저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Effect of Antifibrotic Drug (Pirfenidone) on
TGF-beta Signaling Pathway in Ocular Fbrotic
Disorders
by
Effect of Antifibrotic Drug (Pirfenidone) on
TGF-beta Signaling Pathway in Ocular Fbrotic
Disorders
by
Kihwang Lee
A Dissertation Submitted to The Graduate School of
Ajou University in Partial Fulfillment of the Requirements
for the Degree of Doctor
of Medicine
Supervised by
Jaehong Ahn, M.D., Ph.D.
Major in Medicine
Department of Medical Sciences
The Graduate School, Ajou University
This certifies that the dissertation
of Kihwang Lee is approved.
SUPERVISORY COMMITTEE
Eunhye Joe
Jaehong Ahn
Hongseok Yang
Yun-Hoon Choung
Youn-Hee Choi
- ABSTRACT –
Effect of Anti-fibrotic Drug (Pirfenidone) on TGF- Signaling
Pathway in Ocular Fibrotic Disorders
Purpose: Transforming growth factor-β (TGF- ) plays a key role in transforming retinal
pigment epithelial (RPE) cells into mesenchymal fibroblastic cells, which are implicated in fibrotic disorders of the retina. Herein, the effect of pirfenidone, a novel anti-fibrotic agent, on TGF- 1-mediated fibrogenesis in the human RPE cell line (ARPE)-19 was tested.
Methods: The effect of pirfenidone on the TGF- 1-induced phenotype in ARPE-19 cells
was measured with immunocytochemistry as the change in F-actin. Fibronectin and collagen production was measured with enzyme-linked immunosorbent assay, and cell migration activity was investigated using a scratch assay. Immunoblot analyses of cofilin, Smad protein (Smad) 2/3, p38 mitogen-activated protein kinase, c-Jun N-terminal kinase, and extracellular signal related kinase expression were conducted to elucidate the cell signaling networks that contribute to the anti-fibrotic effect of pirfenidone.
Results: Treatment with TGF- 1 induced typical phenotypic changes such as formation of
stress fiber running parallel to the long axis of cells, and enhanced migration and production of extracellular matrix components such as collagen type I and fibronectin. This fibroblast-like phenotype induced by TGF- 1 was significantly inhibited by pretreatment with
pirfenidone in a dose-dependent manner. Pirfenidone inhibited TGF- signaling by
preventing nuclear accumulation of active Smad2/3 complexes rather than phosphorylation of Smad2/3.
Conclusions: These results collectively provide a rational background for future evaluation
of pirfenidone as a potential anti-fibrotic agent for treating proliferative vitreoretinopathy and other fibrotic retinal disorders.
_________________________________________________________________________
Keywords ; Transforming growth factor- (TGF- ), retinal pigment epithelial (RPE) cells,
TABLE OF CONTENTS
ABSTRACT··· i TABLE OF CONTENTS ··· ii LIST OF FIGURES ··· iv LIST OF ABBREVIATION··· v I. INTRODUCTION ··· 1A. Fibrotic Retinal Diseases ··· 1
B. Retinal Pigment Epithelial Cells··· 2
C. Epithelia-to- Mesenchymal Transition··· 3
D. Transforming Growth Factor – ··· 4
1. Smad-Dependent Signaling ··· 4
2. Smad-Independent Signaling ··· 5
E. Novel Anti-fibrotic Drug (Pirfenidone)··· 5
F. Aims of This Study ··· 8
II. MATERIALS AND METHODS ··· 9
A. MATERIALS··· 9
1.Reagents··· 9
B. METHODS··· 9
1. Cell Cuture ··· 9
2. Enzyme-linked Immunosorbent Assay ··· 9
3. Immunocytochemistry··· 10
4. Cell Migration Assay··· 10
5. Immunoblot analysis··· 10
D. Pirfenidone blocks TGF-β1-induced nuclear translocation but not phosphorylation
of Smads··· 27
IV. DISCUSSION ··· 33
A. Pirfenidone and TGF-β1 induced fibroblastic phenotypes in ARPE-19 cells··· 33
B. Pirfenidone and TGF-β1-induced expression of extracellular matrix components in ARPE-19 cells··· 35
C. Possible molecular mechanisms of pirfenidone for the inhibitory action for TGF-β1-induced fibrogenesis in ARPE-19 cells··· 36
V. CONCLUSION ··· 41
REFERENCES ··· 42
LIST OF FIGURES
Fig 1. Chemical structure of pirfenidone··· 7 Fig 2. Pirfenidone inhibited TGF-β1-induced morphological changes and actin
rearrangement in ARPE-19 cells··· 13 Fig 3. Pirfenidone inhibited TGF-β1-induced morphological changes in ARPE-19 cells
in a dose dependent manner··· 15 Fig 4. Pirfenidone inhibited TGF-β1-induced actin rearrangement in ARPE-19 cells··· 17 Fig 5. Pirfenidone and hydroxylfasudil had little effect on TGF-β1-induced increase
in the cell surface area··· 19 Fig 6. Pirfenidone had little effect on TGF-β1-induced increase in the cell surface area···· 21 Fig 7. Pirfenidone inhibited TGF-β1-induced expression of extracellular matrix··· 23 Fig 8. Pirfenidone inhibited TGF-β1-induced expression of extracellular matrix
with different time periods··· 24 Fig 9. Pirfenidone inhibited TGF-β1-induced migration of ARPE-19 cells··· 25 Fig 10. Activation of p38 MAPK, cofilin, and Smad 2/3 by TGF- β1 in ARPE-19 cells···· 28 Fig 11. Pirfenidone had no effect on TGF-β1-induced phosphorylation of MAPK··· 29 Fig 12. Pirfenidone inhibited TGF-β1-induced signal transduction··· 31 Fig 13. TGF- β1 signaling pathway and their possible role in the regulation of
ECM production in ARPE-19 cells··· 37 Fig 14. Mechanism of action of pirfenidone in ARPE-19 cells··· 39
LIST OF ABBREVIATION
AMD, age-related macular degeneration AKT, protein kinase B (PKB)
CDC42, cell division control protein 42 homolog CNV, choroidal neovascularization
COX, cyclooxygenase ECM, extracellular matrix
EMT, epithelial-to-mesenchymal transition ERK, extracellular signal-related kinase FGF, fibroblast growth factor
HSP, heat shock protein IFN, interferons
IL, interleukin
IPF, idiopathic pulmonary fibrosis JNK, c-Jun N terminal kinase
MAPK, mitogenactivated protein kinase
NADP, nicotinamide adenine dinucleotide phosphate NLRP3, NOD-like receptor pyrin domain containing 3 PED, retinal pigment epithelial detachment
PG, prostaglandin
PVR, proliferative vitreoretinopathy
RAC, ras-related C3 botulinum toxin substrate RhoA, ras homolog gene family, member A ROCK, rho kinase
PI3K, phosphoinositide 3-kinase RNA, ribonucleic acid
RPE, retinal pigment epithelial cells ROP, retinopathy of prematurity
RT-PCR, reverse transcription polymerase chain reaction TGF- 1, transforming growth factor- 1
TGF R, TGF receptor TNF, tumor necrosis factor ZO-1, zonula occludens 1
I. INTRODUCTION
A. Fibrotic Retinal DiseasesFibrotic diseases of the posterior segment of the eye include proliferative vitreoretinopathy (PVR), retinopathy of prematurity (ROP), diabetic retinopathy, and age-related macular degeneration (AMD). ROP is disease affecting the retinas of premature infants. Abnormal proliferation of blood vessels resulting in retinal detachment may lead to blindness. The abnormal fibrovascular proliferation may regress, but progression is more likely and results in fibrous tissue that contracts and produces detachment. The key pathologic change, retinal neovascularization, has several features in common with the other proliferative retinopathies such as diabetic retinopathy. They appear to be associated with local ischemia and the subsequent development of neovascularization.
Diabetic retinopathy is a major problem throughout the world. It is fundamental dysfunction of blood glucose metabolism due to changed insulin production or activity. It is clinically manifested by a microangiopathy involving the retinal circulation. There are permeability abnormalities which lead to leakage within and beneath the retina and lipid accumulation. Capillary closure or ischemia sets the occasion for vasoproliferation at the nerve and on the surface or the retina into the vitreous. Secondary hemorrhage, fibrosis, and tractional detachment evolve, all leading to severe vision loss and even blindness.
