Celastrol induces paraptosis-like cell death via
mitochondrial Ca
2+overload in breast cancer cells
by
A Reum Lee
Major in Molecular Medicine
Department of Biomedical Sciences
Celastrol induces paraptosis-like cell death via
mitochondrial Ca
2+overload in breast cancer cells
by
A Reum Lee
A Dissertation Submitted to The Graduate School of Ajou University in Partial Fulfillment of the Requirements
for the Degree of
Master of Biomedical Sciences
Supervised by
Kyeong Sook Choi, Ph.D.
Major in Molecular Medicine
Department of Biomedical Sciences
This certifies that the dissertation
of A Reum Lee is approved.
SUPERVISORY COMMITTEE
심사위원장 윤 계 순 인
심
사
위
원
최
경 숙 인
심
사
위
원
이
종 수 인
심
사
위
원
김
유 선 인
The Graduate School, Ajou University
June, 21st, 2013
i
- Abstract -
Celastrol induces paraptosis-like cell death via mitochondrial Ca
2+overload in breast cancer cells
Various cancer cells are resistant to chemotherapeutic drugs-induced apoptosis. Thus, cancer cells that have acquired resistance to apoptosis need novel strategies for inducing non-apoptotic cell death. Paraptosis is a kind of non-apoptotic cell death and characterized by extensive vacuolization due to dilation of mitochondria and the endoplasmic reticulum (ER). However, the regulatory mechanisms that control paraptotic events are not yet fully understood. Recently, we found that celastrol, a triterpene extracted from the Chinese “Thunder of God Vine”, induced paraptosis accompanied by dilation of mitochondria and the ER in MDA-MB 435S and MCF-7 breast cancer cells. Inhibition of protein synthesis by cycloheximide blocked celastrol-induced vacuolation and subsequent cell death indicating that protein synthesis is required for this process. Generation of reactive oxygen species and mitochondrial Ca²⁺ overload was shown to act as a critical early signal in celastrol-induced paraptosis-like cell death contributing to the dilation of mitochondria/ER and subsequent paraptotic cell death. Inhibition of mitochondrial Ca2+ uniporter employing ruthenium red and IP₃ receptor employing 2-APB very effectively blocked celastrol-induced paraptosis-like
ii
cell death. Taken together, our results suggest that Ca²⁺ overload into the mitochondria via mitochondrial Ca2+ uniporter and IP3 receptor critically contribute to
celastrol-induced paraptosis-like cell death.
iii
TABLE OF CONTENS
ABSTRACT ---ⅰ TABLE OF CONTENT --- ⅲ LIST OF FIGURES --- v I. INTRODUCTION --- 1II. MATERIALS AND METHODS --- 6
A. Chemicals and antibodies --- 6
B. Cell culture --- 7
C. Measurement of cellular viability --- 7
D. Western blotting --- 7
E. mRFP-GFP-LC3 transfection--- 8
F. Immunocytochemistry --- 8
G. Establishment of the stable cell lines in the fluorescence specifically mitochondria or the endoplasmic reticulum --- 9
H. Transmission electron microscopy --- 9
I. Measurement of ROS levels--- 9
J. Measurement of mitochondrial superoxide anion levels --- 10
K. Measurement of cytosolic and mitochondrial Ca2+ levels --- 10
III. RESULTS --- 11
1.Celastrol induces non-apoptotic cell death and partial apoptosis in human cancer cells- 11 2. Celastrol induces non-autophagic cell death in breast cancer cells --- 16
iv
3. Celastrol induces the swelling of mitochondria and the ER in breast cancer cells --- 23
4. Release of Ca2+ from the ER via IP 3 receptor and mitochondrial Ca2+ influx via uniporter are critical for celastrol-induced paraptosis --- 37
5. ROS generation is also important for celastrol-induced paraptosis --- 50
IV. DISCUSSION --- 60
V. REFERENCES --- 64
v
LIST OF FIGURES
Fig. 1. Effects of celastrol on the viability of human cancer cells --- 12
Fig. 2. Celastrol induces caspase-independent cell death in human cancer cells --- 13
Fig. 3. Effects of celastrol on the activation of caspase in MDA-MB 435S cells --- 14
Fig. 4. Morphological changes in celastrol-treated cancer cells --- 15
Fig. 5. Effects of 3-MA, bafilomycin or chloroquine on celastrol-induced cell death --- 18
Fig. 6. Effects of 3-MA, bafilomycin or chloroquine on celastrol-induced vacuolation --- 19
Fig. 7. The expression of autophagy-associated proteins in breast cancer cells treated with celastrol --- 20
Fig. 8. Celastrol does not induce autophagic flux --- 21
Fig. 9. Expression of mRFP-GFP-LC3 in breast cancer cells treated with celastrol --- 22
Fig. 10. Swelling of mitochondria and the ER is induced by celastrol treatment --- 26
Fig. 11. Electron microscopic observation of breast cancer cells treated with celastrol --- 27
Fig. 12. Celastrol induces the expression levels of PDI in MDA-MB 435S cells --- 28
Fig. 13. Protein synthesis is required for celastrol-induced cell death --- 29
Fig. 14. Cycloheximide blocks the celastrol-induced vacuolation of mitochondria and the ER --- 30
Fig. 15. Cycloheximide blocks the celastrol-induced cell death in human cancer cells --- 31
Fig. 16. Celastrol induces ER stress in breast cancer cells --- 32
Fig. 17. Celastrol inhibits proteasome activity in breast cancer cells --- 33
vi
Fig. 19. Effects of the inhibition of MAP kinases on celastrol-induced cell death --- 35 Fig. 20. Alix is down-regulated by celastrol treatment in MDA-MB 435S cells --- 36 Fig. 21. Celastrol significantly increases mitochondrial Ca2+ levels in MDA-MB 435S cells (FACS analysis) --- 40 Fig. 22. Celastrol markedly increases mitochondrial Ca2+ levels in MDA-MB 435S cells
(Fluorescence microscopy) --- 41 Fig. 23. Celastrol markedly increases mitochondrial Ca2+ levels in human cancer cells (Fluorescence microscopy) --- 42 Fig. 24. Effects of various Ca2+ antagonists on celastrol-induced cell death --- 43 Fig. 25. Mitochondrial uniporter- or IP3R-mediated Ca2+ influxes are critical for the
celastrol-induced increase of mitochondrial Ca2+ in breast cancer cells --- 44 Fig. 26. Mitochondrial uniporter- or IP3R-mediated Ca2+ influxes are critical for the
celastrol-induced dilations of mitochondria/the ER and subsequent cell death in breast cancer cells --- 45 Fig. 27. Mitochondrial Ca2+ overload acts as a signal for celastol-induced paraptosis of
breast cancer cells --- 46 Fig. 28. Activation of IP3R employing adenophostin A accelerates celastrol-increased
mitochondrial Ca2+ levels --- 47 Fig. 29. Activation of IP3R employing adenophostin A accelerates celastrol-induced cell
death --- 48 Fig. 30. Effects of RR or 2-APB on celastrol-induced cell death in NAC blocks the celastrol
vii
Fig. 31. Generation of ROS by celastrol in breast cancer cells --- 52
Fig. 32. Celastrol significantly increases mitochondrial superoxide levels in breast cancer cells --- 53
Fig. 33. Effects of antioxidants on celastrol-induced cell death --- 54
Fig. 34. Antioxidant blocks the celastrol-induced vacuolation of mitochondria/the ER in breast cancer cells --- 55
Fig. 35. Effects of RR or 2-APB on celastrol-generated ROS --- 56
Fig. 36. Effects of NAC on celastrol-induced mitochondrial Ca2+ levels --- 57
Fig. 37. NAC blocks the celastrol-induced paraptosis signals in breast cancer cells --- 58
1
I. INTRODUCTION
Breast cancer is the leading cause of cancer-related mortality among women worldwide, and 30%-40% of breast cancer patients will develop metastatic disease (Al-Ejeh F et al., 2013). Choice of the treatment, such as mastectomy, lumpectomy, radiation therapy and chemotherapy, in breast cancer usually depends on the type of cancer and whether the cancer has spread outside of the breast to the rest of the body (Early Breast Cancer Trialists’ Collaborative Group et al., 2006; Clarke M et al., 2005). In most cases, chemotherapy is most effective, either as an adjuvant or neo-adjuvant therapy, when combinations of more than one chemotherapeutic drug are used together. Various cancer cells are resistant to chemotherapeutic drugs-induced apoptosis. Thus, cancer cells that have acquired resistance to apoptosis need novel strategies for inducing non-apoptotic cell death, such as autophagic cell death, cell death through mitotic catastrophe, or paraptosis. Non-apoptotic cell death may have considerable merit for the treatment of in breast cancer cells which have acquired resistance to apoptosis (Morse DL et al., 2005; Bröker LE et al., 2005).
