저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Characterization of Genetically Modified
Mesenchymal Stem Cells with a Retroviral
Vector Expressing a Suicide Gene
by
Jin Sung Park
Major in Neuroscience
Department of Biomedical Sciences
The Graduate School, Ajou University
Characterization of Genetically Modified
Mesenchymal Stem Cells with a Retroviral
Vector Expressing a Suicide Gene
by
Jin Sung Park
A Dissertation Submitted to The Graduate School of
Ajou University in Partial Fulfillment of the Requirements for The
Degree of Ph.D. in Neuroscience
Supervised by
Haeyoung Suh-Kim, Ph.D.
Sung-Soo Kim, Ph.D.
Major in Neuroscience
Department of Biomedical Science
The Graduate School, Ajou University
This certifies that the dissertation of
Jin Sung Park is approved
SUPERVISORY COMMITTEE
Young-Don Lee
Haeyoung Suh-Kim
Sung-Soo Kim
Young Sook Son
Byoung-Hyun Min
The Graduate School, Ajou University
June, 21th, 2013
-ABSTRACT-
Characterization of Genetically Modified Mesenchymal Stem
Cells with a Retroviral Vector Expressing a Suicide Gene
Human mesenchymal stem cells have emerged as attractive cellular vehicles to deliver therapeutic genes for ex-vivotherapyof diverse diseases because they have the capability to migrate into tumor or lesion sites. Previously, we showed that mesenchymal stem cells could be utilized as a cellular vehicle to deliver a bacterial cytosine deaminase suicide gene to brain tumors. Here, we assessed whether transduction with a retroviral vector encoding cytosine deaminase gene might alter the stem cell property of mesenchymal stem cells. Mesenchymal stem cells were transduced at passage 1 and cultivated up to passage 11. We found that proliferative and differentiation potential, chromosomal stability and surface antigenicity of mesenchymal stem cells were not altered by retroviral transduction. The results indicate that retroviral vectors can be safely utilized for deliver suicide genes to mesenchymal stem cells for ex-vivo therapy. We also found that the single retroviral transduction was sufficient for the sustainable expression up to passage 10. The long-lasting expression of transduced gene warrantsmanufacturing the transduced mesenchymal stem cells tractable and manageable for allogeneic transplantation.Keywords: mesenchymal stem cell, retrovirus, suicide gene, gene therapy, ex-vivo therapy, safety.
TABLE OF CONTENTS
ABSTRACT ---i
TABLE OF CONTENTS --- ii
LIST OF FIGURES --- iii
LIST OF TABLE--- iv
INTRODUCTION --- 1
Part.1.Establishment of Optimal Culture Conditions for Mass Production of ClinicalGrade, Human Mesenchymal Stem Cells Introduction I --- 7
Materials and Methods I--- 10
ResultsI --- 14
Discussion I --- 26
Part. 2. Comparative study of MSCs with genetically modified MSCs Introduction II --- 30Materials and Methods II --- 3 3 Results II --- 38
Discussion II --- 46
REFFERENCES --- 49
LIST OF FIGURES
Fig. 1.The anti-cancer effects of MSCs genetically modified with CD --- 6
Part. 1.Establishment of Optimal Culture Conditions for Mass Production of Clinical Grade, Human Mesenchymal Stem Cells Fig.1. Analysis of MSC yields at passage 0 --- 17
Fig.2. Donor dependent growth of MSCs --- 18
Fig.3. HLA-DR expression (+bFGF condition) --- 20
Fig.4. HLA-DR and CD90 expressions depending on the culture conditions --- 22
Fig.5. Proliferation of MSCs depending on passages and bFGF addition --- 24
Fig.6. Optimal condition for production of MSCs --- 25
Part. 2. Comparative study of MSCs with genetically modified MSCs Fig.1. No effects of retroviral transduction on the MSC proliferation capability --- 39
Fig.2. No effects of retroviral transduction on mutipotency and surface antigen profiles --- 40
Fig.3.In vitro cytotoxic effects of MSC/CD in combination with 5-FC --- 42
Fig.4. Sustainable CD expression in MSC/CD after long-term culture --- 43
LIST OF TABLES
Part. 1.Establishment of Optimal Culture Conditions for Mass Production of Clinical Grade, Human Mesenchymal Stem Cells
Table.1. Donor screening --- 15 Table.2. Surface marker expressions depending on donors and bFGF addition --- 19
Part. 2. Comparative study of MSCs with genetically modified MSCs
INTRODUCTION
General features of mesenchymal stem cells (MSCs)
From the end of the 1960s to the beginning of the 1970s, a Soviet scientist, Alexander Friedenstein, discovered a population of adherent cells in bone marrow (BM) that could differentiate into osteoblast, chondrocytes, and hematopoietic stromal supportive cells. These cells with fibroblast shapes could form clonal colonies when seeded at a low density, which suggested the presence of precursor cells, the colony-forming unit-fibroblasts (CFU-F)(Friedenstein AJ, 1968; Owen and Friedenstein, 1988). Because the cells were capable of differentiating into various lineages of the mesoderm, they were named mesenchymal stem cells. Thesedays, these cells are preferentially named as multipotent mesenchymal stromal cells. The stemness status and the long-term self-renewal potential of MSCs are not yet definitely defined(Philippe et al., 2010).
Recent researches have shown that MSCs can be isolated from almost all of the postnatal organs including adipose tissue(Al-Nbaheen et al., 2012), dental pulp(Razieh Karamzadeh, 2012), oral mucosa and gingiva(Zhang QZ, 2012), fetal liver (Joshi et al., 2012), peripheral blood (Jaber Lyahyai, 2012), synovium (Helen Dry, 2013), periosteum(Agatha H. Kisiel, 2012), lung (Sinclair et al., 2013)and spleen (Hegyi et al., 2010). Wherever the tissue location, MSCs should satisfy three criteria proposed by the Mesenchymal and Tissue Stem Cell Committee of the International Society for Cellular Therapy (ISCT) (Dominici et al., 2006):
l MSCs must be adherent to plastic under standard tissue culture conditions using tissue culture flasks;
l MSCs must express certain cell surface markers such as CD73, CD90, and CD105, and lack expression of other markers including CD45, CD34, CD14, or CD11b, CD79alpha or CD19 and HLA-DR surface molecules as measured by flow cytometry;
l MSCs must have the capacity to differentiate into osteoblast, adipocytes, and chondroblasts under standard in vitrodifferentiating conditions.
MSCs as therapeutic drugs
MSCs have been intervened in over two hundred cases for the treatment of various diseases (http://www.clinicaltrials.gov) because of their biological characteristics associated with therapeutic effects. Currently, the following four properties are considered the most important: (1) the ability to home to sites of inflammation following tissue injury when injected intravenously (2) the ability to differentiate into various cell types (3) the ability to secrete multiple bioactive molecules capable of stimulating recovery of injured cells and inhibiting inflammation (4) the lack of immunogenicity and the ability to perform immunomodulatory functions. These four aspects are combined and overlapped to reduce inflammation and improve tissue repair (Wang et al., 2012)
For example, 69 myocardial infarction patients who had received autologous bone marrow MSCs(BMSCs) or saline showed significant improvement of left ventricular function compared with control group (Chen et al., 2004) and 8 patients with liver cirrhosis were injected by autologous MSCs and their liver functions were statistically improved as verified by the model of end-stage liver disease core by 24 weeks after injection(Kharaziha et al., 2009). In addition, 4 x 107 MSCs were injected into 33 patients undergoing multiple system atrophy (MSA) and the patients showed significant improvement in neurological deficits compared with the placebo group during 360-day period in a phase II randomized trial(Lee et al., 2012). Furthermore, Osiris therapeutics treated 32 acute Graft-versus-Host Disease (GvHD) patients using unrelated and unmatched donor-derived BMSCs in combination with corticosteroids and 94% of them had an initial response to MSCs as well as 77% of them showed complete remission(Kebriaei et al., 2009). The allogeneic MSCs manufactured by Osiris therapeutics have been also exploited in other clinical trials for Crohn's disease (phaseIII), acute myocardial infarction (phaseII), Type1 diabetes mellitus (phaseII), and pulmonary disease (phaseII) and successfully approved for market authorization with the name of Remestemcel-L in Canada for treatment of GvHD in 2012.
Genetic modification
There are non-viral and viral vector systems for gene delivery, but all gene therapy applications depend on the factor that the genetic material needs to be delivered across the cell membrane and ultimately to the cell nucleus and each of the delivery systems has some advantages and disadvantages(Nayerossadat et al., 2012).
Non-viral system is generally based on plasmids with diverse transfection methods including either chemical methods, such as cationic liposomes and polymers, or physical methods, such as gene gun, electroporation, particle bombardment, ultrasound utilization, and magnetofection. (Gehl, 2003; Madeira et al., 2010; Nayerossadat et al., 2012). Even though their main advantages are large gene capacity, safe handling, cost-effectiveness and low immunogenicity, they have drawbacks such as low efficiency and difficulty in transfecting target cells. Despite their low efficiency, they are use in over 25.1% of the clinical trials using gene therapy(Vargas et al., 2012).