AMD is another major cause of severe visual loss in older adults (Bressler et al., 2011). AMD patients have drusen or retinal pigment epithelial abnormalities or both at posterior pole of retina, mainly at fovea (Bressler et al., 1988). Approximately 10% of AMD patients develop the neovascular form of AMD (Ferris et al., 1984). Neovascular AMD can be characterized with choroidal neovascularization (CNV) and associated features such as retinal pigment epithelial detachment (PED), retinal pigment epithelial tears, fibrovascular disciform scarring, and vitreous hemorrhage (Bressler et al., 1988). CNV, in most cases, is accompanied by fibrous tissue (Grossniklaus and Green, 1998). The outer retina is deteriorated slowly by the fibrovascular or fibroglial tissue.
PVR is the most common cause of failed reattachment of a primary rhegmatogenous retinal detachment, and occurs when the proliferation of retinal pigment epithelial cells (RPE) creates traction on the retina (Machemer, 1977). This usually leads to recurrent rhegmatogenous retinal detachment (Ryan, 1985). PVR is characterized by proliferation of RPE cells, glial cells, and inflammatory cells on the retinal surface and within the vitreous gel (Campochiaro, 1997). The RPE cells in vitreous gain contractile properties, and exert traction forces directly on the retina or through the vitreous gels. Breakdown of blood retinal barrier increases cytokine release in the vitreous and causes more inflammation.
B. Retinal Pigment Epithelial Cells (RPE)
RPE, which are normally located in the external cell layer of the retina, are the most critical contributors to the development of fibrotic diseases of the eye (Bochaton-Piallat et al., 2000; Nagineni et al., 2003). RPE, specialized neuro-ectodermally derived pigmented cells, are located between the neurosensory retina and the vascular choroid, and form a monolayer. The RPE plays an important function which is essential for normal outer retinal physiology. Most importantly, the RPE participates in the visual cycle, and phagocytes shed disc from photoreceptor outer segments. Tight junctions of RPE form the outer blood-retinal barrier, and modulate bidirectional ion and metabolic transport between the retina and choroid. RPE also secrete a large variety of factors and signaling molecules including Transforming growth factor- 1 (TGF- 1). Lastly, RPE transport water out of the subretinal space so that the neurosenrosy retina detachment would not occur.
The RPE is a primary site of pathology in fibrotic diseases of the posterior segment of the eye, including AMD and PVR. Hypoxia, inflammation, and mechanical insults cause RPE cells to undergo a process known as the epithelial-to-mesenchymal transition (EMT), and to
and severe tissue fibrosis of the heart, lung, liver, and kidney (Iwano et al., 2002; Zeisberg et al., 2007; Thiery et al., 2009).
C. Epithelia-to- Mesenchymal Transition (EMT)
EMT is an essential role for morphogenesis during early stages of prenatal development and it has become apparent that EMT can be abnormally activated and developed in adult tissues during disease processes such as cancer and fibrosis (Thiery, 2002). EMT describes a temporary and reversible phenotypical change in which well organized, tightly connected epithelial cells transdifferentiate into disorganized and motile mesenchymal cells. In the process of converting from an epithelial cell into a mesenchymal cell, the cells lose epithelial characteristics such as their structural polarity and their cell-to-cell-basement membrane contact, and become migratory mesenchymal cells. These processes are mediated by expression of cell surface molecules, cytoskeletal reorganization, and extracellular matrix (ECM) components and activation of transcription factors (Lee et al., 2006; Kalluri and Weinberg, 2009). More specifically, this process is characterized by disruption of tight junctions between epithelial cells due to down regulation and delocalization of tight junction proteins E-cadherin, zonula occludens 1, occludin, cytokeratin and etc. This is followed by loss of apical-basal cell polarity, dramatic remodeling of the cytoskeleton, and the formation of actin stress fibers. Concomitantly, cells acquire mesenchymal features such as spindle-shaped, fibroblast-like morphology and express mesenchymal components including N-cadherin, vimentin, -Catenin, fibronectin, and alpha smooth muscle actin (Thiery and Sleeman, 2006). EMT has been classified into three subtypes based on the functional effects. Type I EMT involves epiblasts transitioning into motile mesenchymal cells and is associated with forming trophoblastic germ layers. The primary mesenchyme is reinduced to secondary epithelium by mesenchymal-epithelial transition. Type II EMT is activated in condition of inflammation and wound healing. Type II EMT is seen when secondary epithelial cells or endothelial cells transition to resident tissue or inflammation-induced fibroblasts. Type III EMT is detected in the secondary epithelia associated with cancerous tissues, and involves epithelial carcinoma cells transitioning to metastatic tumor cells. Among numerous growth factors, cytokines, hormones, extracellular signals which induce EMT, TGF- 1, the
profibrotic cytokine, can be considered as a the main pathogenic inducer of EMT (Okada et al., 1997; Yang and Liu, 2001).
D. Transforming Growth Factor
(TGF)-The pro-fibrotic cytokine, TGF- , controls numerous biological functions such as apoptosis, immune system, stem cell maintenance, development, migration, cell cycle, and
regulation of the immune system. In particular, transforming growth factor- (TGF- )
signaling has been considered a key effector of the EMT, and is known to induce EMT of RPE cells, transforming RPE cells into mesenchymal cells in vitro (Connor et al., 1989; Carrington et al., 2000; Hinton et al., 2002). Active TGF- binds to its type II receptor, which recruits and phosphorylates type I receptors. Smad-dependent (Phanish et al., 2006) and Smad-independent signal transduction pathways such as JNK, ERK, p38 MAPK and RhoA are involved in EMT induced by TGF- (Hayashida et al., 1999).
1. Smad-Dependent Signaling
The active TGF- 1 ligand initially binds TGF RII followed by recruitment of the TGF RI at the plasma membrane. The conformational switch of TGF RI by the heteromeric receptor-ligand complex allows activated TGF RI to communicate with R-Smads (Smad2/3) through their MH2 domain, resulting in their phosphorylation (Lo et al., 1998). This phosphorylation event initiates the formation of a heterotrimeric complex between phosphorylated R-Smads (Smad2/3) and co-Smad (Smad4). R-Smad and co-Smad can translocate into and accumulate in the nucleus where they act as transcription factors to modulate gene expression (Massague et al., 2005). The activity of TGF- signaling is balanced by a negative feedback loop mediated by the inhibitory Smad7 (Nakao et al., 1997). Unlike R-Smad, Smad7 stays in the nucleus under basal conditions, but it translocates to the
2. Smad-Independent Signaling
TGF- manipulates abundant intracellular signaling pathways in addition to Smads to regulate numerous biological functions. These non-Smad pathways induce various branches of MAP kinase, Rho-like GTPase signaling pathways, and
phosphatidylinositol-3-kinase/AKT pathways. TGF- promotes activation of extracellular signal-related kinase
(ERK) signaling, the c-Jun N terminal kinase (JNK) and p38- mitogen activated protein kinase (MAPK) pathways to regulate apoptosis or cell migration, depending on cellular context (Hartsough and Mulder, 1995; Liao et al., 2001). The PI3K/Akt pathway has been implicated in mediating actin reorganization and cell migration. Certain TGF- functions, such as rearrangement of cytoskeletal organization, cell polarity, and cell migration, are mediated by the Rho-like family of small GTPases (Jaffe and Hall, 2005). TGF- can rapidly activate the Rho-like GTPases, including RhoA, Cdc42, and Rac pathways, in a Smad2/3-independent manner, to promote EMT and control dynamic cytoskeletal organization (Bhowmick et al., 2001).
