal cell death. A. Cresyl violet staining of ischemic WT and TG mouse
brains. In non-ischemic status, neuronal cells in the hippocampus looks smaller in TG group compared to WT group. Magnification, 100×. B. Cell count of cresyl violet-positive neuronal cells in hippocampal CA1 between ischemia WT and TG groups with cerebral ischemia. TG mice showed significantly more neuronal cell death than WT mice. Data are presented as the mean ± standard error of the mean. *P = 0.0135 vs. ischemia WT. C. Cleaved caspase-3 immunoreactivity was increased in the
15
hippocampal CA3 region in the ischemia TG group. D. Cell count of the 3,3′-diaminobenzidine (DAB)-positive cleaved caspase-3-positive cells in the hippocampal CA3 region between the WT and TG groups with cerebral ischemia. There were no significant differences in the number of these cells across ischemia groups.
16
C. Changes in Alzheimer’s pathology 1. Amyloid plaques
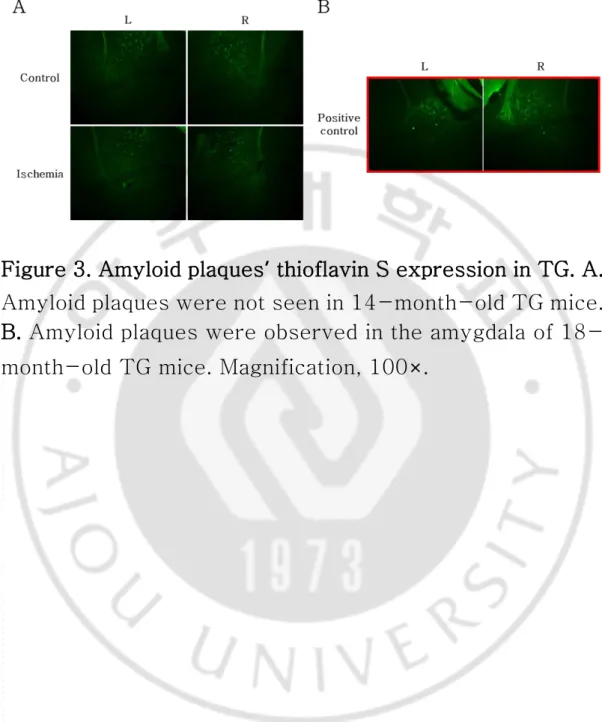
The thioflavin S-positive signal was evaluated in the cortex, hippocampus, and amygdala region. Amyloid plaques were not obviously observed in 14-month-old TG mice with or without cerebral ischemia (Fig. 3A). However, the thioflavin S-positive signal reflected the extracellular accumulation of amyloid aggregates in the amygdala of 18-month-old mice. (Fig. 3B).
17
Figure 3. Amyloid plaques’ thioflavin S expression in TG. A. Amyloid plaques were not seen in 14-month-old TG mice. B. Amyloid plaques were observed in the amygdala of 18-month-old TG mice. Magnification, 100×.
18
2. Amyloid precursor protein
The APP (Y188) signal was evaluated in the cortex, amygdala, and hippocampal CA1 regions. Positive signals were obtained in both TG mice but not observed in both WT mice. In the ischemia group in particular, atrophy was observed in the ischemic lesion (Fig. 4A-C). In the ischemia TG group, the intensity of Y188 staining tended to be reduced, but the number of Y188-positive cells in hippocampal CA1 was not significantly different between the control and ischemia groups (Fig. 4D). Double-staining for Y188 and NeuN confirmed that the Y188 signal was derived from neuronal amyloid (Fig. 4E).
20
Figure 4. Amyloid precursor Y188 expression. A-C. The amyloid precursor Y188 was expressed in TG mouse brains. A. Cortex B. Amygdala C. Hippocampal CA1. D. There were no significant differences in the number of Y188-positive cells between control and ischemia TG groups’ hippocampal CA1. E. Merged images of Y188 and NeuN immunostaining confirms that Y188 staining reflects neuronal amyloid. Magnification, 400×.