Viral vector systems using retrovirus, adenovirus, adeno-associated virus (AAV), herpes virus, pox virus, human foamy virus or lentivirus have been used in numerous clinical trials(Aiuti et al., 2007; Alexander et al., 2007; Bushman, 2007; Huang et al., 2011). All viral vector genomes have been modified by deleting some areas of their genomes so that their replication becomes deranged and it makes them safer, but the system has some problems, such as their marked immunogenicity, toxin production, their limitation in transgenic capacity size, while they represent much higher efficiencies to transduce tissue or cells than non-viral systems (Anson, 2004; Gardlik et al., 2005; Bushman, 2007).
Adenoviral vectors can be utilized of transferring both dividing and nondividing cells and deliver large DNA particles (up to 38kb) with low host specificity and high titer. However, their gene expression is too short term because they would not integrate into the host genome, and gene therapy by adenoviral vectors has caused serious bad side effects including acute immunologic responses and even death of some patient(Marshall, 1999; Teramato et al., 2000; Raper et al., 2003). Adenoviral vectors may be employed when transient transgene expression is desired, but its immunogenicity is still an issue.
AAV can infect both dividing and nondividing cells with low host specificity and high tier like adenoviral vectors but because of having some deficiency in their replication and pathogenicity, are safer than adenoviral vectors. The major disadvantages of AAV are complicated process of vector production and their limited transgene capacity of the particles (up to 4.8kb)(Teramato et al., 2000; Kamimura et al., 2011; Nayerossadat et al., 2012).
Retroviral vectors can accept approximately 8 kb of an exogenous gene sequence and integrate into host genome in only dividing cells, which transmitting sustainable expression of transgene to next generation with single transduction. However, the main limitations of retroviral vectors are their low titer, immunogenic problems, and the risk of insertion, which could possibly cause oncogene activation or tumor-suppressor gene inactivation (Kamimura et al., 2011; Nayerossadat et al., 2012). Lentiviral vectors, a subclass of retroviruses, can infect both dividing and nondividing cells and have the advantages of high-efficiency of infection, long-term stable expression of a transgene, low immunogenicity. They have extensively used for ex vivo gene transfer in central nervous system due to strong tropism for neural stem cells with no significant immune responses and no unwanted side effects (Lachmann and Efstathiou, 1997; Goss et al., 2001; Federici et al., 2009).
MSCs as drug delivery system
MSCs have been emerged as cellular vehicles due to the tumor tropic ability, expression of genetically engineered proteins, and utilization for the delivery of therapeutic gene products, compatibilityin vivoand non-immunogenicity to the host. Genetically modified MSCs have been used for improvement in hematopoietic engraftment following myeloablative transplantation regimens, the targeted delivery of antitumor factors by secretion of growth factors and cytokines, the correction of defective genes in inherited disorders(Menon et al., 2008).
For instance, the pancreatic duodenal homeobox-1(PDX-1) transduced MSCs differentiated into functional insulin-producing cells which produced euglycaemia in streptozotocin-induced diabetic mice (Karnieli et al., 2007; Li et al., 2007) andMSCs
expressingβ-glucuronidase (GUSB) retained normal trafficking ability in nonobese diabetic severe combined immunodeficient mucopolysaccharidosis type VII (MPSII) mice(Meyerrose et al., 2008). In addition,Interferon (INF) –α and β(Nakamizo et al., 2005; Ren et al., 2008), Interleukin-2 2) (Stagg et al., 2004), Interleukin-12 (IL-12)(Elzaouk et al., 2006), CX3CL1(Xin et al., 2007), conditionally replicating adenoviruses (CRAds)(Komarova et al., 2006; Sonabend et al., 2008), NK4(Kanehira et al., 2007), Tumor necrosis factor related apoptosis inducing ligand(TRAIL)(Mohr et al., 2008)and cytosine deaminase (CD)(Kucerova et al., 2008; Chang et al., 2010) were introduced to MSCs and the genetically engineered MSCs were injected to treat cancers.Relative studies have shown that by genetic manipulation of MSCs, either to over express target receptors or by introduction of exogenous genes for expression and secretion of a desired therapeutic factor, the migration efficiency of specific tumor cells can be improved.
Cytosine deaminase (CD) /5-fluorocytosine (5-FC)
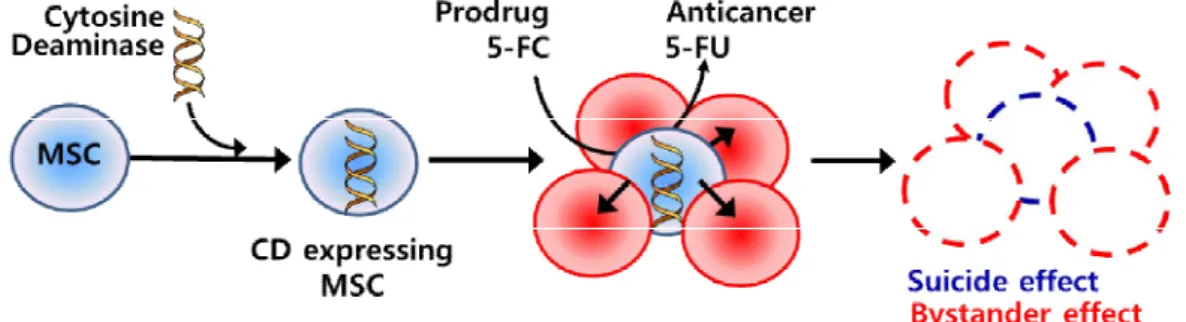
CD is an enzyme found in bacteria and fungi which can convert nontoxic prodrug 5-fluorocytosine (5-FC) intothe highly cytotoxic 5-fluorouracil (5-FU) through deamination.5-FU forms metabolites that inhibit thymidylate synthase or act as false base in DNA and RNA, thereby killing replicating cells. Because 5-FC therapy has been approved by the Food and Drug Administration for central nervous system anti-fungal therapy, its side effects are known and 5-FU is regularly used in the treatment of other types of cancer. 5-FU can be easily move in and out cellular membranes, which convey cytotoxicity to neighboring cells to those expressing CD (Longley et al., 2003; Chen et al., 2007; Chang et al., 2010). BecauseMSCs migrate toward cancer cells and CD generates cytotoxic 5-FU in the presence of 5-FC, CD expressing MSCs can kill not only themselves (suicide effect) but also neighboring cancer cells (bystander effect). The cytotoxic processesvia genetically modified MSCs with CD are illustrated in Fig.1.
Fig. 1.The anti-cancer effectsof MSCs genetically modified with CD
The purpose of this study
In this study, we produced BM-derived MSCs in a KGMP compliantfacility and proved the safety and stability of genetically modified MSCs. In Part 1, we tested whether MSCs retained the stem cell properties to proliferate and differentiate into mesodermal lineage cell types following long-term cultivation. We found that under our experimental conditions, MSCs could be expanded in vitro in a large quantity without losing stem cell properties nor expressing HLA-DR. We also found that bFGF induced HLA-DR expression in MSCs when added to the early BM culture.However, the addition of bFGF in the later period could not induce the HLA-DR expression in MSCs that had been cultivated in the absence of bFGF and thus,were devoid of HLH-DR. In Part 2, we tested the safety of MSCs that had been transduced with a retrovial vector encoding a bacterial CD gene. We also testedwhether the proliferation and differentiation potential of MSCs as well as their CD expression were stably maintained after long-term expansion. We found that naïve MSCs and the genetically modified MSCs/CD cells were similar with respect to proliferation, differentiation potentials, and surface antigenicity. We also found that a single transduction with the retroviral vector was sufficient for stable expression of the CD gene in MSC. Our results suggest that retroviral vectors provide efficient tools to deliver suicide genes to MSCs for ex-vivo gene therapy of cancers.