E. Novel Anti-fibrotic Drug (Pirfenidone)
All the evidence stated above suggest that targeting TGF-β signaling provides new insights for developing novel therapeutic interventions (Sato et al., 2003; Pennison and Pasche, 2007). Pirfenidone (5-methyl-1-phenyl-2-[1H]-pyridone), a small compound with combined anti-inflammatory and anti-oxidative action, is also known for its anti-fibrotic action in various in vitro systems and experimental animal models of lung, kidney, and liver fibrosis (Gurujeyalakshmi et al., 1999; Garcia et al., 2002; Oku et al., 2002; Cho et al., 2007) (Fig. 1). Solomon Margolin, an American pharmacologist, discovered pirfenidone in the course of a synthetic study. The main action of pirfenidone was initially focused as an anti-inflammatory agent. After demonstrating a consistent anti-fibrotic effect in several animal models of pulmonary fibrosis, the focus was shifted to developing pirfenidone as an anti-fibrotic drug for the treatment of IPF. Pirfenidone inhibited TNF-α production in a septic shock model in mice (Cain et al., 1998; Oku et al., 2002). Furthermore, pirfenidone increased the production of IL-10, which is also known as human cytokine synthesis inhibitory factor, in a septic shock model. Pirfenidone effectively inhibited the increase of
powerful pro-fibrotic cytokines such as transforming growth factor (TGF)- 1 and basic fibroblast growth factor (bFGF) in bleomycin-induced lung fibrosis in mice (Oku et al., 2008). In the same model, pirfenidone also prevented the decrease of anti-fibrotic cytokine IFN-γ. A recent report suggested that pirfenidone blocked the formation of a multi-protein complex known as the NLRP3 (NOD-like receptor pyrin domain containing 3) inflammasome (Artlett, 2013). This led to attenuation of innate immunity by decreasing the expression of pro-inflammatory IL-1β and Il-18 (Wang et al., 2013). Reduction in oxidative stress was considered to be another mechanism for the anti-fibrotic effect exerted by pirfenidone. Pirfenidone reduced the oxidative stress caused by toxic hydroxyl radicals in bleomycin-induced lung fibrosis in mice via inhibition of NADPH oxidase isoform 4 (Misra and Rabideau, 2000). Pirfenidone exerted its anti-fibrotic effect through inhibition of HSP 47, a collagen-specific molecular chaperon, resulting in a decrease in collagen synthesis in TGF-β1-induced lung fibroblasts (Kakugawa et al., 2004; Hisatomi et al., 2012). Furthermore, pirfenidone might suppress EMT (Hisatomi et al., 2012; Yang et al., 2013). In eyes, proliferation, migration, and collagen contraction of in human Tenon’s fibroblasts was inhibited by pirfenidone (Yang et al., 2013; Lee et al., 2014). It has been reported that IL-1β induced tissue inhibitors of metalloproteinase-1/collagen levels and cyclooxygenase (COX-2)/ prostaglandin (PG)E2 levels were effectively attenuated by pirfenidone without significant toxicity at the concentration used, indicating the fibrotic and anti-inflammation effects of this agent in thyroid associated ophthalmopathy (Kim et al., 2010; Choi et al., 2013). Lastly, clinical trials have shown that pirfenidone not only extended survival time but it also improved pulmonary function in patients with idiopathic pulmonary fibrosis (Nagai et al., 2002; Azuma et al., 2005). In October 2014, pirfenidone was approved for patients with idiopathic pulmonary fibrosis (IPF) by the Food and Drug Administration (FDA) in the United States.
F. Aims of This Study
Fibrotic diseases of the posterior segment of the eye are a devastating disease that causes blindness. Vision loss and blindness impose a great social and economic burden on individuals and society. For years, there have been no effective medical treatment and the results of surgical management were very poor for the diseases. Pirfenidone, the novel anti-fibrotic drug, have been approved by FDA for patients with IPF, and could be a potential anti-fibrotic agent for treatment of fibrotic retinal disorders in near future.
The specific aims of this study is to analyze and collect the effect and the mechanism of pirfenidone in RPE, which considered as a primary site of pathology in major causes of blindness, to provide a rational background for future evaluation of this drug. For these specific aims,
1. I generated TGF-β1-induced EMT in human retinal pigment epithelial cells line (ARPE-19).
2. I investigated the anti-fibrotic action of pirfenidone by evaluating morphological changes, migration activities, and synthesis of extracellular matrix in TGF-β1-induced ARPE-19 cells.
3. I investigated the molecular mechanisms of pirfenidone for the inhibitory action for TGF-β1-induced fibrogenesis in ARPE-19 cells.
II. MATERIALS AND METHODS
A. Materials1. Reagents
ARPE-19 cells obtained from the American Type Cell Culture (ATCC, Manass, VA) (Dunn et al., 1996). Pirfenidone was purchased from Sigma (St. Louis, MO). Human recombinant TGF-β1 was purchased from R&D Systems (Minneapolis, MN). Specific pharmacological inhibitors of p38 MAPK (SB202190) and Rho (hydroxyfasudil) were obtained from Calbiochem (La Jolla, CA). Antibodies specific to β-actin, N-cadherin, cofilin, phospho-cofilin (Ser3), smad protein (smad) 2/3, phospho-Smad2/3 (Ser465/467), p 38 MAPK, phospho-p38 (Thr180/Tyr182), JNK, phosphor-JNK (Thr183/Tyr185), Erk 1/2, phosphor-Erk1/2 (Thr202/Tyr204), poly (ADP-ribose) polymerase (PARP), and α-tublin were purchased from Cell Signaling (Beverly, MA). Rhodamine-labeled phalloidin and propidium iodide were purchased from Molecular Probes (Eugene, OR).
B. Methods
1. cell cuture
ARPE-19 cells were maintained in Dulbecco’s modified Eagle’s minimum essential medium (DMEM)/F-12 medium (1:1 mixture of DMEM and Hank’s balanced salt solution [HBSS]; Grand Island, NY) supplemented with 10% fetal bovine serum, 100 U/ml penicillin G, 100 mg/l streptomycin, and 2 mmol/l Lglutamine in a humidified incubator at 37 °C under 5% CO2 in 95% air as described previously (Itoh et al., 2007).
2. Enzyme-linked immunosorbent assay
ARPE-19 cells were incubated on 6-well plate in the absence or presence of pirfenidone for 1 h and then treated with TGF-β1 (10 μg/l) for an additional 48 h. All of the cultures contained the same concentration of dimethyl sulfoxide. The supernatants were processed for collagen type I C-terminal peptide and fibronectin enzymelinked immunosorbent assay (ELISA) kits (Takara, Tokyo, Japan) according to the protocol provided by the manufacturer. The color reaction was measured at 450 nm. Collagen type I C-terminal peptide and
fibronectin protein values were normalized by the protein concentration of the total cell lysates.
3. Immunocytochemistry
ARPE-19 cells were cultured in four-well multi-chamber and then supplemented with TGF-β1(10 μg/l) for 48 h in the absence or presence of pirfenidone or hydroxyfasudil. Next, the cells were rinsed for 3 min in 1×phosphate buffered saline (PBS, 137 mmol/l NaCl, 2.7 mmol/l KCl, 10 mmol/l Na2HPO4, 2 mmol/l KH2PO4, pH 7.4), fixed in 5% paraformaldehyde for 30 min, and permeabilized with 0.2% Triton (Sigma, Calbiochem, CA) in PBS for 20 min. The cells were then incubated for 1 h with rhodamine-labeled phalloidin (diluted 1:100). After being washed with PBS, the cells were mounted with FluorSave reagent (Calbiochem) and analyzed with a confocal microscopy (Carl Zeiss, Gottingen, Germany).
4. Cell migration assay
Cell migration was evaluated by assaying the closure of a liner defect produced in a cell monolayer culture as described previously (Song et al., 2010). The defect was generated in a confluent culture of ARPE-19 cells by scraping with a micropipette tip. The cells on 6-well plate were treated with TGF-β1 in the absence or presence of various pharmacological inhibitors. After 48 h, the cells were analyzed with a phase contrast microscopy. Migration distance was determined using i-Solution (iMTechnology, Seoul, Korea), and the shortest distance between the cells that had moved into the wounded region and their respective starting points was determined.
6. Statistical analysis
The data are presented as the mean ± standard deviation (SD). The level of significance for comparisons between samples was determined with one-way analysis of variance (ANOVA) with Tukey’s honest significant difference post-hoc test using InStat software (GraphPad Software Inc., San Diego, CA).
III. RESULTS
A. Pirfenidone inhibits TGF-β1 induced fibroblastic phenotypes in ARPE-19 cells
To investigate the effect of pirfenidone on the TGF-β1-induced EMT, it was first examined whether the TGF-β1-induced morphological changes were affected by pirfenidone. Treatment with TGF-β1 induced prominent morphological changes in ARPE-19 cells, including elongated and spindle-like shapes, which were noticeably suppressed by pretreatment with pirfenidone or SB202190 or hydroxyfasudil, a Rho kinase inhibitor (Fig. 2A and 3). It is noted that the morphologic change induced by TGF-β1 was inhibited by pirfenidone in a dose dependent manner (Fig. 3). After 48h of treatment with 250mg/l and 500mg/l of pirfenidone, TGF-β1 was notably inhibited. Next, cytoskeletal reorganization by staining for F-actin in response to TGF-β1 was examined. As the cells began to form spindle-like processes upon TGF-β1 stimulation, the distribution of F-actin was arrayed in a series of linear and parallel stress fiber-like structures. Stress fiber formation was severely disorganized and failed to develop into more mature and spindle-like structures in the presence of pirfenidone or hydroxyfasudil (Fig. 2B and 4). The inhibitory effect of pirfenidone on stress fiber forming was also dose dependent (Fig. 4). Cells treated with TGF-β1 exhibited up to a fivefold increase in cell surface area compared to unstimulated control cells, which is consistent with a previous report (Lamouille and Derynck, 2007) (Fig. 5 and 6). Pretreatment with hydroxyfasudil alone increased cell surface area and inhibited the TGF-β1-induced increase in the cell surface area; while, pirfenidone had little effect on cell size (Fig. 5 and 6).