Since the first descriptions of programmed cell death mechanisms, which date back to the mid-1960s, several attempts have been made to classify cell death subroutines based on morphological characteristics (Galluzzi L et al., 2012). Thus, in 1973 Schweichel and Merker proposed a classification of several cell death modalities, including ‘type I cell death’ associated with heterophagy, ‘type II cell death’ associated with autophagy and ‘type III cell death’, which was not associated with any type of digestion,
2
corresponding to apoptosis, autophagic cell death and necrosis, respectively (Schweichel JU et al., 1973; Galluzzi L et al., 2007).
A novel non-apoptotic programmed cell death (PCD) process designated paraptosis (from para= next to or related to, and apoptosis) was described by Sperandio S (Sperandio S et al., 2000). Paraptosis, a type of non-apoptotic cell death, has been reported to be induced extensive cytoplasmic vacuolization, swelling of endoplasmic reticulum (ER) and/or mitochondria, without the characteristic apoptotic features of pyknosis, DNA fragmentation or caspase activation (Sperandio S et al., 2000; Wyllie AH et al., 2001). Also this form of PCD is characterized by a requirement for new gene transcription and translation (Sperandio S et al., 2004). And the insulin-like growth factor 1 receptor (IGFIR), epidermal growth factor (EGF), and TAJ/TROY were shown to induce paraptosis (Sperandio S et al., 2000; Fombonne J et al., 2006; Wang Y et al., 2004). Recent reports have shown that paraptosis can be inhibited by the expression of AIP-1/Alix (Sperandio S et al., 2004; Valamanesh F et al., 2007). However, the mechanisms underlying paraptosis, in particular the signals responsible for triggering mitochondrial and ER dilation, have not yet been fully determined.
Celastrol is pharmacologically active compound derived from the Chinese medicinal plant Tripterygium wilfordii, which is used to treat autoimmune diseases, chronic inflammation, asthma, and neurodegenerative disease for years (Allison AC et al., 2001; Canter PH et al., 2006; Brinker AM et al., 2007). Recently, celastrol has been found to inhibit the proliferation, induce apoptosis, and suppress invasion/migration and angiogenesis in a wide variety of tumor models in vitro and in vivo (Huang Y et al.,
3
2008; Pang X et al., 2010; Lu Z et al., 2010; Chen G et al., 2011; Rajendran P et al.,
2012). Also, celastrol was found to induce paraptosis and autophagy in HeLa cells, A549 cells, and PC-3 cells (Wang WB et al., 2012). The efficacy of celastrol to modulate the expression of various key mediators of tumorigenesis such as pro-inflammatory cytokines, adhesion molecules, potassium channels, nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB), TGF-activated kinase 1 (TAK1), C-X-C chemokine receptor type 4 (C-X-CXC-X-CR4), vascular endothelial growth factor receptor (VEGFR), signal transducer and activator of transcription 3 (STAT3), proteasome, and heat shock response has been reported previously (Sethi G et al., 2007; Huang Y et al., 2008; Morita H et al., 2008; Salminen A et al., 2010; Yadav VR et al., 2010; Kannaiyan R et al., 2011a; Kannaiyan R et al., 2011b; Kannaiyan R et al., 2011c). Also, as celastrol is moved into clinical studies, it is important to gain a better understanding of its target and mechanism (Chen Get al., 2011).
Many cellular stresses can cause induction of cell death via mitochondrial dysfunction or endoplasmic reticulum stress (Mariño G et al., 2004). A critical role of mitochondria in the regulation of cell death is now well established. Mitochondria are associated with energetic metabolism (Chacinska A et al., 2009) and also with the first source of reactive oxygen species (ROS) formation and the most susceptive ROS target (Kumari U et al., 2013). ROS are derived from oxygen (O2) that can readily oxidize other
molecules. Most intracellular ROS are derived from superoxide (O2−⋅), which is
generated by the one electron reduction of O2. Superoxide is converted to hydrogen
4
functions of the ER are affected by various intracellular and extracellular stimuli, so called ER stress, which includes inhibition of glycosylation, reduction of disulfide bonds, calcium depletion from the ER lumen, impairment of protein transport to the Golgi apparatus, and expression of mutated proteins in the ER (Kadowaki H et al., 2004). Disruption of ER homeostasis (ER stress) activates an integrated signal transduction pathway termed the unfolded protein response (UPR), mediated by three ER transmembrane sensors: inositol-requiring enzyme 1α (IRE1α), PKR-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6α (ATF6α) (Ron D et al., 2007). UPR induces global changes in gene expression to restore ER homeostasis (Huber AL et al., 2013). In the unstressed ER, BiP/GRP78 binds to the ER luminal domains of the transducers and keeps them inactive in sequestration (Szegezdi E et al., 2006). Upon sensing the accumulation of misfolded/unfolded proteins, BiP/GRP78 dissociates from its clients and translocates to the ER lumen to help protein folding (Bertolotti et al., 2000; Liu CY et al., 2003). Once released from sequestration, PERK and IRE1 are serine/threonine kinases activated by auto-phosphorylation, while ATF6 acts a transcription factor (signaling the unfolded protein response from ER). When the survival response fails to adapt under prolonged ER stress, cell will eventually trigger apoptosis. However, it has been shown recently that ER stress can also induce non-apoptotic cell death, although the underlying mechanism has not been elucidated (Yorimitsu T et al., 2006).
Ca2+, which is a major second messenger in cellular signaling, is involved in many signal pathways to regulate cellular proliferation, differentiation, autophagy, and cell
5
death including apoptosis. The intracellular Ca2+ concentration is finely regulated by several mechanisms, among them ionic channels, the endoplasmic reticulum Ca2+ -ATPase (SERCA), the plasma membrane calcium pump (PMCA), and the mitochondrial Ca2+ transport (Pimentel AA and Benaim G, 2012). The mitochondria and ER were well known to be major reservoirs of intracellular Ca2+ (Pivovarova NB et al., 2002). Especially, mitochondrial Ca2+ was reported to be increased via the ryanodine receptor (RyR) and IP3 receptor channels in ER (Amador FJ et al., 2013; Zhang D et al., 2013).
However, the regulatory mechanisms that control the Ca2+ flux contributing particularly the dilation of mitochondria and ER in paraptotic events are unclear.
In this study, we show that celastrol-induced paraptosis is associated with ER stress in breast cancer cells. Furthermore, we found that celastrol triggers ROS production and mitochondrial Ca²⁺ overload via mitochondrial Ca²⁺ uniporter and IP3 receptor critically
contribute to the paraptotic cell death in cancer cells. Therefore, celastrol treatment may offer an attractive strategy for the treatment of breast cancer, which are resistant to pro-apoptotic therapeutics.
6
II. MATERIALS AND METHODS
A. Chemicals and antibodies
Celastrol, 3-methyladenine (3-MA), bafilomycin A1, N-acetylcysteine (NAC), cycloheximide (CHX), ethylene glycol tetraacetic acid (EGTA), 1,2-bis(o-aminophenoxy)ethane-N,N,N’N’-tetraacetic acid acetoxymethyl ester (BAPTA-AM), ruthenium red, and leupeptin were purchased from Sigma (St. Louis, MO). Rhod-2-AM, chloromethyl-H2DCFDA (CM-H2DCFDA), MitoSOX-Red, calcein acetosymethyl ester
(calcein-AM) and ethidium homodimer (EthD-1) were purchased from Molecular Probes (Carlsbad, CA). Caspases inhibitors benzyloxy-carbonyl-Val-Ala-Asp-(OMe) fluoromethyl ketone (z-VAD-fmk) was from R&D systems (Minneapolis, MN). 2-Aminoethosxydiphenyl borate (2-APB), SB203580, PD98059, U0126, and SP600125 were obtained from Calbiochem (San Diego, CA). Dantrolene was obtained from Alexis Biochemicals (San Diego, CA). The following antibodies were used: monoclonal anti-β-actin (Abcam); anti-PARP (Epitomics Inc.); anti-p62, Cathepsin D, and Cathepsin L (BD biosciences pharmingen); anti-NBR1 (Abnova); anti-ATG7 (Abgent); anti-Noxa (Calbiochem); anti-ubiquitin, ATF4, and Mcl-1 (Santa Cruz Biotechnologies, Santa Cruz, CA); anti-CHOP (GADD153), phospho-ERK1/2, total ERK1/2, phospho-JNK, total JNK, AIP-1/Alix, and LC3B (Cell Signaling, Beverly, MA); anti-caspase-3, caspase-8, caspase-9, and KDEL (Stressgen, BC, Canada); anti-c-FLIP (NF6) (Alexis, San Diego, CA); HRP-conjugated anti-rabbit IgG and HRP-conjugated anti-mouse IgG (Molecular Probes).