Part. 1.Establishment of Optimal Culture Conditions for Mass
Production of Clinical Grade, Human Mesenchymal Stem Cells
Introduction I
Human Leukocyte antigens (HLA)
Organs transplanted between major histocompatibility complex (MHC) identical individuals are readily accepted, whereas organs transplanted between MHC antigen-mismatched individuals are rejected in the absence of immunosuppressive therapy (Michon et al., 1953; Merrill et al., 1956). The World Health Organization Nomenclature Committee has named HLA to the human MHC in 1968 (Chinen and Buckley, 2010). Human Leukocyte antigens (HLA) molecules from a donor are recognized by the recipient’s immune system triggering an alloimmune response, when human transplant is performed. HLA complexes are divided into 3 classes (I, II, and III) on the basis of their tissue distribution, structure, and function. Class I MHC antigens are present on all nucleated cells and are each composed of a 45-kd α heavy chain encoded by genes of the HLA-A, B, or C loci on chromosome 6 and associated noncovalently with a 12k-d protein, β2-microbglobuin, encoded by a gene on chromosome 15(Chinen and Buckley, 2010). MHC class II antigens have a more limited tissue distribution and are expressed only on B lymphocytes, activated T lymphocytes, monocytes, macrophages, Langerhans cells, dendritic cells, endothelium, and epithelial cells. Class II molecules are heterodimers composed of noncovalently associated α and β polypeptide chains encoded by genes of the HLA-D region. There are 3 major class II proteins designated, HLA-DP, HLA-DQ, and HLA-DR. Class I MHC molecules present cytoplasm-derived peptides, or intracellular parasites, principally viruses, whereas MHC class II molecules bind peptides derived from extracellular protein. HLA class I and II molecules are recognized by CD8 and CD4 positive T cells, respectively (Ayala Garcia et al., 2012). In transplantation immunology, the major impact in graft loss comes from the effects of HLA-B and –DR antigens(Opelz et al., 1991). HLA-DR mismatch effect is the most important in the first 6 months after transplantation, the HLA-B effect emerges in the first 2 years, and HLA-A
mismatches have a deleterious effect on long-term graft survival (Zantvoort et al., 1996; Morris et al., 1999; Opelz et al., 1999; Takemoto et al., 2000). Cells expressing HLA molecules stimulate T cells directly only if they possess appropriate costimulatory molecules such as CD90 (B7-1), CD86 (B7-2) or CD40. Allogeneic cells can also activate T cells through an indirect pathway where their HLA antigens are presented by professional antigen presenting cells (APC). A remarkable unique feature of MSCs is that they are considered to be immunoprivileaged as they express low levels of cell-surface HLA class I molecules whereas HLA class II, CD40, CD80, and CD86 are not detectable on the cell surface(Pittenger, 1999; DelaRosa and Lombardo, 2010).
HLA-DR expression via bFGF in MSCs
The frequency of MSCs is so low; ranging from 0.01% to 0.001% in BM and 2% in adipose tissue that they should be expanded ex vivo until reached to an adequate number and then identified by phenotypic analysis (Campagnoli, 2001; Vallee et al., 2009; Sabatino et al., 2012). In this reason, to manufacture MSCs for clinical trials, one of the pivotal steps is scale-up because approximately 2 x 106 / kg are required to treat a patient (Hare et al., 2012; Binato et al., 2013).
In order to increase proliferation capacity of MSCs , basic fibroblast growth factor (bFGF) is widely used because bFGF drive MSCs into S phase in cell cycle, resulting in decline of doubling time and apoptotic cells and further enhancement of proliferation (Ramasamy et al., 2012). On the other hand, Sotiropoulou research team found that bFGF not only increases the proliferation rate but also upregulates HLA-class I and induce low HLA-DR in MSCs(Sotiropoulou et al., 2006). Furthermore, Tarte team detected the expression of HLA-DR reaching 19.2 ± 17 % in bFGF-supplemented MSCs and 11.8 ± 6 % in platelet lysate-supplemented MSCs (Tarte et al., 2010). This is likely because bFGF, Platelet-Derived Growth Factor (PDGF) and Insulin-like growth factor-I (IGF-I) stimulate proliferation via the activation of the ERK and AKT signaling pathways and ERK controls the induction of Class II major histocompatibility complex Transcription Activator (CIITA), a major determinant of HLA-DR transcription(Bocelli-Tyndall et al., 2010; Shirakihara et al., 2011). Sotiropoulou and Tarte presented that
upregulation of HLA-DR through bFGF did not induce in vitro allogeneic T cell responses in in vitro mixed lymphocyte reaction assy. Nevertheless, there was no correlation between in vitro suppression by MSCs in mixed lymphocyte cultures and outcome in MSC therapy for the treatment of 31 patients bearing acute GvHD(von Bahr et al., 2012). Thus, HLA-DR expression still remains as a severe problem when the culture of MSCs is supported with bFGF.Here we report the way to expand MSCs without HLA-DR induction with a clinical scale in a Good Manufacturing Practice (GMP) facility and standardized process for manufacture of therapeutic MSCs, which encompasses donor testing, two passage-expansion, banking and quality control in our GMP facility.
Materials and Methods I
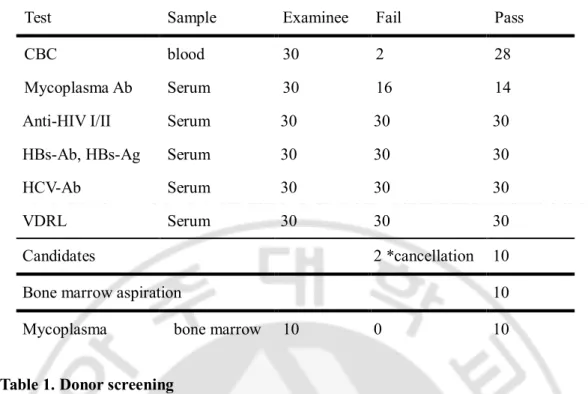
1. Donor screening and bone marrow aspiration
Peripheral bloods from 30 males aged from 20 to 30 were collected and tested about 2 weeks before BM donation for complete blood counts (CBC) and specific human pathogens, such as human immunodeficiency virus I/II (HIVI/II), hepatitis B and C virus (HBV, HCV), mycoplasma, and syphilis as listed in table I with approval of the Institutional Review Board of Ajou University, Medical Center. 20 - 30ml BMs of 10 healthy donors were aspirated from iliac crests and they are collected in 4 or 5 heparin tubes. Polymerase chain reaction (PCR) was conducted to check out mycoplasma contamination from bone marrow aspirates (BMAs) in a heparin tube and rest of them were transferred to GMP facility. BMAs and MSCs derived from 10 donors were compartmentalized with the number from 11 to 20 by donors. For instance, BMA and MSC of first donor were named BMA11 and 11F respectively.
2. Manufacturing BM-derived MSCs
All cell culture manipulations were performed in the Ajou University Center for Cell Therapy located in Kyonggi-Bio Center (Suwon, Korea) and quality control tests were carried out in Department of Anatomy, Ajou University.
3. Mycoplasma test of BM
Mycoplasma tests were conducted with e-Myco™ Mycoplasma PCR Detection kit(Intronbiotechnology, Sungnam, Korea). Briefly, 1ml of BMA was washed out with PBS and resuspended with 500 μl of PBS. Washed BM was heated at 95 ℃ for 10 min and centrifuged for 2min at 13,000 rpm and heated supernatant was used as templates. After adding 10 μl of sterile water and template to e-Myco™ Mycoplasma PCR Detection tubes, PCR was performed according to the manufacturer's instruction.
4. Isolation and cultivation of MSCs
DPBS(welgene, Daegu, Korea). BMA11 was filtered through 70 μm mesh(BD Biosciences, San Jose, CA) and other BMs were used without filtration. 5ml of diluted BMA were carefully loaded on 5ml of ficoll paque premium (1.077 g/ml; GE Healthcare, UK) and centrifuged at 1800rpm for 30 minutes. Mononuclear cells(MNCs) were washed out with DPBS and plated at a density of 1 x 107 to 1.5 x107 per T175 flask in Dulbecco’s modified Eagle’s medium (DMEM, Hyclone, Logan, Utah) supplemented with 10% MSC qualified fetal bovine serum (FBS) originated from Canada, 100 U/ml penicillin, and 100 mg/ml streptomycin(P/S) (Invitrogen, Grand Island, NY). Once confluence had been reached, adherent cells at passage(p) 0 were cryopreserved using Controlled Rate Freezer (CRF) (SY-Lab, Vienna, AUS). For further expansion, P/S had been excluded but 10 ng/ml basic fibroblast growth factor (bFGF, Dong-A Pharmaceutical Co.,Youngin, Korea or Peprotech, Rocky Hill, NJ) had supplied since passage 1 depending on experimental conditions. Fresh culture medium was fed every 2 days and cells were harvested at 70% of confluency to be passaged with the interval 6 to 7 days. Cell plating density had maintained as 1.5 x105 per T175flask in the long-term culture.
5. Cryopreservation and thawing
MSCs were resuspended in a pre-chilled cryoprotectant solution composed of 90% growth medium and 10% dimethyl sulfoxide (DMSO, Bioniche Pharma, Galway, Ireland) at a density of 1.2 x 106/ml. MSCs were packaged into units of 1.2 x 106 viable cells on pre-chilled Lab Armor Beads (Invitrogen), cryopreserved using CRF(SY-Lab), and stored in a liquid nitrogen storage tank. The level of liquid nitrogen was inspected regularly. In order to thaw the cells, frozen cells were rapidly thawed and plated in a T175flask and plated again at a density of 1.5 x105 per T175 flask next day. Cellular viability was measured through trypan blue exclusion whenever harvesting cells.