Cofilin, a small actin-binding protein, is involved in cell mobility and invasion via controlling actin polymerization (Wang et al., 2007). Phosphorylation of cofilin is responsible for TGF-β1-induced actin polymerization, which can be blocked by pretreatment
and morphological changes are mediated by the RhoA pathway and these events are significantly suppressed by pirfenidone.
Figure 2. Pirfenidone inhibited TGF-β1-induced morphological changes and actin rearrangement in ARPE-19 cells. A: ARPE-19 cells were incubated in the absence or
presence of pirfenidone (500 mg/l) or hydroxyfasudil (10 μmol/l) for 1 h, treated with TGF-β1 (10 μg/l) for an additional 48 h, and visualized with a phase contrast microscopy. The data shown are representatives of at least four independent experiments. Magnification, 100×. Scale bar = 20 μm. B: Cells were incubated in the absence or presence of pirfenidone (500 mg/l) or hydroxyfasudil (10 μmol/l) for 1 h, treated with TGF-β1 (10 μg/l) for an additional 48 h, and stained with rhodamine-labeled phalloidin for F-actin and fluorescein isothiocyanate (FITC) - conjugated antibodies for N-cadherin. The data shown are representatives of at least three independent experiments. Magnification, 400×. Scale bar = 20 μm. C: Cells were incubated in the absence or presence of pirfenidone (500 mg/l) for 1 h and then treated with TGF-β1 (10 μg/l) for varying time periods. The total cell lysates were subjected to immunoblot analysis for phospho-cofilin, cofilin, and β-actin. The data shown are representatives of at least two independent experiments. Control: untreated, PFD: pirfenidone, fasudil: hydroxyfasudil.
Figure 3. Pirfenidone inhibited TGF-β1-induced morphological changes in ARPE-19 cells in a dose dependent manner. ARPE-19 cells were incubated in the absence or
presence of pirfenidone (50, 100, 250, and 500 mg/l) or SB202190 (10 μmol/l) for 1 h, treated with TGF-β1 (10 μg/l) for an additional 48 h, and visualized with a phase contrast microscopy. The data shown are representatives of at least four independent experiments. Magnification, 100×.Control: untreated, PFD: pirfenidone, fasudil: hydroxyfasudil.
Figure 4. Pirfenidone inhibited TGF-β1-induced actin rearrangement in ARPE-19 cells. Cells were incubated in the absence or presence of pirfenidone (500 or 1000 mg/l) or
hydroxyfasudil (10 μmol/l) for 1 h, treated with TGF-β1 (10 μg/l) for an additional 48 h, and stained with rhodamine-labeled phalloidin for F-actin. The data shown are representatives of at least three independent experiments. Magnification, 400×. Control: untreated, PFD: pirfenidone, fasudil: hydroxyfasudil.
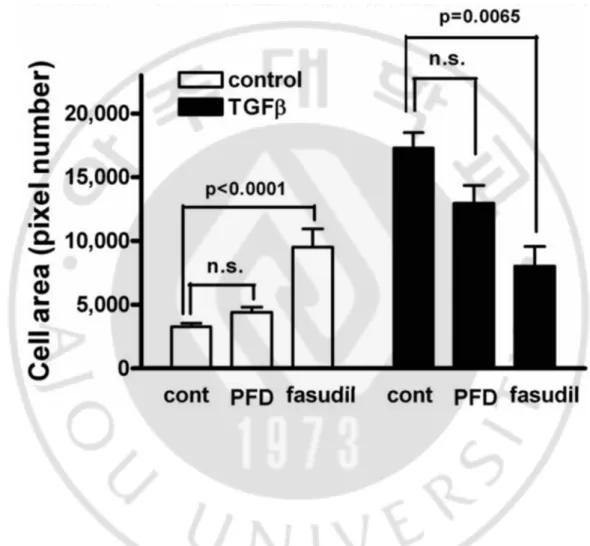
Figure 5. Pirfenidone and hydroxylfasudil had little effect on TGF-β1-induced increase in the cell surface area. Cells were incubated in the absence or presence of
pirfenidone (PFD, 500 mg/l) or hydroxyfasudil (10 μmol/l) for 1 h, treated with TGF-β1 (10 μg/l) for an additional 48 h, and stained with rhodamine-labeled phalloidin for F-actin. Cell size was measured as fold area increase to unstimulated cells. Control: untreated, PFD: pirfenidone, fasudil: hydroxyfasudil.
Figure 6. Pirfenidone had little effect on TGF-β1-induced increase in the cell surface area. Cells were incubated in the absence or presence of pirfenidone (PFD, 500 mg/l) or
hydroxyfasudil (10 μmol/l) for 1 h, treated with TGF-β1 (10 μg/l) for an additional 48 h, and stained with FITC-conjugated antibodies for N-cadherin. Cell size was measured as the number of pixels in the cell boundary indicated by N-cadherin staining. Control: untreated, PFD: pirfenidone, fasudil: hydroxyfasudil.
B. Pirfenidone suppresses the TGF-β1-induced expression of ECM components in ARPE-19 cells
As the next step, the effect of pirfenidone on the basal and TGF-β1-induced synthesis of collagen type I and fibronectin, which are considered as the major ECM components of fibrosis and one of the important features of EMT, was analyzed. Treatment with TGF-β1 increased the expression of collagen type I by ARPE-19 cells up to eightfold. Since RhoA and p38 MAPK are known to be involved in TGF-β-induced ECM production (Itoh et al., 2007; Song et al., 2010), the inhibitory effect of pirfenidone on TGF-β-induced ECM secretion compared with pharmacological inhibitors of RhoA and p38 MAPK was tested. Pretreatment with pirfenidone, hydroxyfasudil, or SB202190 significantly suppressed the TGF-β1-induced secretion of collagen type I, while the same treatment alone had a minimal effect on the basal level of collagen type I synthesis in ARPE-19 cells (Fig. 7A and 8). Parallel results were obtained for fibronectin synthesis; however, hydroxyfasudil had little effect on TGF-β1-induced fibronectin production (Fig. 7B and 8). These results are in accordance with previous findings production (Zhang et al., 1998; Itoh et al., 2007; Lee et al., 2008; Song et al., 2010), indicating the differential involvement of RhoA and p38 MAPK pathways in the TGF-β1-induced secretion of the ECM components.
C. Pirfenidone abrogates the TFG-β1-induced migration of ARPE-19 cells
The effect of pirfenidone on TGF-β1-induced migratory activity, another important phenotype of the EMT was tested. As expected, treatment with TGF-β1 significantly enhanced the migration of cells 48 h after wounding. In contrast, pre-incubation with pirfenidone, SB202190, or hydrofasudil had significant inhibitory effects on cell migration, and blocked the closure of the defect produced in monolayer cell sheets even in the
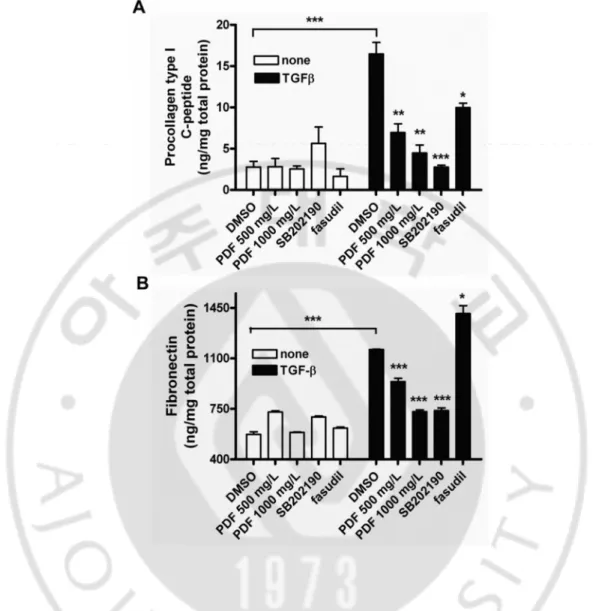
Figure 7. Pirfenidone inhibited TGF-β1-induced expression of extracellular matrix.
Cells were incubated in the absence or presence of SB202190 (10μmol/l), hydroxyfasudil (10 μmol/l), or varying doses of pirfenidone for 1 h and then treated with TGF-β1 (10 μg/l) for an additional 48 h. The supernatants were assayed with ELISA for the level of collagen type I (A) or fibronectin (B). Control: untreated, PFD: pirfenidone, fasudil: hydroxyfasudil. Samples significantly different from the control sample treated with DMSO were indicated with symbols (*, n=5, error bar indicates SD, p<0.05; **, p<0.01; ***, p<0.001).