7 B. Cell culture
The MDA-MB 435S and MCF-7 human breast cancer cell lines, RKO and DLD-1 human colon cancer cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA). MDA-MB 435S, MCF-7, RKO, and DLD-1 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics (GIBCO-BRL, Grand Island, NY). Cells were maintained in a humidified atmosphere containing 5% CO2 at 37℃. Cell culture passage number less than five was used in the present
study. Celastrol (>98% purity, Sigma) was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 20 mM and stored at -20℃. This stock solution was diluted to the required concentration when need.
C. Measurement of cell viability
Cell viability was assessed by double labeling of cells with 2 μM calcein-AM and 4 μM EthD-1. The calcein-positive live cells and EthD-1-positive dead cells were visualized using a fluorescence microscope (Axiovert 200M using Axiovision Release 4.4 and Axiocam HRM digital camera; Zeiss, Oberkohen, Germany) and counted.
D. Western blotting
Cells were washed in PBS and lysed in boiling sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (6.25 mM Tris [pH 6.8], 1% SDS, 10% glycerol, and 5% β-mercaptoethanol). The lysates were boiled for 5 min, separated by SDS-PAGE, and transferred to an Immobilion membrane (Millipore, Bredford, MA,
8
USA). After blocking nonspecific binding sites for 1 h using 5% skim milk, membrane were incubated for 2 h with specific Antibodies. Membranes were then washed three times with TBST and incubated further for 1 h with horseradish peroxidase-conjugated anti-rabbit, -mouse antibody. Visualization of protein bands was accomplished using ECL (Advansta).
E. mRFP-GFP-LC3 transfection
MDA-MB 435S cells were transfected with the plasmid encoding mRFP-GFP-LC3 for 24 h and treated with 2 μM celastrol. Then, observed under a fluorescence microscopy using Zeiss filter sets #10 and #20.
F. Immunocytochemistry
After treatments, cell were fixed with 50% MeOH plus 50% Acetone for 5 min at -20℃ and blocking in 5% BSA in PBS for 30 min. Fixed cells were incubated overnight at 4℃ with primary antibody [anti-PDI (1:200, rabbit, stressgen), anti-COX-II (1:200, mouse, invitrogen)] diluted in PBS and then washed three times in PBS and incubated for 1 h at room temperature with rabbit Alexa Fluor 488 (1:200, Molecular Probes). Or anti-mouse Alexa 594 was used as a secondary antibody (Molecular Probes). Next, cells were washed with PBS. Slides were mounted with ProLong Gold antifade mounting reagent (Molecular Probes) and cell staining was visualized with a fluorescence microscope (Axiovert 200M, Carl Zeiss).
9
G. Establishment of the stable cell lines in the fluorescence specifically mitochondria or endoplasmic reticulum
To establish the stable cell lines expressing the fluorescence specifically in mitochondria or the ER, MDA-MB 435S cells were transfected with the pEYFP-Mito of pEYFP-ER vector (Clontech Laboratories, Mountain View, CA). Stable cell lines over-expressing pEYFP-Mito of pEYFP-ER (YFP-Mito of YFP-ER) were selected with the complete media containing 500 μg/mL G418 (Calbiochem, San Diego, CA). Images of mitochondria or ER were obtained from the fluorescence microscopy using a Zeiss filter set #10.
H. Transmission electron microscopy
Cells were prefixed in Karnovsky’s solution (1% paraformaldehyde, 2% glutaraldehyde, 2 mM calcium chloride, 0.1 M cacodylate buffer, pH 7.4) for 2 h and washed with cacodylate buffer. Post-fixing was carried out in 1% osmium tetroxide and 1.5% potassium ferrocyanide for 1 h. After dehydration with 50-100% alcohol, the cells were embedded in Poly/Bed 812 resin (Pelco, Redding, CA, USA), polymerized, and observed under electron microscope (EM 902A, Zeiss, Oberkohen, Germany).
I. Measurement of ROS
Cells were exposed to 2 μM celastrol for 2, 4, 6, and 8 h. The cells were stained with 5 μM CM-H2DCF-DA for 30 min at 37℃ in the dark. After being washed with PBS or
10
cytometry using CellQuest software (Becton Dickinson, San Jose, CA).
J. Measurement of mitochondrial superoxide anion
Generation of superoxide was determined using a previously established fluorescence microscopy or flow cytometry technique. Celastrol-treated cells were stained with 2.5 μM MitoSOX red for 20 min and samples were observed under a fluorescence microscope equipped with Zeiss filter set #20 (excitation; band pass (BP) 546 nm, emission; BP 575-640 nm).
K. Measurement of cytosolic and mitochondrial Ca²⁺ levels
To measure cytosolic Ca²⁺ levels, treated cells were incubated with 2.5 μM Fluo-3-AM at 37℃ for 20 min, washed with HBSS, and analyzed immediately by flow cytometry. To measure mitochondrial Ca²⁺ levels treated cells were incubated with 2.5 μM Rhod-2-AM at 4℃ for 30 min, washed with HBSS, further incubated with HBSS at 37℃ for 20 min, and then analyzed by flow cytometry. To confirm the mitochondrial localization of the Rhod-2 probe, YFP-Mito cells were loaded with 2.5 μM Rhod-2-AM in HBSS for 30 min at 4℃. The cells were then washed with HBSS, and visualized by the fluorescence microscopy using Zeiss filter sets #10 and #20.
11
III. RESULTS
1. Celastrol induces non-apoptotic cell death and partial apoptosis in human cancer cells
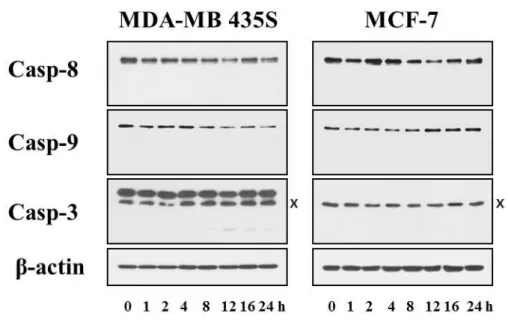
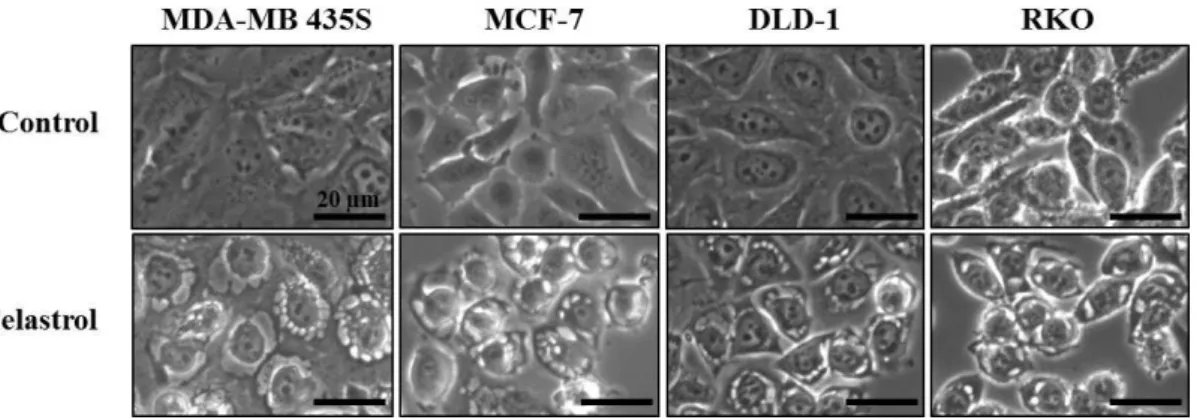
In this study, we investigated the anti-cancer effect of celastrol in breast and colon cancer cells. When we treated MDA-MB 435S, MCF-7 breast cancer cells, DLD-1, RKO colon cancer cells with various doses of celastrol for 24 h, the cell viability assay using calcein-AM and EthD-1, to detect live and dead cells, respectively showed that celastrol dose dependently increased cell death in these cells (Fig. 1). Since celastrol was previously shown to induce apoptosis (Chadalapaka G et al., 2012), we examined whether the celastrol-indcued cell death could be blocked by the inhibition of caspases. However, pretreatment with z-VAD did not alter the celastrol-induced death of these cells (Fig. 2). Furthermore, the proteolytic processing of caspase-8, and -9 was not detected both in MDA-MB 435S and MCF-7 cells (Fig. 3). The proteolytic processing of caspase-3 was minimally observed only in MDA-MB 435S cells, whereas caspase-3 is not expressed in MCF-7 cells (Jänicke RU, 2009). These results suggest that caspase-mediated apoptosis is not a major death mode in these cells treated with celastrol. When we further observed the changes in the cellular morphologies, marked vacuolation, but not apoptotic morphologies such as cellular shrinkage, fragmentation, and blebbing, was commonly detected in these cells undergoing celastrol-indcued cell death (Fig. 4).