6. Flow cytometry analysis
To measure the expression of surface antigen, MSCs were harvested with 0.25% Trypsin/EDTA (Invitrogen) or Accumax (Innovative Cell Technologies, San Diego, CA)
and resuspended in PBS containing 0.1% BSA as previously described(Jin Sung Park, 2013). Cells were stained with fluorochrome-conjugated antibodies against STRO-1, HLA-ABC, HLA-DR, CD34, CD45, CD90, CD105, CD11b, CD29, CD49a, CD73, CD117, CD140b and isotype controls (Biolegend, San Diego, CA or BD Biosciences) for 15 min at room temperature. After washing with PBS containing 0.1% BSA, cells were analyzed using BD FACS Aria (BD Biosciences, San Jose, CA) with Flowing software (Turku Centre for Biotechnology, Finland). All assays included isotype controls.
7. Colony Forming Unit-Fibroblast (CFU-F) assay
100 -150 μl of MNCs isolated from BMs were seeded on 100 mm tissue culture dishes (BD Biosciences) in the growth medium. After 7 to 14 days of culture, medium was removed and cells were stained with 2.3% crystal violet solution (Sigma, St. Louis, MO) after fixation with 4% PFA. The numbers of colonies with more than 50 cells were scored following washing and drying. Results are presented as the number of colonies formed per 1 ml of BM.
8. Growth kinetics
In order to measure the effects of bFGF on proliferation, MSC8 was used, which is BMSC as previously described control cell (Sung Soo Kim1 and Young Don Lee1, 2008). They had been cultured with bFGF from p1 to p3 and without bFGF at p4. Duplicates of MSC8 at p5, p7, and p9 were seeded on 12 well plates with a density of 4 x 103 per well in the presence or absence of bFGF and the numbers of viable cells were counted without passaging using heamocytometer and trypan blue exclusion assay from day 1 to day 9 after plating. To test the effect of supplement depending on a brand, MSC-qualified FBS was compared with Australia (AUS)-originated one (Serana, Bunbury WA, Australia) and Peprotech bFGF used for clinical grade production was compared with research grade one kindly gifted by Dong-A Pharmaceutical Co.
Experimental data were analyzed by Sigma Plot statistical software. The significance between 2 groups was evaluated by linear regression or paired t-test. Linear regression was adjusted to evaluate the correlations among initial quantities of MSCs p0, MNCs and CFU-F and paired t-test was applied to assess statistical significance on doubling times of MSCs at between p1 and p5 to p9 by bFGF supplement. p < 0.05 was considered significant.
Results I
Screening of healthy donors
Blood samples were collected from 30 males and a total of 10 donors were enrolled into bone marrow aspiration. Bloods were tested to screen healthy donors before BMA as listed in Table.1. Of these 30 volunteers, 2 blood samples were against normal criteria in CBC, which showed slightly higher values regarding mean corpuscular hemoglobin and white blood cells than normal ones. Unexpectedly, 16 blood samples were positive for mycoplasma and some of them were suggested to be treated in Ajou medical center. Of 12 examinees except for these 18, 2 canceled BMA processes because of their private situations. Accordingly, 10 donors were found to be eligible for BMA and MSC culture. BMA was confirmed by PCR as mycoplasma-free and transferred to GMP facility to expand MSCs (Table1).
Test Sample Examinee Fail Pass
CBC blood 30 2 28
Mycoplasma Ab Serum 30 16 14
Anti-HIV I/II Serum 30 30 30
HBs-Ab, HBs-Ag Serum 30 30 30
HCV-Ab Serum 30 30 30
VDRL Serum 30 30 30
Candidates 2 *cancellation 10
Bone marrow aspiration 10
Mycoplasma bone marrow 10 0 10
Table 1. Donor screening
Blood samples of 30 volunteers were tested and 12 examinees passed 6 criteria including complete blood count(CBC), infections with mycoplasma, human immunodeficiency virus (HIV), Hepatitis B (HBs-Ab, HBs-Ag), Hepatitis C(HCV), and syphilis(VDRL). 2 volunteers cancelled donation. 10 bone marrows were aspirated from healthy donors and negative for mycoplasma in PCR.
Analysis of the MSCs depending on donors
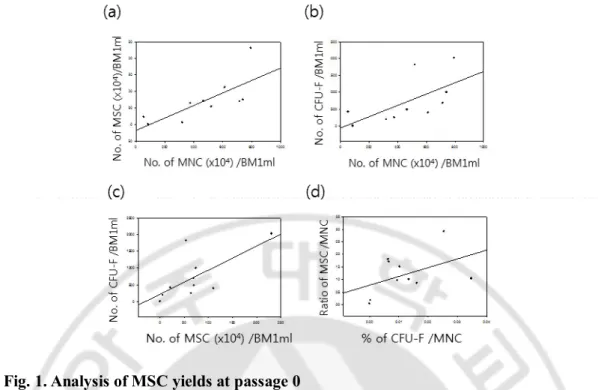
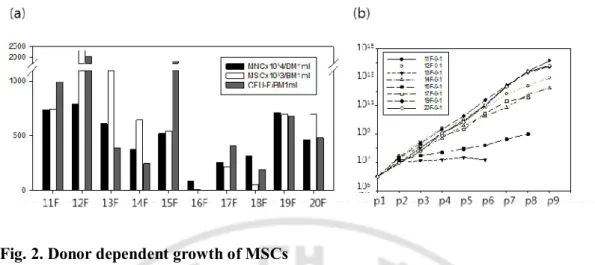
The numbers of MNCs, MSCs, and CFU-Fs were calculated at day 0, 12 to 17 and 7 to 14 respectively after a BMA with the units generated from 1 ml of BM, which ranged from 8.6 x 105 to 7.9 x 106 (average 48.9 ± 7.3 x 105) in MNCs, from 3 x 103 to 2.3 x106 (average 7 ± 2 x 105) in MSCs and from 0 to 2020 (average 722 ± 217) in CFU-Fs with donor variations among 10 BMAs. There were significant correlations between the numbers in MSCs and MNCs (p=0.014, R2= 0.553), MSCs and CFU-Fs (p=0.028, R2=0.474), and MNCs and CFU-F (p=0.049, R2=0.402) in linear regression models (Fig.1a-c). However, the frequencies of MSCs per MNC were not dependent on the frequency of CFU-Fs at p0 (p=0.080, R2=0.207), which indicates that the individual growth rates of MSCs are not identical and it is likely to be donor variations (Fig. 1d). Sequentially, we further proliferated 8 kinds of MSCs (11F, 12F, 13F, 14F, 15F, 17F, 19F and 20F) by p9. 16F and 18F were ruled out of long-term culture because all parameters including the numbers of MNC, MSC and CFU-F at p0 were extremely low (Fig. 2a). As expected, 8 kinds of the MSCs showed very different growth rates and proliferation capacities during long-term culture supplemented with bFGF. 11F, 19F, 20F highly proliferated by 1.3 x1014, 5.3 x 1013 and 6.2 x 1013 from 1 x 106 respectively while 13F, 15F, 17F terminated their growths at p3, p3 and p7 with the numbers of 1.3 x 107, 3.0 x 107 and 2 x 1011 from 1 x 106 respectively(Fig. 2b). There were no significant differences of donor ages, BMA location and culture conditions because all MSCs were derived from posterior iliac crests of twenties and expanded with the interval of 7 days in same culture medium. Therefore, only difference of MSC yields was resulted from individual donors.
Fig. 1. Analysis of MSC yields at passage 0
(a) 10 of MSC p0 with 70 % confluency were harvested between 12 and 17 days after MNC plating and positively correlated with the number of MNCs isolated by ficoll density gradient centrifugation method(P=0.014). (b) CFU-F assay was conducted from each MNC plating. The more MNC, the more CFU-F(P=0.049). (c) the higher number of MSC p0 showed higher number of CFU-F(P=0.028). (d) The frequency of MSCs in MNCs was not dependent on the percentage of CFU-F per MNC(p=0.080). Statistical analysis was performed with Sigma plot and P value means statistical correlation between two factors in linear regression.
Fig. 2. Donor dependent growth of MSCs
(a) 10 MSCs at p0 were expanded from their MNCs and compared with the number of MNC and CFU-F per 1ml of BMA. There were significant differences among donors. (b) 8 MSCs at p1 were cultured by p9. The growths of 3 MSCs(13F, 15F, 17F) were terminated before passage 9 while 3 MSCs(11F, 19F, 20F) showed the great proliferation capacities in the long term culture.