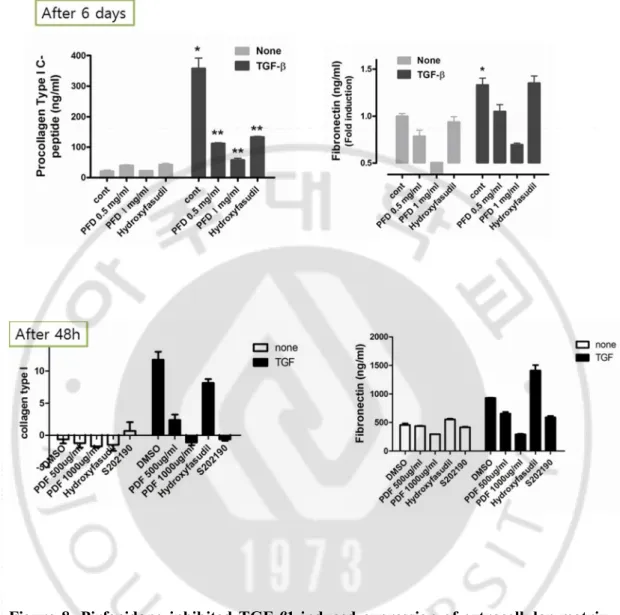
Figure 8. Pirfenidone inhibited TGF-β1-induced expression of extracellular matrix with different time periods. Cells were incubated in the absence or presence of
Figure 9. Pirfenidone inhibited TGF-β1-induced migration of ARPE-19 cells. Cells
were incubated in the absence or presence of varying doses of pirfenidone, SB202190 (10 μmol/l), or hydroxyfasudil (10 μmol/l) for 1 h and then scratched with a 200-μl micropipette tip to form a cell-free (wounded) area. The cells were then incubated in the absence or presence of TGF-β1 (10 μg/l) for an additional 48 h. The vertical axis represents the number of cells that migrated into the wounded region. Stacked bars display the portion of migratory cells classified by migration distance (the gray bar indicates the number of cells that migrated a moderate distance, and the black bar indicates the number of cells that migrated the longest distance). Samples significantly different from the control sample treated with DMSO were indicated with symbols (*, error bar indicates standard deviation (SD), p<0.05; **, p<0.0001; n=10).
D. Pirfenidone blocks TGF-β1-induced nuclear translocation but not phosphorylation of Smads
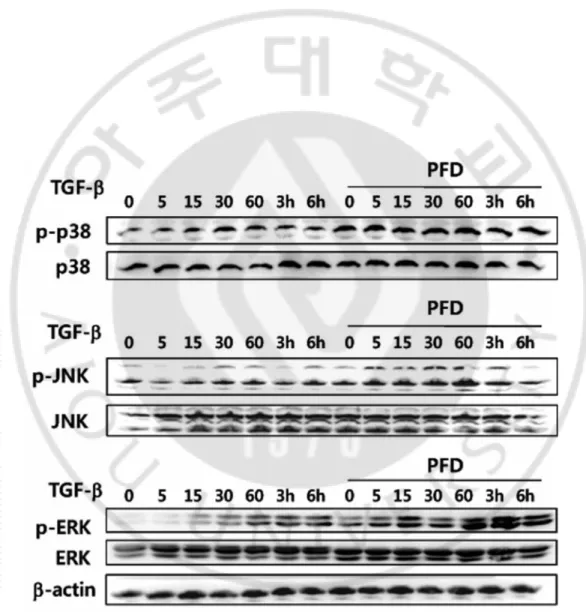
Since pirfenidone abrogated TGF-β1-induced EMT-like phenotypic changes, the Smad and MAPK signaling pathways responsible for the TGF-β1-induced EMT was further investigated. Firstly, the levels of p38 MAPK and Smad 2/3 expression in ARPE-19 cells treated with TGF-β1 were examined by Western analyses, using anti p38 MAPK, anti-phospho-p38 MAPK, anti-Smad 2/3, and anti phospho-Smad2/3. TGF-β1 increased the phosphorylation of p38 MAPK within 30 minutes after stimulation, and its activation persisted for 24 hours (Fig. 10). Western blotting showed that Smads2/3 were phosphorylated within 30 min after exposure to TGF-β1 and remained activated throughout the interval examined up to 24 h, although the level of phosphorylation was less than that seen at 30 min to 1h (Fig 10).
Even though MAKPs such as p38, ERK, and JNK were phosphorylated in a time-dependent manner upon TGF-β1 treatment, pretreatment with pirfenidone had no effect on TGF-β1-induced MAPK phosphorylation (Fig. 11). Contrary to the expectation, pre-incubation with pirfenidone had little effect on the TGF-β1-induced phosphorylation of Smad2/3 (Fig. 12A). Phosphorylated Smads are known to be translocated into the nucleus for activating or repressing responsible genes (Hill, 2009). To determine the effect of pirfenidone on the nucleocytoplasmic shuttling of Smads, the immunoblot analysis using nuclear extracts from the cells treated with TGF-β1 in the absence or presence of pirfenidone was performed. Treatment with TGF-β1 induced the nuclear translocation of phosphorylated Smad2/3; while pretreatment with pirfenidone abrogated TGF-β1-induced nuclear localization of the Smads (Fig. 12B). The blockage of TGF-β1-induced nuclear translocation of the Smads was confirmed with immunocytochemistry (Fig. 12C).
Figure 10. Activation of p38 MAPK, cofilin, and Smad 2/3 by TGF- β1 in ARPE-19 cells. Lysates from cells incubated in the presence of exogenous TGF- β1 (10 μg/l) for the
indicated times were subjected to Western blot analysis using various antibodies. Bands corresponding to the phosphorylated form of p38 MAPK, Smad 2/3, and cofilin and total p38 MAPK, total Smad 2/3, and total cofilin were detected.
Figure 11. Pirfenidone had no effect on TGF-β1-induced phosphorylation of MAPK.
ARPE-19 cells were incubated in the absence or presence of pirfenidone (500 mg/l) for 1 h, and then treated TGF-β1 (10 μg/l) for varying time periods. Total cell lysates (50 μg total protein) were subjected to immunoblot analysis for p38 MAPK, JKN, ERK, and β-actin.
Figure 12. Pirfenidone inhibited TGF-β1-induced signal transduction. A: ARPE-19
cells were incubated in the absence or presence of pirfenidone (500 mg/l) for 1 h, then treated with TGF-β1 (10 μg/l) for varying time periods, and total cell lysates were subjected to immunoblot analysis for phosphor-Smad and Smad2/3. The data shown are representative of three independent experiments. B: Nuclear extracts from the cells incubated in the absence or presence of pirfenidone (500 mg/l) for 1 h and then treated with TGF-β1 (10 μg/l) for an additional 30 min were subjected to immunoblot analysis for Smad2/3. PARP was used for a positive control for nuclear compartment; while α-tubulin was used for a positive control for cytosolic fraction. C: Cells were incubated in the absence or presence of pirfenidone (500 mg/l) for 1 h, then treated with TGF-β1 (10 μg/l) for 30 min, and stained with antibody against phospho-specific Smad2/3 and secondary antibody conjugated with FITC (green). Nucleus was counter-stained with propidium iodide (red). Scale bar=20 μm. The data shown are representative of three independent experiments.
IV. DISCUSSION
In the present study, the strong inhibitory effect of pirfenidone on the TGF-β1-induced EMT in ARPE-19 cells was demonstrated, based on ability of pirfenidone to suppress cytoskeletal organization, ECM synthesis, and cellular migration. In addition, it was delineated that the downstream signaling pathways responsible for the TGF-β1-induced EMT, especially the nucleocytoplasmic shuttling of phosphorylated Smads, were blocked by pirfenidone. Although the antifibrotic efficacy of pirfenidone is well established, this is the first study to describe the molecular mechanisms responsible for the biologic activities of pirfenidone in a human RPE cell line.