21
3. Tau protein
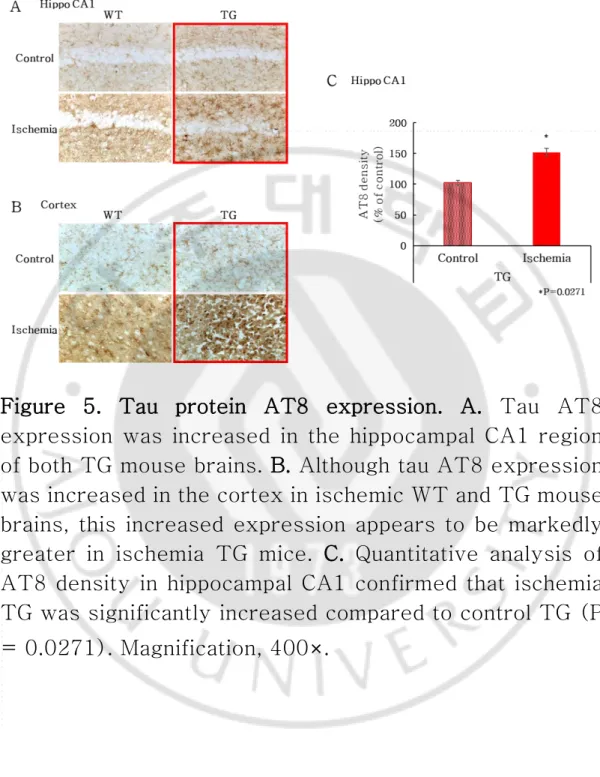
Staining with AT8 allowed the observation of tau protein. Tau-positive signals were evaluated in the cortex and hippocampus. Positive cells increased in the ischemia WT group compared with the control WT group. The ischemia TG group also showed an increase in positive cells compared with the control group (Fig. 5A-B). The density of control/ischemia TG was measured, and the density of ischemia TG was found to be significantly increased (P = 0.0271) (Fig. 5C). These results indicate that ischemia caused hyperphosphorylation of tau leading to activation of glia cells, resulting in the staining of microglia and astrocytes to confirm glial changes.
22
Figure 5. Tau protein AT8 expression. A. Tau AT8 expression was increased in the hippocampal CA1 region of both TG mouse brains. B. Although tau AT8 expression was increased in the cortex in ischemic WT and TG mouse brains, this increased expression appears to be markedly greater in ischemia TG mice. C. Quantitative analysis of AT8 density in hippocampal CA1 confirmed that ischemia TG was significantly increased compared to control TG (P = 0.0271). Magnification, 400×.
23
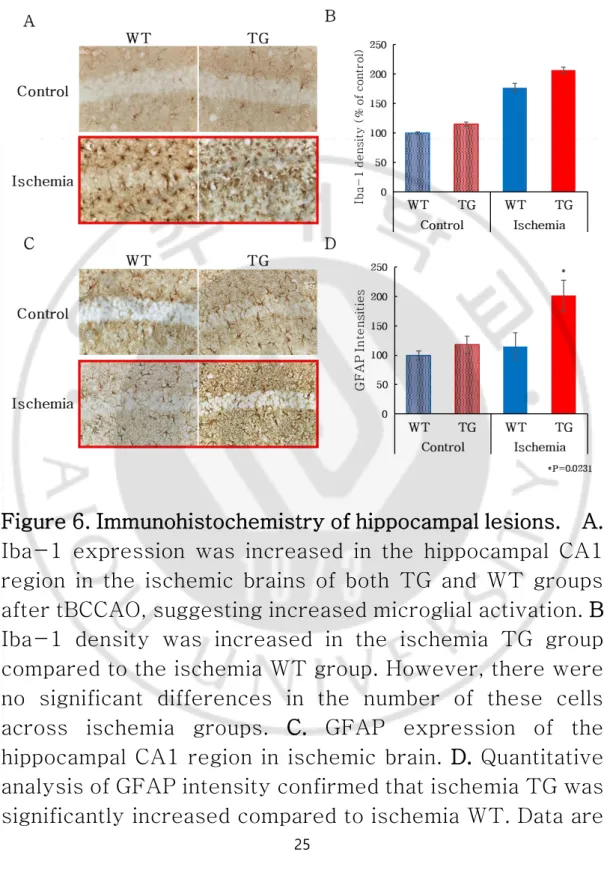
D. Glial changes
Immunohistochemistry experiments were conducted to investigate differences in neuropathological changes. Iba-1 expression in WT control and TG hippocampi were not significantly different between WT and TG group with and without cerebral ischemia, respectively. Nevertheless, the density of Iba-1 positive cells in mice with ischemia was significantly greater than in those without. WT mice exposed to ischemia demonstrated activation of microglia in the hippocampus while microglia were activated in the cortex as well as in the hippocampus of TG mice exposed to ischemia. Iba-1 expression density was quantified to compare Iba-1 expression in each group. The staining density for Iba-1 was significantly increased in both WT and TG mice after ischemia, as compared to that in control WT and TG mice. However, the amount did not differ between TG and WT mice exposed to ischemia (Fig. 6A-B). In the hippocampi of control WT and TG mice, normal astrocyte morphology was observed. In contrast, in WT mice exposed to ischemia, we observed astrogliosis in a region showing signs of cell death. We also observed a cavitation phenomenon in astrocyte-rich regions also showing signs of cell death in TG mice. The GFAP intensity results showed a significant difference between WT and TG groups for brain ischemia (P = 0.0231). Thus, mice
24
neuropathologic changes than were control mice, and TG mice were more vulnerable than WT mice (Fig. 6C-D).