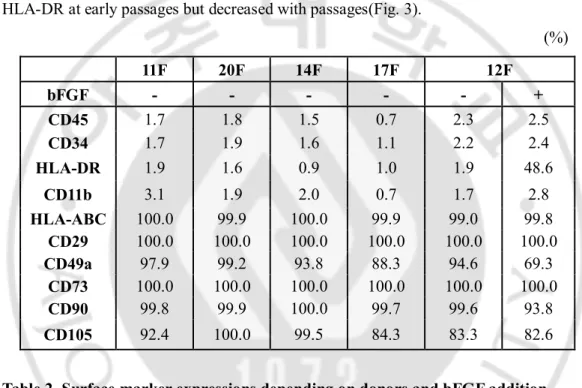
According to International Society for Cellular Therapy (ISCT), there are three criteria to define MSC which are adherence to plastic, specific surface antigen expression and multipotent differentiation potential. For the second standard, MSC must express CD105, CD73, CD90 and lack expression of CD45, CD34, CD14 or Cd11b, CD79α or CD19 and HLA class II (Dominici et al., 2006). To test the differences among donors in the aspect of MSC quality, phenotypical analysis was conducted in 5 MSCs at p1 or p2 by flow cytometry (Table2). There was a critical difference not among donors but between culture conditions. HLA-DR was induced when cultured with bFGF in 12F. In the presence of bFGF, not only 12F but also 11F, 14F, 17F, 19F, 20F were all positive for HLA-DR at early passages but decreased with passages(Fig. 3).
(%) 11F 20F 14F 17F 12F bFGF - - - - - + CD45 1.7 1.8 1.5 0.7 2.3 2.5 CD34 1.7 1.9 1.6 1.1 2.2 2.4 HLA-DR 1.9 1.6 0.9 1.0 1.9 48.6 CD11b 3.1 1.9 2.0 0.7 1.7 2.8 HLA-ABC 100.0 99.9 100.0 99.9 99.0 99.8 CD29 100.0 100.0 100.0 100.0 100.0 100.0 CD49a 97.9 99.2 93.8 88.3 94.6 69.3 CD73 100.0 100.0 100.0 100.0 100.0 100.0 CD90 99.8 99.9 100.0 99.7 99.6 93.8 CD105 92.4 100.0 99.5 84.3 83.3 82.6
Table 2. Surface marker expressions depending on donors and bFGF addition
MSCs were expanded in the presence or absence of bFGF and analyzed through flow cytometry. They did not show significant differences in surface marker expressions but HLA-DR was positive during expansion supplemented with bFGF.
Fig. 3. HLA-DR expression (+bFGF condition)
6 of MSCs were positive for HLA-DR but negative for CD45 when supplemented by bFGF. However, HLA-DR expressions decreased with passages in 5 of MSCs(11, 12, 14, 17, 20F). Some of them became negative for HLA-DR at p8(11F, 17F, 20F) while 19F were still positive for HLA-DR even at p8. All assays included isotype controls.
HLA-DR induction depending on culture conditions in MSCs.
Since MSCs derived from BM have a limitation to proliferate in the long term culture by prolonged doubling time with passages, many researchers have used bFGF for enrichment of MSCs which is one of the powerful mitogen to increase telomere length and drive cells to stay in S phase(Bianchi et al., 2003; Lapi et al., 2008). However, Sotiropoulou and Pratsinis reported that bFGF supplement upregulated HLA-DR on MSCs consistent with our data. To test whether bFGF differentially induces HLA-DR depending on the passages, we compared 2 types of cells, which one had not been exposed to bFGF at early passage and did not express HLA-DR in the absence of bFGF and the other had been expanded in the presence of bFGF at early passages from p1 to p2. The former expressed HLA-DR at p3 by bFGF addition (Fig. 4a upper panel) but did not induce it by bFGF addition at p4(Fig. 4a lower panel). In contrast, the latter rapidly became negative for HLA-DR through bFGF deprivation just for 1 passage (Fig. 4b upper panel) and maintained absence of the expression at p5 but did induce it again at p5 by bFGF addition even after 2 further passaging without bFGF (Fig. 4b lower panel) and HLA-DR expression remained at p7. These responses of MSCs caused by bFGF were consisted with several culture media tests containing 2 kinds of bFGF including animal free one, 2 kinds of serum including MSC-qualified one (data not shown). This implies that the exposure of MSCs to bFGF at early passage results in HLA-DR induction and the expression of HLA-DR could be controlled.
Fig. 4. HLA-DR and CD90 expressions depending on the culture conditions
(a) MSCs expanded in the absence of bFGF for early 3 passages did not induce HLA-DR after bFGF addition. (b) HLA-DR expression in MSCs had been exposed to bFGF for early 2 passages rapidly disappeared after bFGF depravation but it was induced by bFGF even after maintained without bFGF for 2 further passages. CD90 was utilized as a positive marker for MSCs and arrow means passaging.
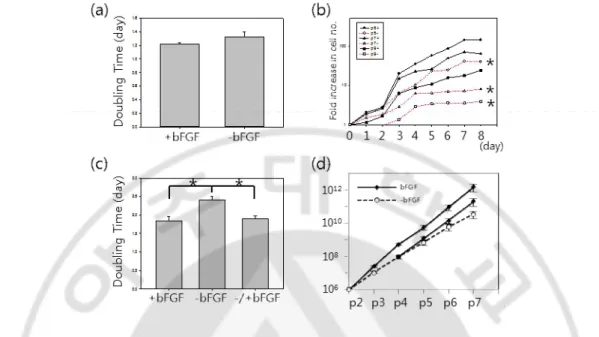
bFGF guarantees extended proliferation capacity in the long term culture.
Even though bFGF induces HLA-DR that is a hot issue in cell therapy, why do we consider adding it into culture of MSCs? To address this question, the response of MSCs to bFGF depending on passages was evaluated. On one hand, 11F, 12F and 14F at p1 were expanded in the presence(+) or absence(-) of bFGF. Doubling times were calculated with plating numbers, harvesting numbers and culture days as 1.21 ± 0.22 days and 1.32 ± 0.79 days on average in +bFGF and -bFGF group respectively(Fig. 5a) and there was no significant difference between 2 groups at p1 (p=0.254). On the other hand, the proliferation rates in +bFGF groups at late passages were statistically higher than -bFGF. MSC8 at p5, p7, p9 were seeded on well plates and counted for 9 days. The cells were exponentially proliferated in +bFGF groups and especially grown fast between day 2 and 3 while the cells in -bFGF groups were significantly less grown than +bFGF group and had retarded since day 6 or 5(Fig. 5b). As a result, bFGF increased the proliferation capacity of MSCs and the effect of bFGF on their growths was great at late passages. To confirm this, 11F cultured with bFGF until p2 had been expanded with bFGF (+bFGF) or without bFGF (-bFGF) for further passages and bFGF had been added into -bFGF group (-/+bFGF) from p4. The numbers of cells from p5 to p8 were measured and doubling times were calculated as averages in 3 groups. As expected, the averages of doubling times from p5 to p8 were 1.84 ± 0.11 days, 2.40 ± 0.11 days and 1.90 ± 0.07 days in +bFGF, -bFGF and -/+bFGF group respectively (Fig. 5c). The cumulative numbers of cells among 3 groups were calculated in 11F and 14F. MSCs p7 could be obtained from 1 x106 MSCs p2 with 1.5 x 1012 ± 7.5 x 1011, 3 x 1010 ± 1.7 x1010 and 2 x 1011 ± 9.8 x1010 in +bFGF, - bFGF and -/+bFGF group respectively on average (Fig. 5d). The extended doubling time at late passages in bFGF were compensated by bFGF supplement in -/+bFGF. The high standard errors were resulted from donor dependent growth rates. In addition, we compared 2 kinds of FBS and bFGF in this experiment. The accumulative fold increases in the number of 12F supplemented with Canada-origin FBS or AUS-origin FBS for 16 days from p1 were 5033 folds and 5067 folds, respectively (p=0.205) and the average fold increases in cell number were 21.1 ± 5.8 folds or 22.6 ± 5.4 folds in MSCs (MSC8 p5, 11F p3, 11F p7, 14F p3, 14F p7) supplemented with research grade
bFGF or certified bFGF for 1 passage respectively (p=0.375) (data not shown). FBSs and bFGFs did not show differences in MSC expansion.
Fig. 5. Proliferation of MSCs depending on passages and bFGF addition
(a) MSC cultures isolated from 3 donors were supplemented with bFGF(+bFGF) or without bFGF(-bFGF) at p1. Doubling times depending on bFGF supplement were not significantly different between 2 groups(P=0.254). (b) MSCs at p5, p7, and p9 were proliferated for 9 days, which were supplemented by bFGF(solid line) or none(dotted line). The growth rates of MSCs cultured without bFGF severely decreased as passage (p5, p= 0.026; p7, p= 0.021; p9, p= 0.014). (c) Proliferation capacity at late passages upon bFGF. MSCs were expanded with bFGF until p2 in 3 groups and double times were calculated from p5 to p8. bFGF was subtracted at p3(bFGF) and supplied at p5 in -/+bFGF. bFGF significantly enhanced the proliferation of +bFGF group(P=0.0119) compared with –bFGF group and compensated prolonged doubling time of –bFGF group in -/+bFGF group(P=0.00799). Statistical analysis was implemented by paired t-test of Sigma plot. (d) 11F and 14F cultured with bFGF until p2 were expanded in the presence(solid line) or absence(dotted line) of bFGF. Decreased growth rate of –bFGF group was recovered through bFGF addition.