A. Pirfenidone and TGF-β1 induced fibroblastic phenotypes in ARPE-19 cells
RPE cells play an important role in the pathogenesis of fibrotic retinal diseases. While TGF-β is accepted to cause the intraocular proliferation of RPE which results in multiple fibrotic retinal diseases, the mechanism involved are not fully understood. It was well established that EMT of ARPE-19 cells was induced by TGF-β treatment and the morphology of cells were changed like fibroblast whereas EGF treatment had little effect on morphology of ARPE-19 cells (Lee et al., 2008). Thus, it is believed that the phenotypes of the ARPE-19 cells can be changed in response to TGF-β. In this study, TGF-β1 induce reorganization of the actin cytoskeleton in ARPE-19 cells, as the F-actin in the thick cortical actin bands of the untreated cells became centrally located actin filaments or stress fibers upon TGF-β1 treatment. Having established TGF-β1 induced fibroblastic phenotypes in ARPE-19 cells, it was examined whether the TGF-β1-induced morphological changes were affected by pirfenidone, and the findings were compared to morphologic changes under the influence of Rho inhibitor which is known to block the key signaling in the TGF-β1-induced cytoskeletal changes and stress fiber formation.
It is well known that TGF-β1 treatment can trigger phosphorylation of ERK, Akt and Smad2/3. But, inhibition of the ERK or Akt signaling pathways failed to change RPE cell morphology when induced by TGF-β1 (Lee et al., 2008). The Rho family members RhoA and Rac1 are stimulated by TGF-β1 in ARPE-19 cells, and the functionally significance of
the activation of Rho was proven by the fact that inhibition of Rho suppressed TGF-β1-induced actin stress fiber formation and morphological changes (Lee et al., 2008). Similarly, Rho inhibition and pirfenidone suppressed the morphological changes induced by TGF-β1 while the inhibitor of p 38 MAPK had no effect. Moreover, Rho Inhibition and pifenidone also suppressed TGF-β1-induced F-actin expression in the current study.
The function of cofilin is to causes actin depolymerization through severing the actin filaments and accelerating the turnover for actin monomers from the pointed end. The inactivation of Cofilin by Rho GTPase blocks disassembling of actin filament and induces cytoskeletal reorganization. The activation of the RhoA protein will activate ROCK, a RhoA kinase, which leads to the stimulation of LIM kinase, which in turn inhibits the protein cofilin by phosphorylating cofilin at serine 3 (Lee et al., 2008). This action is needed for cell motility and other physiological behaviors during EMT. In this study, TGF-β1 treatment enhanced cofilin phosphorylation throughout the course of the experiment. The actin-severing activity of cofilin, therefore, was attenuated by TGF-β1 in ARPE-19 cells. Pirfenidone suppressed the phosphorylation of cofilin when induced by TGF-β1 as expected. Thus, it is safe to assume that pirfenidone blocked the phosphorylation of LIMK by RhoA and/or ROCK in TGF-β1-stimulated ARPE-19 cells and thereby decrease its kinase activity toward cofilin. Suppressed phosphorylation of cofilin loses its ability to stabilizing actin filaments, thereby depolymerize actin filaments.
Cells treated with TGF-β1 showed up to a fivefold increase in cell surface area when compared to the unstimulated control cells, and this result is consistent with a previous report (Lamouille and Derynck, 2007). Pretreatment with hydroxyfasudil alone increased cell surface area and inhibited the TGF-β1-induced increase in the cell surface area; whereas pretreatment of pirfenidone had little effect on cell size and inhibited the TGF-β1-induced increase in the cell surface area. As stated above, the dynamic reorganization of the actin
due to Cdc42/Rac hyperactivity (El-Sibai et al., 2008). The inhibition of Rho or ROCK appears to suppress cell motility in a similar manner, although the phenotypes produced as a result of Rho and ROCK inhibition differ: Rho inhibition led to circumferential expansion under basal conditions, whereas ROCK inhibition resulted in exaggerated growth factor stimulated expansion (El-Sibai et al., 2008). Unbalanced inhibition of Rho by hydroxyfasudil had more dramatic effects on cell morphology than pirfenidone does in this study. These findings collectively suggest that pirfenidone might block ROCK and Cdc42/Rac signaling, since treatment of the cells with pirfenidone induced breakdown of stress fibers without affecting cell size.
Cells during EMT undergo the reorganization of actin cytoskeleton so that the cells can acquire dynamic cell elongation and directional motility, and RhoA facilitates actin stress fibre formation, whereas Rac1 and Cdc42 mainly involve in the formation of lamellipodia and filopodia. In this study, preincubation with pirfenidone, SB202190, or hydrofasudil had significant inhibitory effects on cell migration, and blocked the closure of the defect produced in monolayer cell sheets even in the presence of TGF-β1. The most significant reduction in motility was noted at pirfenidone concentrations of 250 and 500 mg/l. It is believed inhibitory effects on cell migration by pirfenidone was due to the combination of blocking RhoA pathway and reduction of ECM production, whereas inhibitory effects on cell migration by SB202190 (p 38 MAPK inhibitor) was believed to be because of decreased production of ECM rather than blocking the RhoA pathways.
B. Pirfenidone and TGF-β1-induced expression of extracellular matrix components in ARPE-19 cells
In ARPE-19 cells, non-Smad signaling such as Cofilin, p 38, ERK plays a different role in producing ECM. Based on previous reports, blockade of the ERK pathway rather than the RhoA pathway significantly reduced TGF- β1-induced fibronectin accumulation in ARPE-19 cells (Lee et al., 2008). However, blockage of the ERK pathway did not significantly prevent induction of collagen gene expression by TGF-β2 (Kimoto et al., 2004). Meanwhile, p 38 MAPK plays an important role in synthesis of collagen (Kimoto et al., 2004; Saika et al., 2005). In this study, treatment with TGF-β1 increased the expression of collagen type I by
ARPE-19 cells up to eightfold. As expected, pretreatment with pirfenidone, hydroxyfasudil, or SB202190 significantly suppressed the TGF-β1-induced secretion of collagen type I, while the same treatment alone had a minimal effect on the basal level of collagen type I synthesis in ARPE-19 cells. Parallel results were obtained for fibronectin synthesis; however, hydroxyfasudil had little effect on TGF-β1-induced fibronectin production as previously reported (Fig. 13). Nevertheless, recent study which evaluated the effects of Y27632 (ROCK inhibitor) on the mRNA expression levels of ECM in TGF-β-induced- ARPE-19 cells by real-time RT-qPCR revealed that treatment with ROCK inhibitor had almost no effect on the basal mRNA expression levels of fibronectin, but pre-incubation of ARPE-19 with Y27632 significantly reduced the TGF-β-induced mRNA expression levels of fibronectin (Zhu et al., 2013). The discrepancy between two results, in which RhoA was involved in fibronectin synthesis, may be due to different RhoA inhibitor (hydroxyfasudil vs Y27632), different method to detect fibronectin, or different incubation time. Nevertheless, it should be noted that pirfenidone significantly suppressed the TGF-β1-induced secretion of collagen type I and fibronectin, which are considered as the major ECM components of fibrosis and one of the important features of EMT.
C. Possible molecular mechanisms of pirfenidone for the inhibitory action for
TGF-β1-induced fibrogenesis in ARPE-19 cells
Pirfenidone exerted its anti-fibrotic effect through inhibition of HSP 47, a collagen-specific chaperon, resulting in a decrease in collagen synthesis in TGF-β1-induced lung fibroblasts (Nakayama et al., 2008). In animal models of lung fibrosis, pirfenidone also suppressed expression of mRNA and the TGF-β protein (Iyer et al., 1999). Pirfenidone inhibits platelet-derived growth factor-induced proliferation and collagen production in
Figure 13. TGF- β1 signaling pathway and their possible role in the regulation of ECM production in ARPE-19 cells. TGF-β1 promotes ECM production through Smad and
Smad-independent pathway. TGF-β1 activates MAPK pathways, including Erk and p38 pathways, and activation of MAPK pathways causes increased fibronectin synthesis. Meanwhile, RhoA pathway is involved in collagen synthesis. MAPK, mitogen-activated protein kinase
TGFβ induces EMT via direct phosphorylation of RSmad (Smad2/3), or activation of -Smad independent signaling pathways including MAP kinase, Rho GTPase, and PI3 kinase-Akt, resulting in repression of epithelial marker genes and activation of mesenchymal markers (Xu et al., 2009). Current evidence suggests that the EMT can be therapeutically targeted through disrupting TGF-β signaling at different levels: inhibiting TGF-β expression with RNA interference, antagonizing TGF-β ligand activity, inhibiting TGF-β receptor kinase activity by using small-molecule inhibitors, and intervening in Smad activation (Pennison and Pasche, 2007). In particular, nuclear translocation of active Smad complexes and subsequent interactions with the general transcription machinery emerged as crucial steps for therapeutic intervention of TGF-β signaling (Hill, 2009).