25
Figure 6. Immunohistochemistry of hippocampal lesions. A. Iba-1 expression was increased in the hippocampal CA1 region in the ischemic brains of both TG and WT groups after tBCCAO, suggesting increased microglial activation. B. Iba-1 density was increased in the ischemia TG group compared to the ischemia WT group. However, there were no significant differences in the number of these cells across ischemia groups. C. GFAP expression of the hippocampal CA1 region in ischemic brain. D. Quantitative analysis of GFAP intensity confirmed that ischemia TG was significantly increased compared to ischemia WT. Data are
26
presented as the mean ± standard error of the mean. *P = 0.0231 vs. ischemia WT. Magnification, 400×.
27
IV.
DISCUSSION
In this study, we investigated whether AD is particularly vulnerable to brain ischemia by using 3xTG-AD mice triple-transgenic for the APPSwe, tauP301L, and PS1M148V mutations (Sterniczuk, Antle et al. 2010). A significant cognitive decline was observed in the aged mice of the ischemic TG group when we tested their cognitive abilities using the MWM test after inducing cerebral ischemia. Furthermore, observation of the hippocampal CA1 region indicated that cell death was significantly increased in the TG mice as compared to their WT counterparts, indicating that the aged 3xTG-AD mouse is more vulnerable to cerebral ischemia. However, the characteristic AD pathologies did not show such differences.
It has been reported that cerebral hypoperfusion, an early characteristic of the AD brain, is associated with degenerative processes and cognitive decline. This study explored such potential mechanistic associations and studied the relationship between changes in amyloid precursor protein and Tau protein expression and neuropathology in the brains of 3xTG-AD mice. We generated neuropathological changes by inducing transient brain ischemia, which led to cognitive decline and increased levels of neuronal cell death, microglia, and astrocytes in mouse brains of the ischemia TG group. In addition,
28
morphological evidence of cellular atrophy was observed in the ischemia TG group compared to the control TG group. Neuron cells were observed by merging of the double staining for amyloid precursor protein Y188, which proved that amyloid precursor protein Y188 is a neuronal amyloid. Tau protein expression was observed in cells that were also identified as microglia. However, when amyloid beta was observed in the control / ischemia TG group, amyloid beta was not observed. Amyloid beta was observed, however, in 18-month-old TG positive controls.