12
Fig. 1. Effects of celastrol on the viability of human cancer cells. Two breast cancer
cell lines (MDA-MB 435S and MCF-7) and two colon cancer cell lines (DLD-1 and RKO) were treated with celastrol (Cela.) at the indicated concentrations for 24 h. Cellular viability was assessed using calcein-AM and EthD-1 to detect live and dead cells, respectively. Columns, mean of three independent experiments; bars, SE.
13
Fig. 2. Celastrol induces caspase-independent cell death in human cancer cells.
MDA-MB 435S, MCF-7, DLD-1, and RKO cells were pretreated with the indicated concentrations of z-VAD-fmk for 30 min and further treated with 2 μM celastrol for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. Columns, mean of three independent experiments; bars, SE.
14
Fig. 3. Effects of celastrol on the activation of caspase in MDA-MB 435S cells.
MDA-MB 435S cells were treated with 2 μM celastrol for the indicated at time points. Whole cell extracts were prepared and subjected to western blotting using anti-caspase-8, anti-caspase-9, and anti-caspase-3 antibodies. Anti-β-actin antibody was used as a loading control.
15
Fig. 4. Morphological changes in celastrol-treated cancer cells. MDA-MB 435S,
MCF-7, DLD-1, and RKO cells were treated with 2 μM celastrol for 12 h and observed under a phase contrast microscope (magnification, X1000).
16
2. Celastrol induces non-autophagic cell death in breast cancer cells
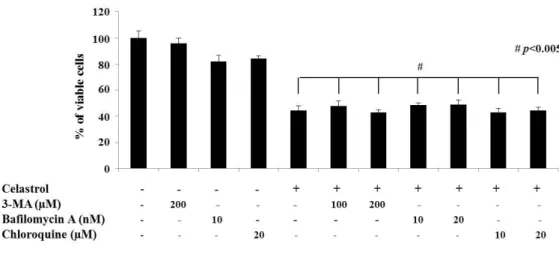
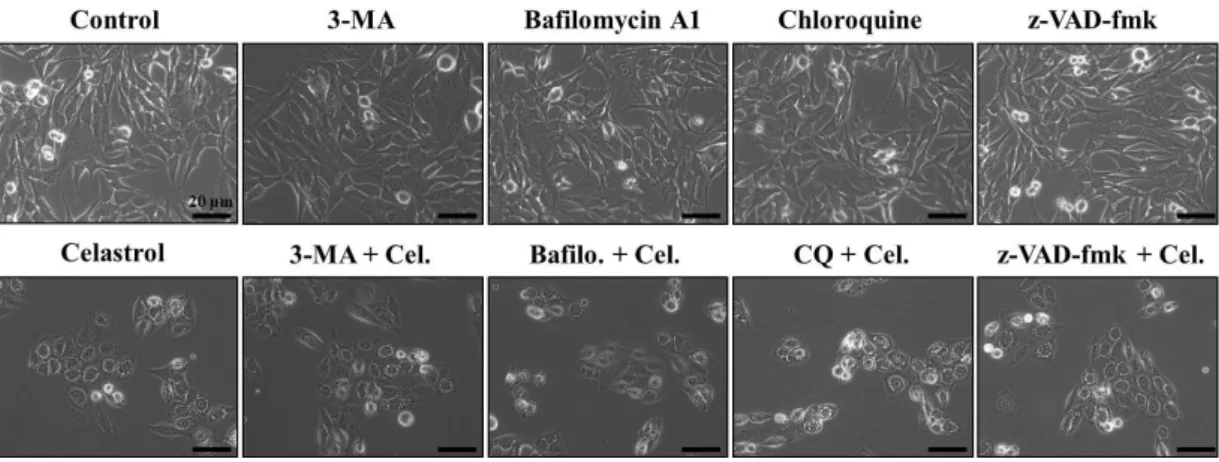
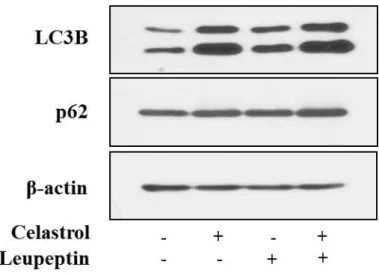
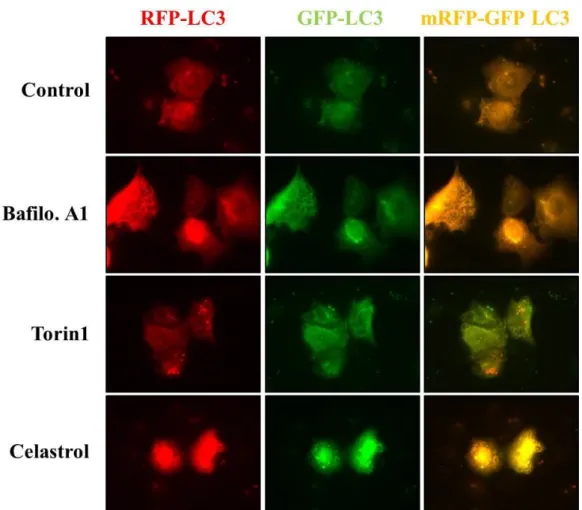
We next examined whether the celastrol-induced vacuolation and subsequent cell death might be associated with autophagy. First, we tested the effects of various autophagy inhibitors on celastrol-induced cell death. However, celastrol-induced cell death was not affected by various autophagy inhibitors, including 3-MA, bafilomycin A, or CQ, (Fig. 5). In addition, pretreatment with these autophagy inhibitors did not affect the celastrol-induced cell death and vacuolation, similar to the effect of z-VAD-fmk (Fig. 6). When we examined the expression of autophagy-associated gene products, we found that celastrol increased the protein levels of both I and II form of LC3B (Fig. 7). While ATG5 protein levels were not altered, the protein levels of ATG7, one of the key proteins important for autophagosome formation (Kang R et al., 2011), and major lysosomal proteases (cathepsin D and cathepsin L) were decreased by celastrol treatment (Fig. 7). Furtheremore, our measurement of autophagic flux activity using leupeptin, a lysosomal inhibitor, showed that celastrol did not further increased the protein levels of LC3B II form (Fig. 8), suggesting that celastrol may not activate autophagy in these cells. Furthermore, time-course experiment showed that the not only LC3B but also p62 and NBR1 (Lamark T et al., 2009) proteins, substrate proteins of autophagy, were progressively accumulated following celastrol treatment (Fig. 7). Moreover, measurement of autophagic flux using mRFP-GFP-LC3 showed that celastrol inhibits autophagy, rather than activation of autophagy, since celastrol induced
17
GFP (+) and RFP (+) LC3 (Fig. 9). These results suggest that the increase in LC3B may be due to inhibition of autophagy, rather than activation of autophagy. Taken together, celastrol inhibits autophagy but its autophagy-inhibiting activity may not be directly associated with celastrol-induced vacuolation and subsequent cell death.
18
Fig. 5. Effects of 3-MA, bafilomycin, or chloroquine on celastrol-induced cell death.
MDA-MB 435S cells were untreated or pretreated with 3-MA, bafilomycin A1 or chloroquine at the indicated concentrations for 30 min and further treated with 2 μM celastrol for 24 h. Cellular viabilities were assessed using calcein-AM and EthD-1.
19
Fig. 6. Effects of 3-MA, bafilomycin, or chloroquine on celastrol-induced vacuolation. MDA-MB 435S cells were untreated or pretreated with 3-MA,
bafilomycin A1 or chloroquine at the indicated concentrations for 30 min and further treated with 2 μM celastrol for 24 h. Cellular viabilities were assessed using calcein-AM and EthD-1. Columns, mean of three independent experiments; bars, SE.
20
Fig. 7. The expression of autophagy-associated proteins in breast cancer cells treated with celastrol. MDA-MB 435S or MCF-7 cells were treated with 2 μM
celastrol for the indicated at time points. Whole cell extracts were prepared and subjected to western blotting using LC3B, p62, NBR1, ATG7, anti-cathepsin D, and anti-anti-cathepsin L antibodies. Anti-β-actin antibody was used as a loading control.
21
Fig. 8. Celastrol does not induce autophagic flux. MDA-MB 435S cells were
pretreated with 20 μM leupeptin and further treated with 2 μM celastrol for 12 h. Whole cell extracts were prepared and subjected to western blotting using LC3B and anti-p62 antibodies. Anti-β-actin antibody was used as a loading control.