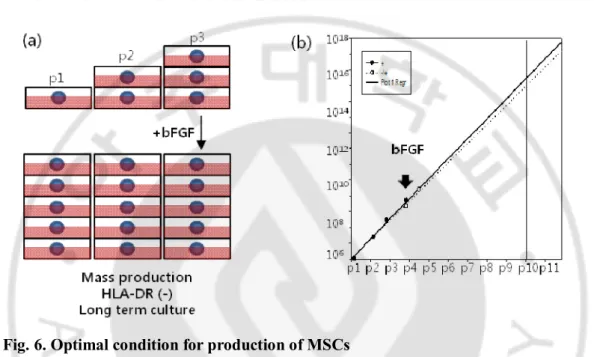
Quality and quantity control via bFGF supplement
The culture of 12F was supplemented with bFGF from p1 or p4. Through the extended linear regression, we estimated the numbers of 12F p10 grown from 1 x 106 cells at p1, which are 3.1 x 107 in bFGF supplied group from p1 and 1.1 x 107 in bFGF supplied group from p4 (Fig. 6b). Through bFGF supplement after p3 culture, mass production of MSCs in the absent expression of HLA-DR could be feasible for clinical application. We illustrated the suggested guide line in Fig. 6a
Fig. 6. Optimal condition for production of MSCs
(a) Suggested guide line for mass production of MSCs. MSCs could be proliferated without the expression of HLA-DR as a large-scale for a long time through bFGF supplement after p3. (b) The estimated growth kinetics in linear regression model. The numbers of MSCs at passage 10 are 3.1 x 1017and 1.1 x 1017 in the groups of +bFGF(solid line) and -/+bFGF(dotted line) respectively as following suggested guide line.
Discussion I
Clinical grade MSCs
For the clinical application of BMSCs, the whole procedures including donor screening, BMA, culture and cryopreservation must be complied with regulation of Good Manufacturing Practice (GMP). However, the original data and proof of concept for cell therapy are generally derived from laboratories, not followed by GMP rules. Therefore, replacement of research grade materials for culture into clinical grade one is the start for manufacturing cells under the GMP regulation (Philippe et al., 2010). Since FBSs and bFGFs did not show differences in proliferation and surface marker expressions of MSCs, we could replace culture medium into certified reagents to produce clinical grade MSCs. We implemented pre-tests of volunteers' blood before BMA. Only examinees passed from the blood tests donated their BMs and individual BMA was transferred into GMP facility after monitoring mycoplasma infection. All of raw materials we used were clinical grades or clearly identified by manufacturers. Standard operating procedures (SOPs) regarding managementand maintenance of facility, devices, take-in materials as well as cell culture procedures were all documented following FDA guideline
Factors affecting the quality and quantity of MSC
The yields of MSCs isolated from BM could be affected by conditions of donors such as age(Mareschi et al., 2006; Katsara et al., 2011; Zaim et al., 2012), gender(Katsara et al., 2011) and disease(Li et al., 2011), BMA method(Fennema et al., 2009; Li et al., 2011), culture method(Santos et al., 2011; Goh et al., 2013; Mizukami et al., 2013; Peter et al., 2013), plating density(Neuhuber et al., 2008; Chen et al., 2009), culture medium(Sotiropoulou et al., 2006; Gottipamula et al., 2013). We aspirated similar volumes of BM from posterior iliac crest with single puncture and all donors were healthy males aged from 21 to 25 years. The method to collect BMA was practically same among donors. For ex vivo expansion of MSCs, we standardized culture protocols in terms of plating density, duration for medium change, the volume of medium and harvest timing and the number of cells for cryopreservation, which providing identical
conditions in cultivation of MSCs. Furthermore, culture methods in comparative studies were so same that the critical influence for MSC quantity was definitely resulted from donor variation in the aspects of frequency in BM, growth rate and sustained proliferation capacity in the long term culture.
On the other hand, the qualities of MSCs depending on donors were remarkably similar in surface marker expressions complied with the criteria of MSCs defined by ISCT. The expression of CD105 in 17F and 12F was slightly low in MSCs at p1 but elevated by over 95 % in both at p3. It is likely due to heterogeneity at p1 as umbilical cord-derived MSCs(Gong et al., 2012). Even though we did not measure the differentiation potentials with values depending on culture conditions or donor variation in this study, 11F and 14F did differentiate into 3 lineages; adipocytes, osteocytes, chondrocytes. According to literatures, the effects of bFGF on differentiation potentials of MSCs grown in bFGF containing medium are still controversial. Many researchers mentioned that bFGF facilitates adiopogenesis (Neubauer et al., 2004; Neubauer et al., 2005; Kakudo et al., 2007), chondrogenesis (Solchaga et al., 2005; Stewart et al., 2007; Cheng et al., 2012) and osteogenesis (Hanada et al., 1997; Sotiropoulou et al., 2006; Jeong et al., 2008; Rider et al., 2008) while it inhibits osteogenesis (Huang et al., 2010; Hughes-Fulford and Li, 2011; Luong et al., 2012), or it does not affect adiopogenesis (Sotiropoulou et al., 2006). Therefore, therapeutic MSCs originated from single donor should be selected for production of clinical grade MSC via comparative analysis.
HLA-DR expression in MSCs
Origination from the patients with inflammatory disease, high cell density, treatment of Transforming Growth factor-β(TGF-β) or interferon-γ (INF-γ) and differentiation could cause HLA-DR induction in MSCs other than bFGF. In the literatures, The mean percentage for HLA-DR was on average 15 ± 10 % in BMSCs derived from 15 patients with myelodysplastic syndromes (Campioni et al., 2006) and HLA-DR was expressed at p1-p3 and decreased with passages in the BMSCs derived from 7 Crhon's disease patients (Bernardo et al., 2009). In addition, INF-γ is known as an inducible factor to promote the expression of HLA-class I and HLA-class II (Thibodeau et al., 2012).
Romieu-Mourez found that IFN-γ induced MHC-class II expression was enhanced by high cell density or serum deprivation and suppressed by TGF-β in mouse MSC (Romieu-Mourez et al., 2007) but did not detected in differentiated human MSCs (Le Blanc et al., 2003). We found that bFGF could induce HLA-DR expression on the surface of MSCs which had been exposed to bFGF at early passage. It might be due to the distinction of original populations of MSCs following the selection by bFGF at early passages. bFGF involves in maintenance of stemness in a variety stem cells including MSCs (Martin et al., 1997; Bianchi et al., 2003; Gotoh, 2009; Coutu et al., 2011) and HLA-DR was induced by bFGF as well as the expression level was decreased with passages (Bocelli-Tyndall et al., 2010) consistent with our results. Moreover, amniotic membrane-derived MSCs expressed HLA-DR in only early passages not late passage (Kim et al., 2007) and one report describes the positivity of HLA-DR on human BMSCs sorted with D7-FIB microbeads, a human fibroblast marker, suggesting that HLA-DR may play a role in hematopoietic cell maturation (Jones et al., 2002). Taken together, bFGF might be more favorable to select HLA-DR+ MSCs at early passages as more primitive and then selected HLA-DR+ cells upon bFGF might lose the characteristics of HLA-DR expression and high proliferative potentials more and more with passages. While MSCs might rarely response to bFGF during such selection period for proliferation (Fig. 5a), once selected, HLA-DR+ cells might get sensitive to bFGF (Fig. 5b-d). Thus, bFGF is required for long-term culture, even though the effect of bFGF in HLA-DR- cells at late passage is not elusive yet.
Mass production and banking
We developed a protocol to expand MSCs with a large-scale through cultivation without bFGF for 3 passages and then propagation with bFGF by clinical lots(Fig. 6a). Since HLA-DR is inducible on MSCs by inflammatory factor and bFGF (Bocelli-Tyndall et al., 2010) and recrudesced by bFGF addition even though HLA-DR was not expressed after bFGF deprivation (Fig. 4), we added bFGF for MSC expansion after 3 passage cultivation. The extended linear regression showed that the number of cells at late passages did not differ between +bFGF and -/+bFGF group (Fig. 6b). Consequently, we
could obtain an adequate number of cells as many as we want.
NIH Bone marrow stromal cell transplantation center was established in 2008 in order to develop a novel cell product for the treatment of a variety of human diseases and disorders. KFDAhas approved tree MSC products for manufactures and sales in 2011 and 2012;Hearticellgram-AMI fortreating heart attacks(FCB-Pharmicell, South Korea), CARTISTEM for treating traumatic degenerative osteoarthritis(Medipost, South Korea), Cupistemfor treating crohn's fistula(Anterogen, South Korea). Even though they showed notable improvements against target diseases, there is a limitation to use widely. They should be promptly delivered to patients as soon as detached from culture wares because they could not be stable for longer than 2 days in liquid state. Eventually, cryopreservation is indispensable. Here we successfully cryopreserved clinical grade MSCs p1 or p2 cultured without bFGF using controlled rate freezing as a master cell bank and they could be expanded more for various purposes.