Cerebral ischemia induces oxidative stress and inflammation in the brain, resulting in the breakdown of the blood–brain barrier (BBB). Consequently, microglia and astrocytes are activated and cause neuronal damage (Kahlson and Colodner 2016). Neuronal cell death occurs in the hippocampal CA1 region, which is vulnerable to ischemia. In addition, cerebral ischemia causes a decrease in CBF, resulting in impaired memory function and exacerbation of dementia. Amyloid beta precursor protein, amyloidogenic fragments, tau-like pathology, and other AD factors can be induced at the site of injury in brain ischemia (Kalaria 2000). Neurodegenerative diseases are observed as signs of cerebrovascular disease. Thus, according to our findings, cognitive impairment in AD is associated with cerebral ischemia and subsequent neuronal pathological changes.
29
In AD, the characteristic neurological changes displayed are often accompanied by additional vascular pathology and features of other neurodegenerative diseases (Kelly, He et al. 2017). According to clinical studies, cerebral vascular occlusion contributes to the pathogenesis of AD, and cerebrovascular disease in AD is more detrimental than cerebral vascular disease in the elderly (Kisler, Nelson et al. 2017). Ischemic stroke has recently been implicated as a factor in the deterioration of AD (Lee, Im et al. 2011). The cognitive impairment resulting from the combination of AD and ischemic stroke is exacerbated compared to ischemic stroke-related or AD-only injury (Santos, Snyder et al. 2017). The complex relationship between AD and ischemic stroke includes cerebral hypoperfusion, energy deficiency, inflammation, dysfunction of capillaries, immune wasting, oxidative stress, and the accumulation of key proteins, including amyloid beta and Tau protein (Pendlebury 2012, Dong, Maniar et al. 2018).
Tauopathies, aggregations of Tau protein, are
characterized by a fibrous inclusion found in neurons and glial cells. Activated microglia and astrocytes and increased levels of proinflammatory molecules are also pathological features of brain regions affected by Tau pathology (Leyns and Holtzman 2017).The occurrence of tauopathy, a major pathologic feature, appears to play a role in nerve inflammation and in aggravation of tau pathology and neurodegeneration (Qiu, Ng et al. 2016). In addition to toxic
30
protein aggregates, activated astrocytes and macrophages, as well as elevated inflammatory marker levels, are other pathological features characteristic of tauopathies (Kovacs 2015). Chronic glial activation may promote the formation of tangles and the reduction of connective compliance, leading to neurodegenerative diseases (Wyss-Coray and Mucke 2002, Ransohoff 2016).
Therefore, our results show that as tau-positive cells increase, astrocytes significantly increase, and tauopathy is affected by astrocytes (Verkhratsky, Olabarria et al. 2010), which is consequently associated with gliosis and neuroinflammation.
Taken together, the results of this study show that ischemia contributes to the cognitive dysfunction of AD and AD mice, who are more vulnerable to ischemic injury, such as apoptosis, necrosis, and neuronal cell death, and exacerbates cognitive deficits associated with this condition.
31
V. REFERENCES
1. Cechetti, F., A. S. Pagnussat, P. V. Worm, V. R. Elsner, J. Ben, M. S. da Costa, R. Mestriner, S. N. Weis and C. A. Netto (2012). "Chronic brain hypoperfusion causes early glial activation and neuronal death, and subsequent long-term memory impairment." Brain Res Bull 87(1): 109-116.
2. De Silva, T. M. and A. A. Miller (2016). "Cerebral Small Vessel Disease: Targeting Oxidative Stress as a Novel Therapeutic Strategy?" Front Pharmacol 7: 61.
3. Delacourte, A., J. P. David, N. Sergeant, L. Buee, A. Wattez, P. Vermersch, F. Ghozali, C. Fallet-Bianco, F. Pasquier, F. Lebert, H. Petit and C. Di Menza (1999). "The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer's disease." Neurology 52(6): 1158-1165.
4. Dong, S., S. Maniar, M. D. Manole and D. Sun (2018). "Cerebral Hypoperfusion and Other Shared Brain Pathologies in Ischemic Stroke and Alzheimer's Disease." Transl Stroke Res 9(3): 238-250.