Smads independent signaling molecules have also been implicated in TGF -mediated EMT, including Erk, PI3K/Akt, RhoA, and cofilin (Hartsough and Mulder, 1995; Liao et al., 2001). Induction of Erk and p38-MAPK phosphorylation by TGF- regulates the expression of genes involved in the remodeling of extracellular matrix and disruption of adherens and tight junctions to facilitate EMT (Hartsough and Mulder, 1995; Liao et al., 2001). However, studies using Smad-binding defective TGF RI constructs that can still mediate MAPK signal indicated that Smads are required for Erk-induced EMT process (Yu et al., 2002). Even though MAKPs such as p38, ERK, and JNK were phosphorylated in a time-dependent manner upon TGF-β1 treatment, pretreatment with pirfenidone had no effect on TGF-β1-induced MAPK phosphorylation in this study. As Smad independent pathway was not responsible for the molecular mechanisms of pirfenidone for the inhibitory action for TGF-β1-induced fibrogenesis in ARPE-19 cells, Smad pathway was further evaluated. Unfortunately, preincubation with pirfenidone had little effect on the TGF-β1-induced
Figure 14. Mechanism of action of pirfenidone in ARPE-19 cells. The mechanism of
action of pirfenidone is by abrogating TGF-β1-induced nuclear localization of the Smads in ARPE-19 cells. Thus, pirfenidone suppressed Smads signaling without affecting phosphorylation of Smad2/3 by TGF-β1. However, there is possibility that pirfenidone has effect on cytoskeletal arrangement by blocking RhoA pathway.
was performed. Treatment with TGF-β1 induced the nuclear translocation of phosphorylated Smad2/3; while pretreatment with pirfenidone abrogated TGF-β1-induced nuclear localization of the Smads. These finding was further confirmed with immunocytochemistry.
Here, it was demonstrated that pirfenidone inhibits TGF-β-activated Smad signaling by preventing nuclear accumulation of phosphorylated Smad2/3, which can suppress Smads signaling without affecting other pathways regulated by TGF-β.
Since the fibrotic transformation of RPE cells is regarded as the main contributor to various fibrotic diseases of the eye (Bochaton-Piallat et al., 2000; Nagineni et al., 2003), the inhibitory action of pirfenidone on TGF-β-induced phenotypic changes of a human RPE cell line provides a rationale for a trial of this potential antifibrotic agent in treating proliferative vitreoretinopathy and other fibrotic retinal disorders. However, these results are based on a single human RPE cell line, and further studies involving primary RPE cell cultures are required.
V. CONCLUSION
Fibrotic diseases of the posterior segment of the eye lead to permanent visual loss due to scar tissue formation by EMT of RPE cells. TGF-β is known to play a key role in the EMT process of RPE cells, which is characterized by morphological change, cytoskeletal rearrangement and accumulation of extracellular matrix (ECM). Herein, It was examined the in vitro effect of pirfenidone, a novel anti-fibrotic agent, on TGF-β1-mediated EMT phenotypic changes in ARPE-19 cells. Treatment with TGF-β1 induced an elongation of cell shape and the formation of stress fiber running parallel to the lone axis of cells, which were significantly inhibited by pirfenidone and hydroxyfasudil, a Rho kinase inhibitor. Moreover, pretreatment of pirfenidone significantly suppressed TGF-β1-induced production of ECM components such as collagen type I and fibronectin by RPE cells, which were differentially dependent on Rho and p38MAP kinase pathways. To elucidate the signaling pathways involved in inhibitory effect of pirfenidone, it was further examined the involvement of Smad and p38 MAPK pathways. Treatment with TGF-β1 induced time-dependent phosphorylation of Smad2/3 and p38MAPK. Pretreatment with pirfenidone had no effect on TGF-β1-induced MAPK phosphorylation and Smad2/3. Treatment with TGF-β1 induced the nuclear translocation of phosphorylated Smad2/3; while pretreatment with pirfenidone abrogated TGF-β1-induced nuclear localization of the Smads. Therefore it was concluded that pirfenidone inhibits TGF-β-activated Smad signaling by preventing nuclear accumulation of phosphorylated Smad2/3, which can suppress Smads signaling without affecting other pathways regulated by TGF-β. In summary, these results collectively provide a rational background for future evaluation of pirfenidone as a potential antifibrotic agent for treating proliferative vitreoretinopathy and other fibrotic retinal disorders.
REFERENCES
1. Artlett CM: Inflammasomes in wound healing and fibrosis. J Pathol 229: 157-167,
2013
2. Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, Taguchi Y, Nagai S,
Itoh H, Ohi M, Sato A, Kudoh S: Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 171: 1040-1047, 2005
3. Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga
CL, Moses HL: Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell 12: 27-36, 2001
4. Bochaton-Piallat ML, Kapetanios AD, Donati G, Redard M, Gabbiani G, Pournaras
CJ: TGF-beta1, TGF-beta receptor II and ED-A fibronectin expression in myofibroblast of vitreoretinopathy. Invest Ophthalmol Vis Sci 41: 2336-2342, 2000
5. Bressler NM, Bressler SB, Fine SL: Age-related macular degeneration. Surv
Ophthalmol 32: 375-413, 1988
6. Bressler NM, Doan QV, Varma R, Lee PP, Suner IJ, Dolan C, Danese MD, Yu E,
Tran I, Colman S: Estimated cases of legal blindness and visual impairment avoided using ranibizumab for choroidal neovascularization: non-Hispanic white population in the United States with age-related macular degeneration. Arch Ophthalmol 129: 709-717, 2011
7. Cain WC, Stuart RW, Lefkowitz DL, Starnes JD, Margolin S, Lefkowitz SS:
Inhibition of tumor necrosis factor and subsequent endotoxin shock by pirfenidone. Int J Immunopharmacol 20: 685-695, 1998
10. Cho ME, Smith DC, Branton MH, Penzak SR, Kopp JB: Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2: 906-913, 2007
11. Choi YH, Back KO, Kim HJ, Lee SY, Kook KH: Pirfenidone attenuates
IL-1beta-induced COX-2 and PGE2 production in orbital fibroblasts through suppression of NF-kappaB activity. Exp Eye Res 113: 1-8, 2013
12. Connor TB, Jr., Roberts AB, Sporn MB, Danielpour D, Dart LL, Michels RG, de
Bustros S, Enger C, Kato H, Lansing M, et al.: Correlation of fibrosis and transforming growth factor-beta type 2 levels in the eye. J Clin Invest 83: 1661-1666, 1989
13. Di Sario A, Bendia E, Macarri G, Candelaresi C, Taffetani S, Marzioni M,
Omenetti A, De Minicis S, Trozzi L, Benedetti A: The anti-fibrotic effect of pirfenidone in rat liver fibrosis is mediated by downregulation of procollagen alpha1(I), TIMP-1 and MMP-2. Dig Liver Dis 36: 744-751, 2004
14. Di Sario A, Bendia E, Svegliati Baroni G, Ridolfi F, Casini A, Ceni E, Saccomanno
S, Marzioni M, Trozzi L, Sterpetti P, Taffetani S, Benedetti A: Effect of pirfenidone on rat hepatic stellate cell proliferation and collagen production. J Hepatol 37: 584-591, 2002
15. Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM: ARPE-19, a human
retinal pigment epithelial cell line with differentiated properties. Exp Eye Res 62: 155-169, 1996
16. El-Sibai M, Pertz O, Pang H, Yip SC, Lorenz M, Symons M, Condeelis JS, Hahn
KM, Backer JM: RhoA/ROCK-mediated switching between Cdc42- and Rac1-dependent protrusion in MTLn3 carcinoma cells. Exp Cell Res 314: 1540-1552, 2008
17. Ferris FL, 3rd, Fine SL, Hyman L: Age-related macular degeneration and blindness
due to neovascular maculopathy. Arch Ophthalmol 102: 1640-1642, 1984
18. Flanders KC: Smad3 as a mediator of the fibrotic response. Int J Exp Pathol 85:
47-64, 2004
19. Garcia L, Hernandez I, Sandoval A, Salazar A, Garcia J, Vera J, Grijalva G, Muriel
P, Margolin S, Armendariz-Borunda J: Pirfenidone effectively reverses experimental liver fibrosis. J Hepatol 37: 797-805, 2002