22
Fig. 9. Expression of mRFP-GFP-LC3 in breast cancer cells treated with celastrol.
MDA-MB 435S cells were transiently transfected with mRFP-GFP-LC3 construct and treated with autophagy inhibitor (10 nM bafilomycin A1) or autophagy activator (1 μM Torin1) or 2 μM celastrol for 12 h. Fluorescent images were observed under a fluorescence microscope.
23
3. Celastrol induces the swelling of mitochondria and the ER in breast cancer cells
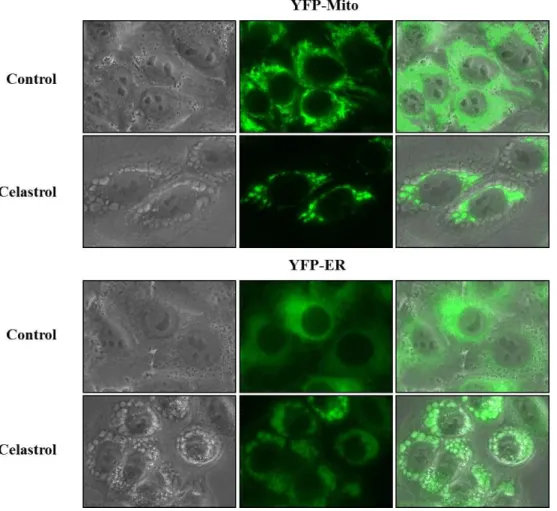
Recently, non-apoptotic cell death called paraptosis has been reported to accompany swelling of the mitochondria and enlargement of the endoplsmic reticulum along with preservation of the nuclear chromatin (Fombonne J et al., 2000; Wang Y et al., 2004). Next, we tested whether these vacuoles are derived from mitochondria or endoplasmic reticulum (ER). To do this, we established the MDA-MB 435S sublines transfected with the Mito-YFP plasmid for mitochondrial labeling and sublines transfected with ER-YFP plasmid for ER labeling. As shown in Fig. 10, the mitochondria in untreated YFP-Mito cells showed an elongated morphology, and the ER in untreated YFP-ER cells exhibited as reticulate structure. After treatment with 2 μM celastrol for 3 h, numerous vacuoles were observed around the nucleus in YFP-Mito cells, the ER fluorescence in YFP-ER cells mainly co-localized with vacuoles scattered at the cellular periphery (Fig. 10). These observations indicate that celastrol-induced vacuoles are originated from both mitochondria and the ER. These observations indicate that celastrol-induced vacuoles originated from both mitochondria and the ER. We further observed the ultrastructure of mitochondria and the ER by electron microscopy. In untreated MDA-MB 435S cells, various sizes of mitochondria with intact cristae were observed and besides the mitochondria, the ER with reticular morphologies was detected. In contrast, in MDA-MB 435S cells treated with 2 μM celastrol, swollen and fused mitochondria were observed around the nuclei and dilated the ER
24
structures undergoing fusion among the ER were surrounded at the cellular periphery (Fig. 11).
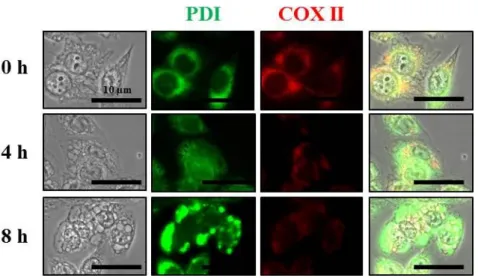
When further performed the immunocytochemistry using the specific antibodies against Complex II (COX II), a mitochondrial protein, and protein disulfide-isomerase (PDI), an ER protein, we found that COX II expression showed many circular patterns of mitochondria around the nuclei at 4 hr of celastrol treatment. At 8 hr, the sizes of these COX II-positive dilated mitochondria were increased but their numbers were reduced (Fig. 12). The expression of pattern of PDI, an ER resident protein, revealed the reticular morphologies in untreated cells, but ring structure, possibly present at the dilated ER membrane at 4 h or celastrol treatment. Interestingly, further enhanced PDI expression was found within much more enlarged ER at 8 h of celastrol treatment, suggesting that these extensive changes in the ER may contribute to ER stress. Previously, paraptosis was shown to require protein synthesis (Schneider D et al., 2004), we tested the effect of cycloheximide on celastrol-induced vacuolation and subsequent cell death (Fig.13 and Fig. 14). We found that pretreatment with cycloheximide almost completely blocked not only the dilation of mitochondria and the ER but also cell death in YFP-Mito and YFP-ER cells (Fig. 14). In addition, pretreatment with cycloheximide almost completely blocked celastrol-induced cell death in these cells. These results are similar to their effect on the viability of MDA-MB 435S cells treated with celastrol (Fig. 15). Taken together, these results indicate that celastrol induces paraptosis that is accompanied by swelling and fusion of respective mitochondria and the ER
25
in these breast cancer cells. Next, we investigated whether celastrol activates paraptotic signals in breast cancer cells. Since PDI expression was increased by celastrol treatment as shown in Fig. 12, we first examined the expression of other ER stress marker proteins. We found that eIF2a phosphorylation was increased and CHOP, ATF4, and KDEL were upregulated, similar to PDI (Fig. 12), indicating that celastrol induces ER stress (Fig. 16). Recently, we have shown that proteasomal inhibition critically contributes to curcumin-indcued paraptosis, leading to ER stress (Yoon MJ et al., 2010). We found that celastrol treatment also accumulated poly-ubiquitinated proteins (Fig. 17), suggesting that celastrol inhibits proteasomal activity. We examined the changes in the activities of ERK and JNK following celastrol treatment. While ERKs showed relatively progressive activation both in MDA-MB 435S and MCF-7 cells, JNKs demonstrated a biphasic activation patterns in these cells treated with celastrol (Fig. 18). An experiment designed to test the functional significance of MAP kinases (MAPK) in this process showed that celastrol-induced cell death in MDA-MB 435S cells. While inhibition of JNK pathway using SP600125 significantly inhibited it, inhibition of ERK pathway using PD98059 partially inhibited celastrol-induced cell death (Fig. 19). In addition, the levels of AIP-1/Alix protein, a known inhibitor of paraptosis (Sperandio S et al., 2004; Valamanesh F et al., 2007), were down-regulated by celastrol treatment in MDA-MB 435S cells (Fig. 20). Taken together, our results indicate that celastrol kills these breast cancer cells via paraptosis as a major death mode.
26
Fig. 10. Swelling of mitochondria and the ER is induced by celastrol treatment.
YFP-Mito cells or YFP-ER cells were treated with 2 μM celastrol for 3 h and then observed under a fluorescence microscope.
27
Fig. 11. Electron microscopic observation of breast cancer cells treated with celastrol. MDA-MB 435S cells were untreated or treated with 2 μM celastrol for 12 h
28
Fig. 12. Celastrol induces the expression levels of PDI in MDA-MB 435S cells.
MDA-MB 435S cells were treated without or with 2 μM celastrol at time points. Immunocytochemistry using anti-PDI and anti-COX II antibodies were performed and the representative images of cells are shown.
29
Fig. 13. Protein synthesis is required for celastrol-induced cell death. MDA-MB
435S cells were untreated or pretreated with cycloheximide (CHX) at the indicated concentrations for 30 min and further treated with 2 μM celastrol for 24 h. Cellular viabilities were assessed using calcein-AM and EthD-1. Columns, mean of three independent experiments; bars, SE.
30
Fig. 14. Cycloheximide blocks the celastrol-induced vacuolation of mitochondria and the ER. YFP-Mito cells or YFP-ER cells were untreated or pretreated with
cycloheximide (CHX) for 30 min further treated with 2 μM celastrol for 3 h and then observed under a fluorescence microscope.
31
Fig. 15. Cycloheximide blocks the celastrol-induced cell death in human cancer cells. MCF-7, DLD-1, and RKO cells were untreated or pretreated with cycloheximide
(CHX) at the indicated concentrations for 30 min and further treated with 2 μM celastrol for 24 h. Cellular viabilities were assessed using calcein-AM and EthD-1. Columns, mean of three independent experiments; bars, SE.
32
Fig. 16. Celastrol induces ER stress in breast cancer cells. Breast cancer cell lines
(MDA-MB 435S and MCF-7) were treated with 2 μM celastrol for the indicated at time points. Whole cell extracts were prepared and subjected to western blotting using anti-phospho eIF2α (p-eIF2α), anti-eIF2α, anti-ATF4, anti-CHOP, and anti-KDEL antibodies. Anti-β-actin antibody was used as a loading control.