5. Fan, Y. L., J. Q. Wan, Z. W. Zhou, L. Chen, Y. Wang, Q. Yao and J. Y. Jiang (2014). "Neurocognitive improvement after carotid artery stenting in patients with chronic internal carotid artery occlusion: a prospective, controlled, single-center study." Vasc Endovascular Surg 48(4): 305-310.
32
6. Ferrer, I., I. Lopez-Gonzalez, M. Carmona, L. Arregui, E. Dalfo, B. Torrejon-Escribano, R. Diehl and G. G. Kovacs (2014). "Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression." J Neuropathol Exp Neurol 73(1): 81-97.
7. Gorelick, P. B., A. Scuteri, S. E. Black, C. Decarli, S. M. Greenberg, C. Iadecola, L. J. Launer, S. Laurent, O. L. Lopez, D. Nyenhuis, R. C. Petersen, J. A. Schneider, C. Tzourio, D. K. Arnett, D. A. Bennett, H. C. Chui, R. T. Higashida, R. Lindquist, P. M. Nilsson, G. C. Roman, F. W. Sellke and S. Seshadri (2011). "Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the
american heart association/american stroke
association." Stroke 42(9): 2672-2713.
8. Hardy, J. and D. J. Selkoe (2002). "The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics." Science 297(5580): 353-356.
9. Jo, W. K., A. C. Law and S. K. Chung (2014). "The neglected co-star in the dementia drama: the putative roles of astrocytes in the pathogeneses of major neurocognitive disorders." Mol Psychiatry 19(2): 159-167.
10. Kahlson, M. A. and K. J. Colodner (2016). "Glial Tau Pathology in Tauopathies: Functional Consequences."
33
Journal of experimental neuroscience 9(Suppl 2): 43-50.
11. Kalaria, R. N. (2000). "The role of cerebral ischemia in Alzheimer's disease." Neurobiol Aging 21(2): 321-330.
12. Kelly, S. C., B. He, S. E. Perez, S. D. Ginsberg, E. J. Mufson and S. E. Counts (2017). "Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease." Acta Neuropathol Commun 5. 13. Kisler, K., A. R. Nelson, A. Montagne and B. V.
Zlokovic (2017). "Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease." Nat Rev Neurosci 18(7): 419-434.
14. Kovacs, G. G. (2015). "Invited review:
Neuropathology of tauopathies: principles and practice." Neuropathol Appl Neurobiol 41(1): 3-23.
15. Lee, J. S., D. S. Im, Y. S. An, J. M. Hong, B. J. Gwag and I. S. Joo (2011). "Chronic cerebral hypoperfusion in a mouse model of Alzheimer's disease: an additional contributing factor of cognitive impairment." Neurosci Lett 489(2): 84-88.
16. Leyns, C. E. G. and D. M. Holtzman (2017). "Glial contributions to neurodegeneration in tauopathies." Mol Neurodegener 12(1): 50.
17. Lull, M. E. and M. L. Block (2010). "Microglial
activation and chronic neurodegeneration."
34
18. Montagne, A., D. A. Nation, J. Pa, M. D. Sweeney, A. W. Toga and B. V. Zlokovic (2016). "Brain imaging of neurovascular dysfunction in Alzheimer's disease." Acta Neuropathol 131(5): 687-707.
19. Moolman, D. L., O. V. Vitolo, J. P. Vonsattel and M. L. Shelanski (2004). "Dendrite and dendritic spine alterations in Alzheimer models." J Neurocytol 33(3): 377-387.
20. Pendlebury, S. T. (2012). "Dementia in patients hospitalized with stroke: rates, time course, and clinico-pathologic factors." Int J Stroke 7(7): 570-581.
21. Qiu, L., G. Ng, E. K. Tan, P. Liao, N. Kandiah and L. Zeng (2016). "Chronic cerebral hypoperfusion enhances Tau hyperphosphorylation and reduces autophagy in Alzheimer's disease mice." Sci Rep 6: 23964.