20. Grossniklaus HE, Green WR: Histopathologic and ultrastructural findings of surgically excised choroidal neovascularization. Submacular Surgery Trials Research Group. Arch Ophthalmol 116: 745-749, 1998
21. Gurujeyalakshmi G, Hollinger MA, Giri SN: Pirfenidone inhibits PDGF isoforms in
bleomycin hamster model of lung fibrosis at the translational level. Am J Physiol 276: L311-318, 1999
22. Hartsough MT, Mulder KM: Transforming growth factor beta activation of
p44mapk in proliferating cultures of epithelial cells. J Biol Chem 270: 7117-7124, 1995
23. Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA,
Topper JN, Gimbrone MA, Jr., Wrana JL, Falb D: The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 89: 1165-1173, 1997
24. Hayashida T, Poncelet AC, Hubchak SC, Schnaper HW: TGF-beta1 activates MAP
kinase in human mesangial cells: a possible role in collagen expression. Kidney Int 56: 1710-1720, 1999
25. Hewitson TD, Kelynack KJ, Tait MG, Martic M, Jones CL, Margolin SB, Becker
GJ: Pirfenidone reduces in vitro rat renal fibroblast activation and mitogenesis. J Nephrol 14: 453-460, 2001
26. Hill CS: Nucleocytoplasmic shuttling of Smad proteins. Cell Res 19: 36-46, 2009
27. Hinton DR, He S, Jin ML, Barron E, Ryan SJ: Novel growth factors involved in the
pathogenesis of proliferative vitreoretinopathy. Eye (Lond) 16: 422-428, 2002
28. Hisatomi K, Mukae H, Sakamoto N, Ishimatsu Y, Kakugawa T, Hara S, Fujita H,
TGF-30. Itoh Y, Kimoto K, Imaizumi M, Nakatsuka K: Inhibition of RhoA/Rho-kinase pathway suppresses the expression of type I collagen induced by TGF-beta2 in human retinal pigment epithelial cells. Exp Eye Res 84: 464-472, 2007
31. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG: Evidence that
fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 110: 341-350, 2002
32. Iyer SN, Gurujeyalakshmi G, Giri SN: Effects of pirfenidone on transforming
growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther 291: 367-373, 1999
33. Jaffe AB, Hall A: Rho GTPases: biochemistry and biology. Annu Rev Cell Dev
Biol 21: 247-269, 2005
34. Kakugawa T, Mukae H, Hayashi T, Ishii H, Abe K, Fujii T, Oku H, Miyazaki M,
Kadota J, Kohno S: Pirfenidone attenuates expression of HSP47 in murine bleomycin-induced pulmonary fibrosis. Eur Respir J 24: 57-65, 2004
35. Kalluri R, Weinberg RA: The basics of epithelial-mesenchymal transition. J Clin Invest 119: 1420-1428, 2009
36. Kim H, Choi YH, Park SJ, Lee SY, Kim SJ, Jou I, Kook KH: Antifibrotic effect of
Pirfenidone on orbital fibroblasts of patients with thyroid-associated ophthalmopathy by decreasing TIMP-1 and collagen levels. Invest Ophthalmol Vis Sci 51: 3061-3066, 2010
37. Kimoto K, Nakatsuka K, Matsuo N, Yoshioka H: p38 MAPK mediates the
expression of type I collagen induced by TGF-beta 2 in human retinal pigment epithelial cells ARPE-19. Invest Ophthalmol Vis Sci 45: 2431-2437, 2004
38. Lamouille S, Derynck R: Cell size and invasion in TGF-beta-induced epithelial to
mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 178: 437-451, 2007
39. Lee J, Ko M, Joo CK: Rho plays a key role in TGF-beta1-induced cytoskeletal rearrangement in human retinal pigment epithelium. J Cell Physiol 216: 520-526, 2008
40. Lee JM, Dedhar S, Kalluri R, Thompson EW: The epithelial-mesenchymal
transition: new insights in signaling, development, and disease. J Cell Biol 172: 973-981, 2006
41. Lee K, Young Lee S, Park SY, Yang H: Antifibrotic effect of pirfenidone on human pterygium fibroblasts. Curr Eye Res 39: 680-685, 2014
42. Liao JH, Chen JS, Chai MQ, Zhao S, Song JG: The involvement of p38 MAPK in
transforming growth factor beta1-induced apoptosis in murine hepatocytes. Cell Res 11: 89-94, 2001
43. Lo RS, Chen YG, Shi Y, Pavletich NP, Massague J: The L3 loop: a structural motif
determining specific interactions between SMAD proteins and TGF-beta receptors. EMBO J 17: 996-1005, 1998
44. Machemer R: Massive periretinal proliferation: a logical approach to therapy. Trans
Am Ophthalmol Soc 75: 556-586, 1977
45. Massague J, Seoane J, Wotton D: Smad transcription factors. Genes Dev 19:
2783-2810, 2005
46. Misra HP, Rabideau C: Pirfenidone inhibits NADPH-dependent microsomal lipid
peroxidation and scavenges hydroxyl radicals. Mol Cell Biochem 204: 119-126, 2000
47. Nagai S, Hamada K, Shigematsu M, Taniyama M, Yamauchi S, Izumi T:
Open-label compassionate use one year-treatment with pirfenidone to patients with chronic pulmonary fibrosis. Intern Med 41: 1118-1123, 2002
48. Nagineni CN, Samuel W, Nagineni S, Pardhasaradhi K, Wiggert B, Detrick B,
Hooks JJ: Transforming growth factor-beta induces expression of vascular endothelial growth factor in human retinal pigment epithelial cells: involvement of mitogen-activated protein kinases. J Cell Physiol 197: 453-462, 2003
49. Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P: Identification of Smad7, a
TGFbeta-51. Nobes CD, Hall A: Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81: 53-62, 1995
52. Okada H, Danoff TM, Kalluri R, Neilson EG: Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol 273: F563-574, 1997
53. Oku H, Nakazato H, Horikawa T, Tsuruta Y, Suzuki R: Pirfenidone suppresses
tumor necrosis factor-alpha, enhances interleukin-10 and protects mice from endotoxic shock. Eur J Pharmacol 446: 167-176, 2002
54. Oku H, Shimizu T, Kawabata T, Nagira M, Hikita I, Ueyama A, Matsushima S,
Torii M, Arimura A: Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol 590: 400-408, 2008
55. Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA: Focal adhesion kinase: a
regulator of focal adhesion dynamics and cell movement. Oncogene 19: 5606-5613, 2000
56. Pastor JC, de la Rua ER, Martin F: Proliferative vitreoretinopathy: risk factors and
pathobiology. Prog Retin Eye Res 21: 127-144, 2002
57. Pennison M, Pasche B: Targeting transforming growth factor-beta signaling. Curr Opin Oncol 19: 579-585, 2007
58. Phanish MK, Wahab NA, Colville-Nash P, Hendry BM, Dockrell ME: The
differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem J 393: 601-607, 2006
59. Ryan SJ: The pathophysiology of proliferative vitreoretinopathy in its management.
Am J Ophthalmol 100: 188-193, 1985
60. Saika S, Yamanaka O, Ikeda K, Kim-Mitsuyama S, Flanders KC, Yoo J, Roberts
AB, Nishikawa-Ishida I, Ohnishi Y, Muragaki Y, Ooshima A: Inhibition of p38MAP kinase suppresses fibrotic reaction of retinal pigment epithelial cells. Lab Invest 85: 838-850, 2005
61. Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A: Targeted disruption of
TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest 112: 1486-1494, 2003
62. Sheridan CM, Occleston NL, Hiscott P, Kon CH, Khaw PT, Grierson I: Matrix metalloproteinases: a role in the contraction of vitreo-retinal scar tissue. Am J Pathol 159: 1555-1566, 2001
63. Shihab FS, Bennett WM, Yi H, Andoh TF: Pirfenidone treatment decreases
transforming growth factor-beta1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity. Am J Transplant 2: 111-119, 2002
64. Shimizu T, Fukagawa M, Kuroda T, Hata S, Iwasaki Y, Nemoto M, Shirai K,
Yamauchi S, Margolin SB, Shimizu F, Kurokawa K: Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int Suppl 63: S239-243, 1997
65. Shimizu T, Kuroda T, Hata S, Fukagawa M, Margolin SB, Kurokawa K:
Pirfenidone improves renal function and fibrosis in the post-obstructed kidney. Kidney Int 54: 99-109, 1998
66. Song S, Kang SW, Choi C: Trichostatin A enhances proliferation and migration of
vascular smooth muscle cells by downregulating thioredoxin 1. Cardiovasc Res 85: 241-249, 2010
67. Thiery JP: Epithelial-mesenchymal transitions in tumour progression. Nat Rev
Cancer 2: 442-454, 2002
68. Thiery JP, Acloque H, Huang RY, Nieto MA: Epithelial-mesenchymal transitions
in development and disease. Cell 139: 871-890, 2009
69. Thiery JP, Sleeman JP: Complex networks orchestrate epithelial-mesenchymal
transitions. Nat Rev Mol Cell Biol 7: 131-142, 2006