Part. 2. Comparative study of MSCs with genetically modified
MSCs
Introduction II
Mesenchymal stem cells (MSCs) have been utilized for the treatment of diverse diseases, including neuropathies such as Parkinson’s disease (Bouchez et al., 2008), Huntington’s disease (Armstrong et al., 2000), multiple sclerosis(Horn et al., 2008; Karussis et al., 2010), amyotrophic lateral sclerosis (Choi et al., 2012), ischemic stroke (Yoo et al., 2008; Perasso et al., 2010), and non-neurological diseases such as myocardial infarction (Amado et al., 2005; Miyahara et al., 2006), and graft-versus-host diseases (Le Blanc et al., 2004). The therapeutic effects of MSCs are ascribed to their paracrine functions that include the secretion of beneficial molecules (Tate et al., 2010; Bai et al., 2012), anti-inflammatory factors (Dao et al., 2011; Jenhani et al., 2011), or extracellular matrix (Aizman et al., 2009). However, a major challenge is how to render MSCs more disease-specific and enhance their paracrine effects. Since MSCs are highly migratory to lesion and tumor sites (Lee et al., 2009), it has been suggested they can be used as cellular vehicles to deliver therapeutic genes to target tissues for ex-vivo therapy and to overcome targeting problems of conventional gene therapy. In order to tailor MSCs to be more disease-specific or to modify them as gene carriers, viral vectors are frequently utilized to introduce therapeutic genes into MSCs.
Previously, we showed that MSCs could be utilized as a cellular vehicle to deliver a cytosine deaminase (CD) gene to brain tumors (Chang et al., 2010). CD genes are naturally expressed in bacteria and fungi, but absent in humans. CD can convert a nontoxic prodrug, 5-fluorocytosine (5-FC) into 5-fluorouracil (5-FU), an anti-cancer drug that has been used for the treatment of gastrointestinal cancers (Longley et al., 2003). Cell membranes are highly permeable to 5-FU, which can enter neighboring cells through simple diffusion and exert cytotoxic effects by interfering with DNA and RNA synthesis (bystander effects). We showed that MSCs infected with a retroviral vector expressing an E. coli CD gene could migrate toward brain tumors and suppress tumor growth through bystander effects (Chang et al., 2010), when animals were systemically
administered with 5-FC. In addition to our efforts, other laboratories have utilized MSCs as cellular vehicles to deliver therapeutic genes including interleukin-12 (Ryu et al., 2011), herpes simplex virus-thymidine kinase (Uchibori et al., 2009), tumor necrosis factor apoptosis ligand (Sasportas et al., 2009), and interferon-β to brain tumors (Nakamizo et al., 2005).
Retroviral vectors are often used to guarantee long-lasting transgene expression. However, these vectors can cause insertional mutagenesis when they integrate into host chromosomes. In clinical trials carried out in Europe, 8 of 9 patients with X-linked severe combined immunodeficiency (SCID-X1) exhibited clinical improvement after receiving an infusion of CD34+ autologous hematopoietic stem cells that were transduced with retroviral vectors carrying the intact γ chain gene. However, some patients developed acute leukemia in subsequent years due to in vivo cloning and expansion of hematopoietic stem cells that carried insertional mutations (Hacein-Bey-Abina et al., 2010). Unlike ex-vivo therapy using hematopoietic stem cells, the lifespan of our CD-expressing MSCs is transient in vivo due to the suicide effects of CD in combination with 5-FC (Chang et al., 2010). Indeed, it has been proposed that suicide genes such as HSV-tk, CD, or inducible caspase-9 can be utilized to ablate abnormal, unwanted cells in vivo and increase the safety of gene and cell therapy (Ramos et al., 2010). Thus, the potential risks of insertional mutagenesis associated with retroviral vectors may not be relevant to CD-expressing MSCs. Nonetheless, it is a prerequisite to ensure the chromosomal stability of genetically modified MSCs before clinical application.
The characteristics and phenotypes of MSCs vary according to the tissue source (bone marrow, adipose tissues, and umbilical cord blood), passage cycle, and culture conditions (Horwitz et al., 2005). One study reported that MSCs could undergo spontaneous malignant transformation upon in vitro cultivation for extended periods of time, although human MSCs tend to be resistant to spontaneous malignant transformation (Achille et al., 2011). Transformed human MSCs with epithelial polygonal morphology emerged between 11-106 weeks after most MSCs cells stopped growing, and the transformed cells grew well in an anchorage-independent manner, similar to cancer cells (Rosland et al.,
2009). For murine MSCs, spontaneous transformation is always accompanied by gross chromosomal alterations (Josse et al., 2010). Therefore, it is necessary to establish safe criteria with respect to the genomic stability of MSC for planning ex-vivo therapy.
Material methods II
1. Isolation and cultivation of MSCs
Human MSCs were originally derived from the iliac crest bone marrow of healthy 10- to 15-year-old donors undergoing bone marrow aspiration for future allogeneic transplantation with approval of the Institutional Review Board of Ajou University, Medical Center, as previously described(Kim et al., 2005). Briefly, mononucleate adherent cells were harvested and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen, Grand Island, NY), and 10 ng/ml basic fibroblast growth factor (bFGF, Dong-A Pharmaceutical Co., Youngin, Korea).
2. Retroviral Transduction
CD-expressing MSC was prepared by transducing MSCs with a retroviral vector encoding CD, as previously described(Chang et al., 2010). The CD gene was cloned from Escherichia coli K12 MG1655 (KRIBB, Daejon, Korea) by PCR (forward primer: 5’-GAA TTC AGG CTA GCA ATG TCG AAT AAC GCT TTA CAA AC-3’; reverse primer: 5’-GGA TCC TCT AGC TGG CAG AC A GCC GC-3’) and then inserted into pFIP plasmid (ViroMed, Seoul, Korea). pFIP/CDretroviral vector was produced in a FLYRD18 packaging cell line expressing the Moloney Murine Leukaemia (MoMLv) virus gag-pol gene and the cat endogenous virus RD114 env gene.Two days after plating of packaging cells in a density of 1.5x106/T75flask, the viral supernatants were collected and syringe-filtered using a 0.45-mm filter. MSCs at passage 1 were plated in a density of 1x105cell/100mm dish and exposed to retrovirus with 20 multiplicity of infection for 8 hrs in the presence of 4 mg/ml polybrene (Sigma-Aldrich, St. Louis, MO) and 10ng/ml bFGF(Chang et al., 2010). Two days later, cells were subcultured and 2mg/ml puromycin (Sigma-Aldrich) was added to the culture for 2weeks. Surviving cells were pooled and maintained by subculturing every 5-7days. To compare growth kinetics of MSCs with MSC/CD, cells were counted by trypan blue exclusion test and plated at adensity of
1,000 cells/cm2for next passage culture. All cell culture medium was replaced with fresh one every 2-3 days.
3. Differentiation
Adipogenic, osteogenic and chondrogenic differentiation were performed as previously described (Pittenger, 1999) with a slight modification. Briefly, cells were plated in a density of 2x104 cells / 3.8cm2 in culture medium and grown to reach confluence. The culture medium was replaced with adipogenic medium supplemented with 0.5mM isobutylmethylxanthine, 60mM indomethacine, 1mM dexamethasone, 10 mg/ml of insulin for 3 weeks or osteogenic medium supplemented with 0.1 μM dexamethasone, 60 mM ascorbic acid, and 10 mM β-glycerophosphate for 5 weeks. Adipogenic differentiation was verified by accumulation of lipid droplets stained with Oil Red O whereas osteogenic differentiation by accumulation of extracellularcalcium crystals stained by Alizarin Red S. Chondrogenic differentiation was induced by cultivating 2x105 cells in pellets in induction medium supplemented with 1% FBS, 6.25 mg/ml insulin, 10 ng/ml TFG-β1, 50 ng/ml ascorbic acid for 6 weeks. Alican Blue was used to stain metachromic extracellular material in the pellet and then Nuclear Fast Red was used for counter staining of chondrocytes.
4. Flow cytometry analysis
To measure the expression of surface antigen, MSCs or CD-expressing MSCs were harvested with 0.25% Trypsin/EDTA(Invitrogen) and resuspended in PBS containing 1%BSA. Cells were stained with fluorochrome-conjugated antibodies against STRO-1, HLA-ABC, HLA-DR, CD34, CD45, CD90, CD105, CD11b, CD29, CD49a, CD73, CD117, and isotype controls (Biolegend, San Diego, CA) for 10minutes at room temperature (RT). After washing with PBS containing 1%BSA, cells were analyzed using BD FACS vantage (BD Biosciences, San Jose, CA) with CellQuestPro software. All assays included isotype controls or negative controls without a primary antibody.