33
Fig. 17. Celastrol inhibits proteasome activity in breast cancer cells. Breast cancer
cell lines (MDA-MB 435S and MCF-7) were treated with 2 μM celastrol for the indicated at time points. Whole cell extracts were prepared and subjected to western blotting using anti-ubiquitin antibody. Anti-β-actin antibody was used as a loading control.
34
Fig. 18. Celastrol induces ERK and JNK activation in breast cancer cells. Breast
cancer cell lines (MDA-MB 435S and MCF-7) were treated with 2 μM celastrol for the indicated at time points. Whole cell extracts were prepared and subjected to western blotting using anti-phospho ERK (p-ERK), anti-ERK, anti-phospho JNK (p-JNK), and anti-JNK antibodies. Anti-β-actin antibody was used as a loading control.
35
Fig. 19. Effects of the inhibition of MAP kinases on celastrol-induced cell death.
MDA-MB 435S cells were pretreated with SP600125 or PD98059 at the indicated concentrations for 30 min and further treated with 2 μM celastrol for 24 h. Cellular viabilities were assessed using calcein-AM and EthD-1. Columns, mean of three independent experiments; bars, SE.
36
Fig. 20. Alix is down-regulated by celastrol treatment in MDA-MB 435S cells.
MDA-MB 435S cells were treated with 2 μM celastrol for the indicated at time points. Whole cell extracts were prepared and subjected to western blotting using anti-Alix antibody. Anti-β-actin antibody was used as a loading control.
37
4. Release of Ca2+ from the ER via IP
3 receptor and mitochondrial Ca2+ influx
via uniporter are critical for celastrol-induced paraptosis
Celastrol induced the dilation of both mitochondria and the ER, major reservoirs of intracellular Ca2+ (Biaqioli M et al., 2008), as shown in Fig.10 and Fig. 11, we next tested whether celastrol-induced paraptosis is associated with the disruption in intacellular Ca2+ homeostasis. Flow cytometry using Fluo-3, a cell-permeable Ca2+-indicator dye, demonstrated that treatment of MDA-MB 435S cells with 2 μM celastrol dramatically increased intracellular Ca2+ levels ([Ca2+]i) with a peak
at 3 h of celastrol treatment (Fig. 21 A). When we further examined whether the mitochondrial Ca2+ levels ([Ca2+]m) are also modulated by celastrol treatment, flow
cytometry using Rhod-2, an indicator dye for mitochondrial Ca2+, showed that celastrol increased [Ca2+]m with a peak at 2 h of celastrol treatment (Fig. 21 B).
Celastrol-induced increase in [Ca2+]m was further confirmed by the fluorescent
microscopy using Rhod-2 in YFP-Mito cells. We showed that mitochondrial Ca2+ levels were dramatically increased in the dilated mitochondria of YFP-Mito cells by celastrol treatment at 2 h, and further showed that these high levels were sustained thereafter (Fig. 22). In addition, the increase in [Ca2+]m was commonly
observed in MCF-7, DLD-1 and RKO cells treated with celastrol (Fig. 23). We next examined whether these increase in Ca2+ levels plays a critical role in celastrol-induced paraptosis. While treatment with each antagonist alone did not affect the viability of MDA-MB 435S cells, treatment with 2 μM celastrol alone
38
for 24 h reduced their viability up to about 43%. Pretreatment with either EGTA (a chelator or extracellular Ca2+) or BAPTA-AM (a chelator of free cytosolic Ca2+) did not appear to affect celastrol-induced cell death in these cells (Fig. 24), suggesting that the simple scavenging of extracellular or intracellular Ca2+ does not alter celastrol-induced paraptosis. Next, we examined the significance of the mitochondrial Ca2+ overload in celastrol-induced paraptosis. During curcumin-induced paraptosis, intracellular Ca2+ levels are increased by curcumin treatment and Ca2+ enters into mitochondria via mitochondrial Ca2+ uniporter, which is inhibited by ruthenium red (RR) (Yoon MJ et al., 2012). We found that pretreatment of MDA-MB 435S cells with RR very effectively and dose-dependently blocked celastrol-induced cell death (Fig. 24), suggesting that uniporter-mediated mitochondrial Ca2+ influx may play an important role in celastrol-induced cell death. Next, we investigated whether the release of Ca2+ from the ER via Ca2+ release channels contributes to celastrol-induced paraptosis. Next, we investigated whether the release of Ca2+ from the ER via Ca2+ release channels contributes to celastrol-induced paraptosis. Our experiments use the specific inhibitors of two major intracellular Ca2+ release receptors, IP3 receptor
(IP3R) (Ascher-Landsgerg J et al., 1999) and ryanodine receptor (RyR) (Marks AR,
1992). We found that celastrol-induced cell death was dose-dependently inhibited by 2-APB, a potent IP3R inhibitor (Powell JA et al., 2001), but not by dantrolene,
an inhibitor of ryanodine receptors (Zhao F et al., 2001) (Fig. 24). We next examined whether the death-blocking effect of RR or 2-APB is due to the
39
inhibition of celastrol-induced mitochondrial Ca2+ influx. We found that either pretreatment with RR or 2-APB inhibited celastrol-induced increase in [Ca2+]m
(Fig. 25), suggesting that their blocking effect on celastrol-induced cell death may be due to the inhibition of release of Ca2+ from the ER via IP3R and subsequent
mitochondrial Ca2+ influx via uniporter. Furthermore, both RR and 2-APB very effectively blocked the celastrol-induced dilation of not only mitochondria but also the ER in YFP-Mito and YFP-ER cells (Fig. 26). Moreover, both RR and 2-APB markedly inhibited celastol-induced accumulation of poly-ubiquitinated proteins and CHOP as well as activation of ERK and JNK (Fig. 27). Importance of IP3
R-mediated Ca2+ release from the ER was further confirmed by use of adenophostin A, an agonist of IP3R. We found that pretreatment of MDA-MB 435S cells with
adenophostin A further increased [Ca2+]m (Fig. 28) and accelerated the
celastrol-induced cell death (Fig. 29). Celastrol-celastrol-induced cell death was commonly inhibited by either 2-APB or RR in MCF-7, DLD-1 and RKO cells treated with celastrol (Fig. 30), suggesting that IP3R-mediated Ca2+ release from the ER and
uniporter-mediated mitochondrial Ca2+ influx are common critical signals in various cancer cells undergoing celastrol-induced paraptosis. Taken together, these results indicate that Ca2+ released from the ER via IP3R may influx into the mitochondria, resulting
in the mitochondrial dilation and subsequent paraptotic events in breast cancer cells.
40
Fig. 21. Celastrol significantly increases mitochondrial Ca2+ levels in MDA-MB
435S cells (FACS analysis). Cells treated with or without 2 μM celastrol for indicated
at time points were stained with 2.5 μM Fluo-3 or 2.5 μM Rhod-2, and processed for FACS analysis. Intensity of Fluo-3 or Rhod-2 fluorescence were assessed and denoted in the graph.
41
Fig. 22. Celastrol markedly increases mitochondrial Ca2+ levels in MDA-MB 435S
cells (Fluorescent microscopy). YFP-Mito cells treated with or without 2 μM celastrol
for 2 h were stained with 2.5 μM Rhod-2 and then observed using phase-contrast and fluorescence microscopy.
42
Fig. 23. Celastrol markedly increases mitochondrial Ca2+ levels in human cancer
cells (Fluorescent microscopy). YFP-Mito cells treated with or without 2 μM celastrol
were stained with 2.5 μM Rhod-2 and then observed using phase-contrast and fluorescence microscopy.
43
Fig. 24. Effects of various Ca2+ antagonists on celastrol-induced cell death.
MDA-MB 435S cells were pretreated with the indicated concentrations of various Ca2+ -associated inhibitors and further treated with or without 2 μM celastrol for 16 h. Cellular viability was measured using calcein-AM and EthD-1. Columns, mean of three independent experiments; bars, SE.
44
Fig. 25. Mitochondrial uniporter- or IP3R-mediated Ca2+ influxes are critical for
the celastrol-induced increase of mitochondrial Ca2+ in breast cancer cells.
MDA-MB 435S cells were pretreated with 5 μM ruthenium red (RR) or 20 μM 2-APB and further treated with 2 μM celastrol for 3 h. Cells were stained with Rhod-2 and processed for fluorescent microscopy. Representative images of cells are shown.
45
Fig. 26. Mitochondrial uniporter- or IP3R-mediated Ca2+ influxes are critical for
the celastrol-induced dilations of mitochondria/the ER and subsequent cell death in breast cancer cells. YFP-Mito and YFP-ER cells were pretreated with 5 μM
46
and then observed under fluorescent and phase-contrast microscopy.