22. Ransohoff, R. M. (2016). "How neuroinflammation contributes to neurodegeneration." Science 353(6301): 777-783.
23. Santos, C. Y., P. J. Snyder, W. C. Wu, M. Zhang, A. Echeverria and J. Alber (2017). "Pathophysiologic
relationship between Alzheimer's disease,
cerebrovascular disease, and cardiovascular risk: A review and synthesis." Alzheimers Dement (Amst) 7: 69-87.
24. Song, B., Q. Ao, Y. Niu, Q. Shen, H. Zuo, X. Zhang and Y. Gong (2013). "Amyloid beta-peptide worsens cognitive impairment following cerebral
ischemia-35
reperfusion injury." Neural Regen Res 8(26): 2449-2457.
25. Sterniczuk, R., M. C. Antle, F. M. Laferla and R. H. Dyck (2010). "Characterization of the 3xTg-AD mouse model of Alzheimer's disease: part 2. Behavioral and cognitive changes." Brain Res 1348: 149-155.
26. Swaab, D. F., M. A. Hofman, P. J. Lucassen, A. Salehi and H. B. Uylings (1994). "Neuronal atrophy, not cell death, is the main hallmark of Alzheimer's disease." Neurobiol Aging 15(3): 369-371; discussion 379-380. 27. Verkhratsky, A., M. Olabarria, H. N. Noristani, C. Y. Yeh and J. J. Rodriguez (2010). "Astrocytes in Alzheimer's disease." Neurotherapeutics 7(4): 399-412. 28. Wyss-Coray, T. and L. Mucke (2002). "Inflammation
in neurodegenerative disease--a double-edged
sword." Neuron 35(3): 419-432.
29. Xu, X., B. Zhang, K. Lu, J. Deng, F. Zhao, B. Q. Zhao and Y. Zhao (2016). "Prevention of Hippocampal Neuronal Damage and Cognitive Function Deficits in Vascular Dementia by Dextromethorphan." Mol Neurobiol 53(5): 3494-3502.
36 -국문요약-
알츠하이머 모델의 뇌 허혈 시 세포 사멸에 대한 취약성
알츠하이머병 (Alzheimer’s disease, AD)은 대부분 뇌혈관 질환의 위험이 증가된 노인들에게 주로 영향을 미치는 질환이다. AD 병리학이 뇌 허혈로 인해 뇌에 미치는 영향에 대한 연구는 잘 알려져 있지 않다. 현재 연구에서 우리는 AD 에 의해 영향을 받는 뇌가 뇌 허혈에 취약하고 뇌 아밀로이드 베타 (Aβ)와 타우 단백질 축적 및 퇴행성 변화를 유도함으로써 더 큰 괴사 및 신경 세포 사멸을 일으킨다는 가설을 세웠다. 본 연구에서는 AD (3xTG-AD)의 형질 전환 (TG) 마우스 모델에서 혈관성 치매 모델로 흔히 사용되는 양측 경동맥 폐색에 의한 일과성 대뇌 허혈의 영향을 연구하였다. 양측 경동맥 폐쇄 15 분 후 재관류를 한 후 야생형 (WT) 마우스와 비교하여 인지 기능 변화, 신경 세포 사멸 정도, 치매 특이성 신경 병리학적 변화를 평가하였다. 행동 연구의 결과는 뇌 허혈에 노출된 TG 마우스가 유사하게 처리된 WT 마우스보다 공간 학습 및 기억에 더 큰 결점을 보였다는 것을 입증하였다. TG 마우스의 인지 손상은 WT 마우스에 비해 뇌 허혈로 인한 더 큰 신경 세포 사멸로 인산 것 일 수 있다. 그러나 유사한 Aβ 축적이 두 군에서 관찰되었다. 허혈성 TG 마우스에서 caspase-3, GFAP 및 Iba-1 의 발현이 증가하였다. 결론적으로, 우리의 결과는 AD 의 신경 병리학이 뇌를 뇌 허혈 / 재관류에 보다 취약하게 만드는 변화를 유발할 수 있음을 시사한다. 핵심어: 알츠하이머병, 뇌 허혈, 3xTG-AD 마우스, 아밀로이드 베타, 타우 단백질, 신경 병리 변화