5. Anti-cancer effects
For in vitro suicide effects, cells were plated at a density of 10,000 cells/well in 12-wellplates, and 24 hrs later, 5-FC (Archimica, UK)was added at the indicated concentrations. MTT(3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazolium bromide, Sigma-Aldrich)assays were performed to measure cell viability on day7. The mediumwas replaced every other day with fresh growth mediumcontaining the indicated concentrations of 5-FC. Thevalues at each 5-FC concentration are expressed relative tothat of untreated cells and presented as the means ±SE. To assess bystander effectsin
vitro, U87MG glioma cells were transduced by a lentiviral vector expressing green
fluorescence protein (GFP) and GFP-positive cells were sorted by FACS. U87MG/GFP cells were cocultured with MSCs or MSC/CD cells in a ratio of 104:104in 12-well plates. Twenty-fourhours later, 5-FC was added to obtain the indicated concentrations,and the medium was replaced every 2 days thereafter.On day 7, fluorescent images of the remaining U87MG/GFP cells were acquired first with a fluorescence microscopy and then the cells were lysed in Passive Lysis Buffer (Promega,Madison, WI). The fluorescence values of the cell lystaes were measured using a fluorometer (Molecular Device, Sunnyvale, CA) and expressed relatively to the value of untreated cells (means ± SE).
6. Immunoassays with anti-CD antibody
E.coli CD was produced as a 48 kilodalton protein in E.coliBL21 using a pET vector and
purified using a Ni-column. Anti-CD rabbit polyclonal antibody was custom-made by Abfrontier (Seoul, Korea). Cells were lysed in RIPA buffer (50 mM Tris; pH 7.4, 1 M NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate) using a standard protocol. 45 μg whole cell lysates of MSC and MSC/CD cells were separated on poly-acylamide gel for western analysis of CD and 30 μg for b-actin. Proteins on the gel were transferred to PVDF membrane, and probed with anti-CD antibody (1:5000) or anti-b-actin antibody (1:5000). Immunoreactivity was visualized using horse radishperoxidase-conjugated anti-rabbit or anti-mouse IgG antibodies (1:5000, Zymed,
San Francisco, CA) and the SuperSignal Chemiluminescence Substrate kit (PIERCE, Rockford, IL).
For immunocytochemistry, cells grown on coverslip were fixed with 4 % PFA for 10 min at RT. To block nonspecific binding, cells were incubated in blocking solution (0.1 % Triton X-100, 0.1 % BSA, 10 % normal horse serum in PBS) for 2 hrs at RT and then in the presence of a polyclonal anti-CD antibody (1:500 diluted in blocking solution) at 4℃ overnight. After washing, cells were reacted with Alexa 488-conjugated anti-rabbit IgG antibodies (1:200) at RT for 30 min. Nuclei were counter stained with Hochest 33258 for 5 min. After washing, fluorescent images were acquired with a fluorescent microscope (Olympus, Shinjuku, Japan).
For flow cytometry analysis, MSC/CD cells were harvested, fixed with 4 % PFA, and resuspended in PBS containing 0.1 % Triton X-100 and 1 % BSA. Cells were incubated with anti-CD antibody (1:500) for 30 minutes at RT and then with Alexa 488-conjugated anti-rabbit IgG antibody. After washing twice, cells were analyzed as described above.
7. Quantitative polymerase chain reaction (qPCR)
Total RNA was isolated from cells using RNAzol B (Tel-Test, Friendswood, TX), and cDNA synthesized from 1 μg of RNA using the First-strand cDNA synthesis kit (Roche, Mannheim, Germany). Amplification was performed using a Taqman universal PCR master mix kit (Applied Biosystems, Foster City, CA) and one tenth volume of the first-strand cDNA reaction mixture using Roto-Gene® Q (Qiagene, Hilden, Germany) and a software provided by the manufacturer. RelativeCD gene expression to GAPDH in MSCs was calculated with Delta-Delta CT relative quantification and presented relatively with respect to the value of MSC at passage 8 (means ± SE). PCRprimers and probes were summarized in Table 1.
Gene Sequences CD Forward Reverse Probe TGATGAGATCGATGACGAGCAGTC GGGTTGGCGACAAAGTTAATACCG 56-FAM/ACACCACGG/ZEN/CAATGCACTCCTATAAC/3IABkFQ GAPDH Forward Reverse GGCCATCCACAGTCTTCTG CAGCCTCAAGATCATCAGCAA
Probe 56-FAM/ATGACCACA/ZEN/GTCCATGCCATCACT Table 1. Primers and probes for quantitative PCR
8. Chromosomal stability and tumorigenicity tests
For chromosome analysis, 20 cells at metaphase were counted after stained with Leishman stainsolution, and 5 cells were analyzed for more detailed karyotyping using CytoVision®. GTG-banding with 450 bands of resolution was conducted in a clinical Cytogenetics Laboratory at Ajou University Hospital (www.ajoumc.or.kr). To measure in
vitro transformation, MSC, MSC/CD and U87MG cells were cultured in soft agar using a
kit according to the manufacturer’s protocol (Millipore, Billerica, MA). Briefly, 24 well plates were coated with 0.8 % agarose in the mixture of distilled water and growth medium, and 500 μlof base agar was added to a well. Cells were plated in 0.4% agarose solution in a density of 1, 250 cells per well suspended and incubated for 2 weeks with addition of 250 μl of growth medium twice a week. To measure in vivo tumorigenicity, we suspended 5x106 cells in 100 μl of PBS conatining 20 % matrigel (BD Biosciences) and subcutaneously innoculated them in Balb/C/nu/nu mice (Nara Biotech, Seoul, Korea). As a positive control, an equal number of U87MG was innoculated. Tumor dimensions were measured with a caliper and the volumes were calculated as π/6 x length x width x height. All animal protocols were approved by the Institutional Animal Care and Use Committee of Ajou University School of Medicine
Results II
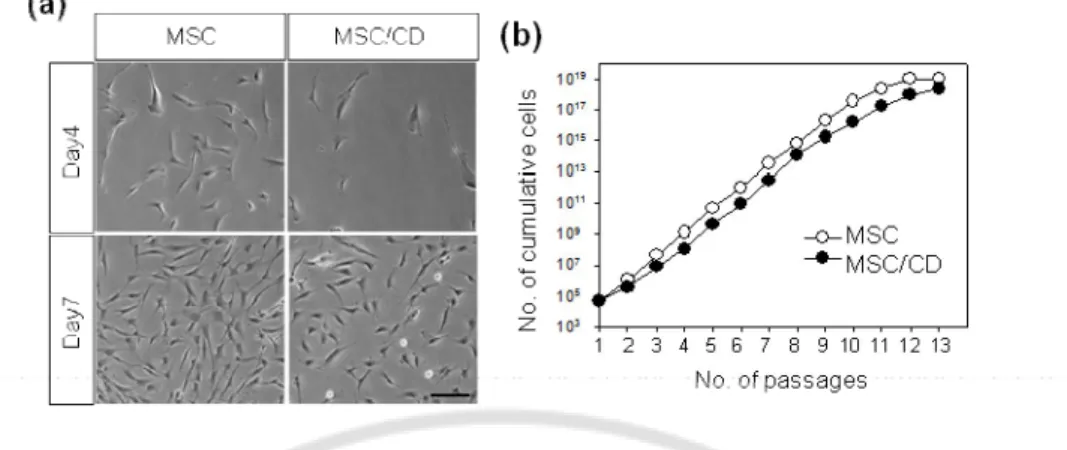
MSCs were isolated from the bone marrow of a human volunteer and expanded in vitro as previously reported(Kim et al., 2008).Retroviral vectors expressing E.coli CD gene generated in FlYRD18 packaging cells were added to MSCs at passage 1 and CD-expressing MSCs (MSC/CD) were selected in the presence of puromycin. In the beginning, the growth rate of MSC/CD cells was temporarily retarded by transduction and selection but soon recovered (Fig. 1a). Once they were grown to confluence at day 7, MSCs and MSC/CD cells were exponentially expanded by approximately 19-fold per every 6-day culture (Fig. 1b) and the growth rates were indistinguishable up to passage 10 between both cell types. The data indicated that either transduction with retroviral vectors or CD expression does not interfere with the proliferation capability of MSCs.
Characterization of MSC/CD cells
According to the ISCT(International Society for Cellular Therapy), MSCs can be defined by three criteria such as adhering to plastic, expressing specific surface antigens, and differentiating into mesodermal multilineages(Dominici et al., 2006). Both naïve MSCs and MSCs/CD adhered to plastic culture dishes on which they exhibited similar fibroblastic morphology (Fig. 2a). Both cell types retained differentiation potential when induced to differentiate into adipocytes, osteocytes, and chondrocytes (Fig. 2a). These MSCs and MSCs/CD cells were identical to express the typical surface antigen of classical MSCs; being positive for STRO-1, HLA-ABC, CD29, CD49a, CD73, CD90 and CD105while being negative for HLA-DR, CD45, CD34, CD11b and CD117 (Fig. 2b). These results indicate that retrovirus-mediated CD expression does not alter the morphology, multipotency, and surface antigenic properties of MSCs.