Fig. 27. Mitochondrial Ca2+ overload acts as a signal for celastrol-induced
paraptosis of breast cancer cells. MDA-MB 435S cells were treated with 5 μM
ruthenium red (RR) or 20 μM 2-APB and/or 2 μM celastrol for 24 h. Whole cell extracts were prepared and subjected to western blotting using ubiquitin, CHOP, anti-phospho ERK (p-ERK), anti-ERK, anti-anti-phospho JNK (p-JNK), and anti-JNK antibodies.
47
Anti-β-actin antibody was used a loading control.
Fig. 28. Activation of IP3R employing adenophostin A accelerates
celastrol-increased mitochondrial Ca2+ levels. MDA-MB 435S cells were pretreated with 10
μM adenophostin A and further treated with 2 μM celastrol for 3 h. Cells were stained with Rhod-2 and processed for fluorescent microscopy. Representative images of cells are shown.
48
Fig. 29. Activation of IP3R employing adenophostin A accelerates celastrol-induced
cell death. MDA-MB 435S cells were pretreated with the indicated concentrations of
adenophostin A and further treated with or without 2 μM celastrol for 16 h. Cellular viability was measured using calcein-AM and EthD-1. Columns, mean of three independent experiments; bars, SE.
49
Fig. 30. Effects of RR or 2-APB on celastrol-induced cell death in human cancer cells. MCF-7, DLD-1, and RKO cells were pretreated with the indicated concentrations
of RR or 2-APB and further treated with or without 2 μM celastrol for 16 h. Cellular viability was measured using calcein-AM and EthD-1. Columns, mean of three independent experiments; bars, SE.
50
5. ROS generation is also important for celastrol-induced paraptosis
Previously, celastrol was shown to induce ROS accumulation in tumor cells, contributing to its cytotoxicity (Chen G et al., 2011) and we recently reported that ROS, particularly mitochondrial superoxide, play a critical role in paraptosis (Yoon MJ et al., 2010). Thus, we tested whether ROS levels are altered during celastrol-induced cell death in MDA-MB 435S cells. Assessment of ROS levels using CM-H2DCF-DA, an
intracellular ROS-indicator dye, showed that celastrol increased cellular ROS levels with a peak at 4 h (Fig. 31). Next, we tested whether intracellular superoxide levels were increased during celastrol-induced paraptosis in breast cancer cells. Staining with MitoSOX-red, a fluorescent probe targeted to the mitochondria and highly selective to oxidation by superoxide (Lieven CJ et al., 2006), demonstrated that mitochondrial superoxide levels with a peak at 4 h in celastrol-treated MDA-MB 435S cells (Fig. 32). Pretreatment with NAC or GSH dose-dependently blocked the celastrol-induced cell death (Fig. 33). Furthermore, pretreatment with NAC effectively inhibited the dilation of mitochondria and the ER in YFP-Mito and YFP-ER cells (Fig. 34). These results suggest that ROS generation critically contribute to the celastrol-induced paraptosis. Next, we further investigated the relationship between mitochondrial Ca2+ and ROS in celastrol-induced paraptosis. We found that pretreatment with either 2-APB or RR also very effectively inhibited the celastrol-induced increase in ROS, similar to the effect of
51
NAC pretreatment (Fig. 35). Very interestingly, pretreatment with NAC also remarkably inhibited celastrol-induced increase in [Ca2+]m, similar to the effect of
2-APB or RR pretreatment (Fig. 36). These results indicated that there exists cross-modulation between mitochondrial Ca2+ influx and ROS generation in celastrol-induced paraptosis. When we further examined the effect of NAC pretreatment on celastrol-induced other paraptotic signals, NAC pretreatment almost completely inhibited celastrol-induced accumulation of poly-ubiquitinated proteins and CHOP, and activation of ERK and JNK (Fig. 37). Taken together, these results suggest that both mitochondrial Ca2+ and ROS act as very initial critical signals in celastrol-induced paraptosis. Next, we investigated whether mitochondrial Ca2+ influx and ROS generation commonly play critical roles in other types of cancer cells undergoing celastrol-induced cell death. We found that pretreatment with NAC significantly and dose-dependently inhibited celastrol-induced cell death in MCF-7, DLD-1, and RKO cells (Fig. 38). Collectively, our results suggest that mitochondrial Ca2+ influx and ROS generation commonly trigger paraptosis in cancer cells undergoing celastrol-induced paraptosis.
52
Fig. 31. Generation of ROS by celastrol in breast cancer cells. MDA-MB 435S cells
were treated with 2 μM celastrol for indicated at time points, exposed to 5 μM CM-H2DCF-DA for 30 min and analyzed by flow cytometry. X axis, fluorescence intensity,
Y axis, relative number of cells. Intensity of CM-H2DCF-DA fluorescence was assessed
53
Fig. 32. Celastrol significantly increases mitochondrial superoxide levels in breast cancer cells. MDA-MB 435S cells were treated with 2 μM celastrol for indicated time
points, exposed to 2.5 μM MitoSOX-red for 20 min and analyzed by flow cytometry. X axis, fluorescence intensity, Y axis, relative number of cells. Intensity of MitoSOX-red fluorescence was assessed and denoted in the graph.
54
Fig. 33. Effects of antioxidants on celastrol-induced cell death. MDA-MB 435S cells
were pretreated with the indicated concentrations of antioxidants (NAC and GSH) for 30 min and further treated with 2 μM celastrol for 16 h. Cellular viability was assessed using calcein-AM and EthD-1. Columns, mean of three independent experiments; bars, SE.
55
Fig. 34. Antioxidant blocks the celastrol-induced vacuolation of mitochondria/the ER in breast cancer cells. YFP-Mito and YFP-ER cells were pretreated with 2 mM
NAC and further treated with 2 μM celastrol for 3 h, and then observed under fluorescent and phase-contrast microscopy.
56
Fig. 35. Effects of RR or 2-APB on celastrol-generated ROS. MDA-MB 435S cells
were pretreated with 1 mM NAC, 20 μM 2-APB, or 2 μM RR and further treated with 2 μM celastrol for indicated concentration, exposed to 5 μM CM-H2DCF-DA for 30 min
and analyzed by flow cytometry. X axis, fluorescence intensity, Y axis, relative number of cells. Intensity of CM-H2DCF-DA was assessed and denoted in the graph.
57
Fig. 36. Effects of NAC on celastrol-induced mitochondrial Ca2+ levels. MDA-MB
435S cells were pretreated with 1 mM NAC, 20 μM 2-APB, or 2 μM RR and further treated with 2 μM celastrol for indicated concentration, exposed to 2.5 μM Rhod-2 and analyzed by flow cytometry. X axis, fluorescence intensity, Y axis, relative number of cells. Intensity of Rhod-2 was assessed and denoted in the graph.
58
Fig. 37. NAC blocks the celastrol-induced paraptosis signals in breast cancer cells.
MDA-MB 435S cells were treated with 2 mM NAC and/or 2 μM celastrol for 24 h. Whole cell extracts were prepared and subjected to western blotting using anti-ubiquitin,
59
anti-CHOP, anti-phospho ERK (p-ERK), anti-ERK, anti-phospho JNK (p-JNK), and anti-JNK antibodies. Anti-β-actin antibody was used as a loading control.
Fig. 38. Effects of NAC on celastrol-induced cell death in human cancer cells.
MCF-7, DLD-1, and RKO cells were pretreated with the indicated concentrations of NAC for 30 min and further treated with 2 μM celastrol for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. Columns, mean of three independent experiments; bars, SE.
60
IV. DISCUSSION
Despite recent improvements in anti-cancer therapies against cancer, inherent or acquired cellular resistance to various pro-apoptotic treatments often leads to therapeutic failure (Moulder S, 2010). Thus, development of novel therapeutic strategy to induce alternative cell death modes may be helpful to overcome resistance of cancer cells to conventional cancer therapy. For this purpose, understanding of the molecular basis of non-apoptotic cell death is essential.
Celastrol is an agent derived from Tripterygium wilfordii and it has been used for autoimmune diseases, chronic inflammation, asthma and neurodegenerative disease for years (Allison AC et al., 2001; Canter PH et al., 2006; Brinker AM et al., 2007). It also inhibits proliferation of cancer cells by inhibiting NF-κB activity and by inducing apoptosis (Kim Y et al., 2011). Moreover, recent paper gives additional evidence that celastrol could also serve as a drug in cancer therapy (Salminen A et al., 2010).
The term “paraptosis” was originally introduced to describe a form of programmed cell death found to be morphologically and biochemically distinct from apoptosis (Sperandio S et al., 2004). Previously, paraptosis was defined mainly according to the morphological criteria, including extensive cytoplasmic vacuolation resulting from progressive swelling of mitochondria and endoplasmic reticulum (Yoon MJ et al., 2010). Paraptosis is not prevented by caspase inhibitors or overexpression of anti-apoptotic
61
Bcl-2-like proteins (Sperandio S et al., 2004; Valamanesh F et al., 2007) and it activates specific members of the mitogen-activated protein kinase family, including MEK-2 and JNK-1 (Sperandio S et al., 2004). However, the regulatory mechanisms that control paraptotic events, particularly the dilation of mitochondria and the ER, are not yet fully understood. Here, we show for the first time that the paraptosis-inducing activity of celastrol contributes to its cytotoxicity against human cancer cells. Typical apoptotic characteristics such as nuclear fragmentation and apoptotic bodies are not observed in celastrol-treated breast cancer cells. In addition, celastrol-induced cell death is not blocked by pan-caspase inhibitors, although caspase-3 is minimally cleaved by celastrol treatment. These results suggest that apoptosis is not a major death in breast cancer cells treated with celastrol. Instead, celastrol induces vacuolation prior to cell death in breast cancer cells and this vacuolation is resulted from the swelling and subsequent fusion of mitochondria or the ER, not from autophagy. As a result, cells at late phase of celastrol treatment contain small numbers of megamitochondria and a substantially expanded ER. These results suggest that these irrecoverable physical changes in mitochondria and the ER leading to their functional loss may be responsible for irreversible cell death. In addition, paraptotic characteristics, such as a requirement for protein synthesis (Sperandio S et al., 2000), negative involvement of AIP/Alix (Sperandio S et al., 2004; Valamanesh F et al., 2007) are commonly observed in MDA-MB 435S cells treated with celastrol. Previously, Sperandio et al. showed that IGFIR-induced paraptosis is mediated by the MEK-2 and JNK (Jun N-terminal kinase)-1 (Sperandio S et al., 2004). In addition, ERK and/or JNK were shown to be associated with paraptosis induced by
62
yessotoxin (Korsenes MS et al., 2011), taxol (Sun Q et al., 2010), and glucocorticoid (Valamanesh F et al., 2007). Insulin-like growth factor I receptor (IGFIR)-induced paraptosis was effectively blocked by inhibition of JNK pathway (Sperandio S et al., 2004), consistent with our results. However, toxol-induced paraptosis in lung carcinoma cells was not inhibited by inhibition of ERK pathway, similar to our results. These results suggest that the dependency of paraptosis on JNK pathway may be important than that of ERK. Or the importance of ERK or JNK in paraptosis may be different depending on the stimuli.
Our results show that 2-APB, inhibitor of IP3R (Brann JH and Fadool DA, 2006),
blocked celastrol-induced vacuolation and subsequent cell death, inhibiting various paraptotic signals, including phosphorylated ERK, phosphorylated JNK, CHOP, and poly-ubiquitinated proteins. Neither Ca2+ chelators (BAPTA-AM and EGTA) nor RyR inhibitor (dantrolene) blocked celastrol-induced vacuolation and cell death. Previously, we have shown that curcumin-induced paraptosis was blocked by ryanodine and ruthenium red, but not by 2-APB (Yoon MJ et al., 2010). These results suggest that Ca2+ release and from the ER and the uniporter-mediated influx are critical for the paraptotic cell death, although either ryanodine receptor or IP3 receptor may be
activated depending on the paraptotic stimuli. Taken together, mitochondrial Ca2+ uniporter and IP3R have a critical role of mitochondrial Ca2+ overload in
celastrol-induced paraptosis. These results suggest that the release from the ER and mitochondrial influx of Ca2+ act initial signal for celastrol-induced paraptosis.
63
Reactive oxygen species (ROS) play an important role in physiological cellular functions by activating several enzymatic cascades and transcription factors (Droge W, 2002). Excessive ROS signals, however, are detrimental, causing Ca2+ overload, mitochondrial depolarization, lipid peroxidation, transcription factor activation and DNA damage, and lead to apoptotic and/or non-apoptotic cell death (Yan Y et al., 2006). Previously, generation of reactive oxygen species (ROS) was shown to be critically involved in celastrol-induced apoptosis (Lee JH et al., 2012). In our study, celastrol induced the generation of intracellular ROS (CM-H2DCF-DA) and mitochondrial
superoxide (MitoSOX-red). Scavenging of ROS by antioxidants (NAC and GSH) blocked not only the vacuolation but also cell death induced by celastrol. In addition, NAC almost completely inhibited celastrol-induced other paraptotic signals, including phosphorylated ERK, phosphorylated JNK, CHOP, and poly-ubiquitinated proteins. Very interestingly, NAC very remarkably inhibited celastrol-increased mitochondrial Ca2+ levels ([Ca2+]m). Also, we found that inhibition of mitochondrial Ca2+ influx using
ruthenium red very effectively blocked celastrol-increased ROS production. Taken together, these results suggest that cross-modulation between mitochondrial Ca2+ overload and ROS generation exists and they act as critical initial signals for celastrol-induced paraptosis.
In summary, our results show that mitochondrial Ca2+ overload and ROS generation by celastrol critically contribute to the dilation of both mitochondria and the ER, leading to paraptosis. These novel findings not only improve the understanding of the mechanistic basis of anti-cancer effects of celastrol via induction of paraptosis but also
64
offer a potential therapeutic strategy for breast cancer, which is resistant to pro-apoptotic therapeutics.
V. REFERENCES
1. Al-Ejeh F, Shi W, Miranda M, Simpson PT, Vargas AC, Song S, Wiegmans AP, Swarbrick A, Welm AL, Brown MP, Chenevix-Trench G, Lakhani SR, Khanna KK. “Treatment of Triple-Negative Breast Cancer Using Anti-EGFR Directed Radioimmunotherapy Combined with Radiosensitizing Chemotherapy and PARP Inhibitor.” J Nucl Med. [E-pub ahead of print], 2013.
2. Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. “Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease.” Prog NeuropsychopharmacolBiol Phsychiatry. 25:1341-1357, 2001.
3. Amador FJ, Stathopulos PB, Enomoto M, Ikura M. “Ryanodine receptor calcium release channels: lessons from structure-function studies.” FEBS J. [E-pub ahead of print], 2013.
4. Ascher-Landsberg J, Saunders T, Elovitz M, Phillippe M. “The effects of 2-aminoethoxydiphenyl borate, a novel inositol 1,4, 5-trisphosphate receptor modulator on myometrial contractions.” Biochem Biophys Res Commun. 264:979-982.
65
5. Bazhenova EN, Saris NE, Zvyaqilskaya RA. “Stimulation of the yeast mitochondrial calcium uniporter by hypotonicity and by ruthenium red.”
Biochim Biophys Acta. 1371:96-100, 1998.
6. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. “Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response.”
Nat Cell Biol. 2:326-332, 2000.
7. Biaqioli M, Prifferi S, Raqqhianti M, Bucci S, Rizzuto R, Pinton P. “Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis.” Cell Calcium. 43:184-195, 2008. 8. Bodalia A, Li H, Jackson MF. “Loss of endoplasmic reticulum Ca2+ homeostasis:
contribution to neuronal cell death during cerebral ischemia.” Acta Pharmacol
Sin. 34:49-59, 2013.
9. Brinker AM, Ma J, Lipsky PE, Raskin I. “Medicinal chemistry and pharmacology of genus Triptertygium (Celastraceae).” Phytochemistry 68:732-766, 2007.
10. Brini M, Carafoli E. “Calcium pumps in health and disease.” Physiol Rev. 89:1341-1378, 2009.
11. Bröker LE, Kruyt FA, Giaccone G. “Cell death independent of caspases: a review.” Clin Cancer Res. 11:3155-3162, 2005.
66
12. Canter PH, Lee HS, Ernst E. “A systematic review of randomized clinical trials of Tripterygium wilfordii for rheumatoid arthritis.” Phytomedicine 13:371-377, 2006.
13. Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. “Importing mitochondrial proteins: machineries and mechanisms.” Cell. 138:628-644, 2009. 14. Chadalapaka G, Jutooru I, Safe S. “Celastrol decreases specificity protein (Sp)
and fibroblast growth receptor-3 (FGFR3) in bladder cancer cells.”
Carcinogenesis. 33:886-894, 2012.
15. Chen G, Zhang X, Zhao M, Wang Y, Cheng X, Wang D, Xu Y, Du Z, Yu X. “Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent cytotoxicity in tumor cells.” BMC Cancer. 11:170, 2011.
16. Clarke M, Collins R, Darby S, Davies C, Elphinstone P, Evans E, Godwin J, Gray R, Hicks C, James S, MacKinnon E, McGale P, McHugh T, Peto R, Taylor C, Wang Y; Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). “Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials.” Lancet. 366:2087-2106, 2005.
17. Crawford KW, Bowen WD. “Sigma-2 receptor agonists activate a novel apoptotic pathway and potentiate antineoplastic drugs in breast tumor cell lines.”